

Высокомолекулярные соединения (серия А), 2022, T. 64, № 1, стр. 61-71
ТЕРМОДИНАМИЧЕСКАЯ СОВМЕСТИМОСТЬ ПОЛИАКРИЛАМИДА С АГАРОЗОЙ. УЧЕТ ВЛИЯНИЯ ЖЕСТКОСТИ ЦЕПИ ПОЛИСАХАРИДА
А. П. Сафронов a, b, Т. В. Терзиян a, А. Манас Кызы c, *, Л. В. Адамова a
a Уральский федеральный университет
620002 Екатеринбург, ул. Мира, 19, Россия
b Институт электрофизики Уральского отделения Российской академии наук
620016 Екатеринбург, ул. Амундсена, 106, Россия
c УНИИМ – филиал ФГУП “ВНИИМ им. Д.И. Менделеева”
620075 Екатеринбург, ул. Красноармейская, 4, Россия
* E-mail: aiganysh9@gmail.com
Поступила в редакцию 10.06.2021
После доработки 22.10.2021
Принята к публикации 09.11.2021
- EDN: VJUQHC
- DOI: 10.31857/S2308112022010072
Аннотация
Значения энтальпии, энергии Гиббса и энтропии смешения полиакриламида с агарозой определены с использованием термодинамических циклов на основе данных микрокалориметрии и изотермической равновесной сорбции. Измерены величины энтальпии растворения в воде и получены изотермы сорбции воды для пленок полиакриламида, агарозы и их смесей в разном соотношении. Для обработки сорбционных данных использована модель сорбции, учитывающая влияние жесткости цепи полисахарида на изменение химического потенциала воды при образовании раствора в процессе сорбции. Показано хорошее соответствие модельных зависимостей экспериментальным изотермам во всей области изменения объемной доли полимера и относительного давления пара. Установлено, что энтальпия смешения полиакриламида с агарозой отрицательна во всем диапазоне составов. Смеси, содержащие менее 50 мас. % агарозы, являются термодинамически несовместимыми и характеризуются положительными значениями энергии Гиббса смешения, в то время как для смесей, содержащих более 50% агарозы, энергии Гиббса отрицательны. Расчетные значения энтропии смешения отрицательны во всем диапазоне соотношений агарозы и полиакриламида, что свидетельствует об упорядочении в данной полимерной смеси. Результаты исследования термодинамической совместимости полиакриламида и агарозы сопоставлены с закономерностями набухания и механическими свойствами гидрогелей на основе полувзаимопроникающих сеток на основе этих полимеров.
ВВЕДЕНИЕ
Полиакриламид (ПАА) представляет собой синтетический полимер акриламида, широко используемый в технологиях получения гидрогелей биомедицинского назначения. Трехмерная ковалентная сетка ПАА обеспечивает эластичность, гидрофильность и отклик материала на изменение внешних условий. Есть два основных недостатка в биомедицинском применении таких гелей: низкая механическая прочность и плохая биосовместимость полимера. Одним из способов преодоления указанных недостатков стало использование природных полимеров как компонентов гелей на основе ПАА [1–4]. В этом случае набухшая в воде химически сшитая сетка ПАА содержит линейные цепи биополимера, которые удерживаются в объеме материала за счет сетки физических связей. Таким образом, в геле сочетаются две взаимопроникающие сетки на основе двух полимеров различной химической природы. Гели со структурой двойной сетки “double network hydrogels – DN hydrogels”, содержащие природные полимеры, активно исследуются [5, 6] как перспективные материалы для создания биосенсоров, систем доставки лекарств, тканевой инженерии и т.п.
Механизм повышения прочности DN-гелей [5, 7] обусловлен сочетанием двух сеток, где прочность связей, обеспечивающих сшивку макромолекул, значительно различается. Химически сшитая сетка синтетического полимера сосуществует с сеткой слабых физических связей биополимера, которые могут обратимо разрываться под действием нагрузки, обеспечивая возможность диссипации механической энергии и сохраняя целостность ковалентной сетки. Природные полимеры, в частности полисахариды, содержат большое количество гидроксильных групп, которые обеспечивают формирование физической сетки водородных связей. Кроме этого, усиливающее действие двойной сетки связывают с межмолекулярным взаимодействием полимерных сеток друг с другом [8], где макромолекулы полужесткоцепного полисахарида оказывают армирующее действие на гель. В этой связи одним из ключевых вопросов, возникающих при изучении механических характеристик DN-гелей, становится вопрос оценки энергетики образования межмолекулярных связей между компонентами, формирующими двойную сетку геля.
Изучение изменения термодинамических функций – энтальпии ΔH, энтропии ΔS и энергии Гиббса ΔG процессов, реализуемых в системе, является фундаментальным подходом, позволяющим получить их количественные параметры, в том числе и для процесса межмолекулярного взаимодействия. Для определения ΔH и ΔG в полимерсодержащих системах разработан и хорошо апробирован подход с использованием термодинамических циклов [9, 10]. Экспериментальное определение изменения энтальпии ΔH основано на проведении непосредственного калориметрического измерения теплового эффекта растворения полимерсодержащей системы. Изменение энергии Гиббса ΔG рассчитывается из данных сорбционных измерений с использованием уравнений теории Флори–Хаггинса, описывающей термодинамику растворов гибкоцепных полимеров. Известно, что полисахариды являются полужесткоцепными полимерами, что ограничивает применение теории Флори–Хаггинса в общеизвестном виде. В работах [11, 12] описана одна из немногочисленных попыток учета конформационного поведения макромолекул в термодинамике растворов. Представляет интерес использование предложенного подхода для модификации уравнений теории Флори–Хаггинса и их применения для описания систем, содержащих полисахариды.
Цель настоящей работы – изучение термодинамических параметров взаимодействия компонентов двойных сеток на основе ПАА и агарозы. Агароза – полисахарид, получаемый экстракцией из красных водорослей. Макромолекулярная цепь агарозы имеет линейное строение и состоит из чередующихся остатков D-галактозы и 3,6-ангидро-L-лактозы, соединенных попеременно β(1 → 4)- и α(1 → 3)-связями [13]. Смеси агарозы с полимерами и сополимерами акрилового ряда, в частности с ПАА, широко используются для разработки материалов различного назначения. Так, описывается применение гибридных биосорбентов для очистки воды [14], компонентов регулирования вязкости косметических и фармацевтических препаратов [15], материалов с памятью формы [16] и гидрогелей различного назначения [2].
Термодинамические функции смешения (ΔH, ΔS, ΔG) ПАА с агарозой были определены в пленках на основе смеси линейных полимеров. Такие системы удобны для экспериментального изучения с применением метода термодинамических циклов. Полученные термодинамические параметры взаимодействия были сопоставлены с данными о степени набухания и результатами механических испытаний DN-гидрогелей, в которых сочетались химически сшитая сетка ПАА и физическая сетка агарозы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ПАА синтезировали методом радикальной полимеризации в растворе. Использовали агарозу марки Agarose hohe-EEO-high производства SERVA (“Feinbiochemica”, Heidelberg, Germany). Молекулярную массу ПАА (2.19 × 105) и агарозы (2.7 × 104) определяли вискозиметрическим методом с использованием констант Марка–Хаувинка: ПАА – К = 0.63 , a = 0.80, вода [17], агароза – K = 7.0 × 10–4, a = 0.72, 0.75 М водный раствор KSCN, 35° [18].
Образцы для сорбционных и калориметрических исследований готовили в виде пленок толщиной 25–30 мкм из 1%-х водных растворов при температуре 95°С, сушили при температуре 70°С на воздухе, затем при остаточном давлении 103 Па при 25°С до постоянной массы.
Сорбцию паров воды ПАА, агарозой и их смесями при 25°С изучали объемным методом на автоматическом анализаторе площади поверхности и пористости ASAP 2020 (“Micromeritics”) c приставкой для сорбции паров. Принцип метода заключается в определении количества сорбированного вещества по уменьшению давления пара сорбата, находящегося в парообразной фазе и занимающего определенный заранее прокалиброванный объем.
Энтальпии растворения полимеров в воде определяли методом калориметрии с использованием микрокалориметра типа Тиана–Кальве (ДАК 1-1) с изотермической оболочкой, термостатированной при 298 ± 0.1 К [19]. В ходе эксперимента в тонкостенную стеклянную калориметрическую ампулу помещали 0.04–0.08 г пленочного образца, ампулу запаивали и опускали в ячейку калориметра, заполненную водой (~5 мл). В процессе калориметрического опыта ампулу разбивали в водной среде и регистрировали термоЭДС, вызываемую тепловым потоком. Энтальпию процесса растворения рассчитывали с учетом константы калориметра, устанавливающей взаимосвязь между мощностью теплового потока и термоЭДС. Абсолютная погрешность калориметрических измерений, по данным электрической калибровки прибора, составляла ±0.02 Дж.
Гели синтезировали реакцией свободнорадикальной полимеризации в водном растворе акриламида в присутствии агарозы и сшивающего агента – метилендиакриламида. Использовали акриламид производства “Sigma-Aldrich” (США). Концентрация мономера в реакционной смеси составляла 1.6 моль/л, метилендиакриламид добавляли в количестве, обеспечивающем 1 сшивку на 300 мономерных звеньев сетки полиакриламида. Концентрация агарозы в реакционной смеси составляла 0.5, 1, 2, 3 и 4 мас. %, что соответствовало 4.0, 7.8, 14.4, 20.2 и 25.2% полисахарида в смеси с ПАА. В качестве инициатора радикальной полимеризации использовали 0.1 М раствор пероксодисульфата аммония. Для синтеза гелей предварительно готовили водный раствор агарозы при температуре 95°С в полиэтиленовой реакционной форме, куда добавляли все необходимые компоненты синтеза и оставляли при повышенной температуре на 60 мин. Готовые гели вынимали из формы и помещали в дистиллированную воду для реализации процесса набухания. Механическим испытаниям подвергались равновесно набухшие гели.
Для определения модуля Юнга гелей были получены деформационные кривые сжатия в водной среде. Использовали образцы цилиндрической формы диаметром и высотой около 10 мм. Нагружение проводили в квазистатическом режиме с оптической регистрацией деформации. Диапазон рабочих напряжений составил от 0 до 6 кПа.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Следует отметить, что термодинамические функции смешения полимеров зависят от их предыстории. В настоящей работе были исследованы смеси ПАА и агарозы, полученные испарением водных растворов при температуре 95°С. В таких условиях макромолекулы агарозы находятся в конформации статистического клубка [20]. В связи с этим все термодинамические функции, представленные далее, относятся к взаимодействию ПАА с агарозой в конформациях неупорядоченных клубков.
На первом этапе оценивали энергетику взаимодействия ПАА с агарозой, которая определяется величиной и знаком энтальпии их смешения в зависимости от соотношения полимеров. Энтальпию смешения полимерной смеси нельзя измерить непосредственно. Для ее расчета использовали термохимический цикл, составленный из следующих процессов [21]:
1) ПАА + агароза = смесь + ΔHm;
2) ПАА + вода (избыток) = раствор ПАА + ΔH1;
3) агароза + вода (избыток) = раствор агарозы + ΔH2;
4) смесь + вода (избыток) = раствор смеси + + ΔH3;
5) раствор ПАА + раствор агарозы = раствор смеси + ΔH*.
В данном цикле стадии 2–5 были проведены в калориметрической ячейке, и величины ΔH1, ΔH2, ΔH3, ΔH* являлись непосредственно измеренными. Полимерная смесь, растворение которой проводится на стадии 4, была приготовлена отдельно. Величина ΔH*, измеренная на стадии 5, принимала значения в пределах экспериментальной погрешности, и при дальнейших расчетах ею пренебрегали. Энтальпию смешения системы ПАА–агароза рассчитывали по уравнению
(1)
$\Delta H = {{\omega }_{1}}\Delta {{H}_{1}} + {{\omega }_{2}}{{H}_{2}} - \Delta {{H}_{3}} + \Delta H{\kern 1pt} *,$Ключевая величина в уравнении (1), которая определяет вид концентрационной зависимости энтальпии смешения полимерной смеси ПАА–агароза, – энтальпия растворения полимерной смеси ΔH3. Эта величина является функцией соотношения ПАА и агарозы в смеси. Ее концентрационная зависимость приведена на рис. 1 (кривая 1).
Рис. 1.
Зависимость энтальпии растворения в воде полимерной смеси ПАА–агароза (ΔH3, кривая 1) и энтальпии смешения ПАА с агарозой (ΔHm, кривая 2) от весовой доли агарозы в смеси. Температура 25°С. Линии проведены лишь как визуальный ориентир. Пунктиром обозначена аддитивная сумма энтальпии растворения ПАА и агарозы при их разном соотношении. Размер точек соответствует погрешности определения энтальпии растворения и смешения: ± 2 Дж/г.
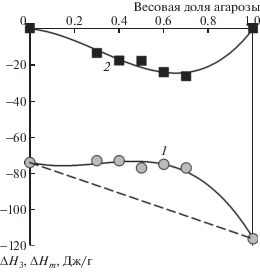
Точки на осях концентрационной зависимости ΔH3 соответствуют следующим величинам: энтальпии растворения ПАА в воде ΔH1 и энтальпии растворения агарозы в воде ΔH2 . Все величины (ΔH1, ΔH2 и ΔH3, принимают отрицательные значения. Энтальпия растворения агарозы в воде более отрицательна, чем энтальпия растворения ПАА. Энтальпии растворения смесей занимают промежуточное положение, причем точки, соответствующие их значениям, лежат выше штриховой линии, обозначающей аддитивную сумму энтальпий растворения ПАА и агарозы при отсутствии взаимодействия между ними.
Значения энтальпии смешения ПАА с агарозой, рассчитанные по уравнению (1), также представлены на рис. 1 (кривая 2). Видно, что во всем диапазоне соотношений ПАА и агарозы энтальпия их смешения отрицательна, что указывает на энергетически сильное взаимодействие их звеньев. Минимум на концентрационной зависимости ΔHm приходится на весовую долю агарозы около 70%. Учитывая разницу в молярных массах звена ПАА (71) и агарозы (306), это отвечает примерному соотношению 2 звена ПАА на 1 звено агарозы. Величина энтальпии смешения при таком соотношении (минимум кривой) составляет около –25 Дж/г, или –6.33 кДж/моль звеньев смеси данного состава. Данная величина свидетельствует об образовании водородных связей между звеньями ПАА и агарозы в их смеси. Можно предположить, что водородные связи формируются при взаимодействии амидных групп звеньев ПАА и гидроксильных групп звеньев агарозы. В составе полимерной цепи агарозы чередуются звенья, содержащие три гидроксильные группы, и звенья, содержащие одну гидроксильную группу. В среднем на два звена агарозы приходится четыре гидроксильные группы. Эти группы могут потенциально образовать водородные связи с четырьмя звеньями ПАА, что и обеспечит соотношение звеньев ПАА : агароза = 2 : 1.
Обратимся теперь к зависимости энергии Гиббса смешения ПАА с агарозой, которая определяет термодинамическое сродство данной полимерной смеси. Энергия Гиббса смешения полимеров рассчитывается по циклу, который аналогичен представленному выше для расчета энтальпии смешения, но вместо величин энтальпии растворения в него входят соответствующие величины энергии Гиббса. В отличие от величин энтальпии растворения ПАА, агарозы и их смесей в воде значения энергии Гиббса не измеряются непосредственно, а рассчитываются на основании изотерм сорбции паров воды для этих систем. Остановимся подробнее на анализе изотерм сорбции паров воды образцами ПАА, агарозы и их смесей, что имеет критически важное значение для расчета энергии Гиббса смешения.
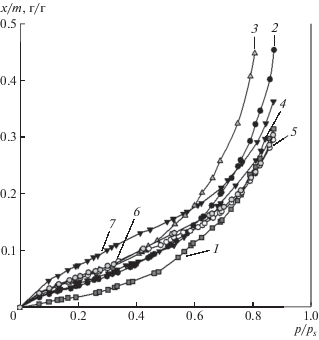
На рис. 2 представлены изотермы сорбции паров воды пленками ПАА, агарозы и их смесей разного состава в координатах: относительное давление паров воды p/ps – масса воды, сорбированная 1 г полимерной пленки (x/m). Видно, что все изотермы имеют вид S-образных кривых с небольшим выпуклым начальным участком в области малых относительных давлений и протяженным вогнутым участком в широком диапазоне более высоких значений относительного давления. Изотермы имеют тенденцию к неограниченному возрастанию при стремлении относительного давления к насыщению. Изотерма для ПАА расположена ниже всех остальных, т.е. сорбция воды ПАА наименьшая в исследованном ряду. Вплоть до относительного давления, равного 0.6, выше других расположена изотерма для агарозы, т.е. вплоть до указанного значения относительного давления сорбция воды на агарозе наибольшая в исследованном ряду. Изотермы для смесей ПАА–агароза занимают промежуточное положение. Для некоторых смесей при относительном давлении выше 0.7 сорбция существенно возрастает и превышает сорбцию на агарозе.
Рис. 2.
Экспериментальные изотермы сорбции паров воды пленками ПАА (1), агарозы (7) и их смесей с соотношением компонентов ПАА : агароза = 70 : 30 (2), 60 : 40 (3), 50 : 50 (4), 40 : 60 (5) и 30 : 70 (6). Температура 25°С. Размер точек соответствует погрешности измерения количества адсорбированной воды ±0.005 г/г.
Хорошо известно, что форма изотермы сорбции паров на образце полимера определяется фундаментальной связью относительного давления паров и химического потенциала растворителя в составе раствора, образующегося при сорбции:
где φ – объемная доля полимера в образующемся в результате сорбции растворе.Для обработки полученных изотерм сорбции мы использовали термодинамическую модель, предложенную в наших предыдущих работах [22–24], которая учитывает стеклообразное состояние полимера при температуре опыта. Применение данной модели связано с тем, что и ПАА, и агароза при 25°С находятся в стеклообразном состоянии. В этом случае изменение химического потенциала растворителя может быть представлено как сумма следующих вкладов:
(3)
$\Delta {{\mu }_{1}} = \Delta {{\mu }_{{1,mix}}} + \Delta {{\mu }_{{1,glass}}} + \Delta {{\mu }_{{1,contr}}}$Здесь $\Delta {{\mu }_{{1,mix}}}$, $\Delta {{\mu }_{{1,glass}}}$ и $\Delta {{\mu }_{{1,contr}}}$ – вклады, обусловленные смешением компонентов при образовании раствора полимера; релаксацией неравновесной стеклообразной структуры полимера в результате адсорбции; сжатием системы при заполнении молекулами растворителя неравновесных вакансий в стеклообразной структуре полимера соответственно.
Вклады $\Delta {{\mu }_{{1,glass}}}$ и $\Delta {{\mu }_{{1,contr}}}$ обусловлены неравновесной рыхлой упаковкой стеклообразной структуры полимера, и для их описания мы использовали следующие уравнения [22, 23]:
(4)
$\Delta {{\mu }_{{1,glass}}} = RT{{g}_{{ads}}}(1 - \varphi _{V}^{0}){{\varphi }^{{\frac{1}{{\varphi _{V}^{0}}} - 1}}}$(5)
$\Delta {{\mu }_{{1,contr}}} = - RT\ln (1 - \varphi _{V}^{{\text{0}}}{{\varphi }^{{\frac{1}{{\varphi _{V}^{0}}} - 2}}}),$Обычно вклад $\Delta {{\mu }_{{1,mix}}}$ в уравнении (3) рассматривается в форме классического выражения теории Флори–Хаггинса [25]. Однако при интерполяции экспериментальных изотерм модельным уравнением (3), включающем классическое выражение для $\Delta {{\mu }_{{1,mix}}}$, наблюдалось существенное отклонение экспериментальных точек от модельной кривой при высоких относительных давлениях пара. В то же время в области малых и средних значений давления пара отклонений экспериментальных точек от расчетной изотермы не было. При расчете энергии Гиббса смешения полимерной смеси значения сорбции при высоких относительных давлениях пара существенным образом сказываются при интегрировании изотермы по уравнению Гиббса–Дюгема для расчета химического потенциала полимера (см. ниже). Следовательно, интерполяция экспериментальных значений должна быть проведена с максимальной точностью.
В связи с этим для вклада $\Delta {{\mu }_{{1,mix}}}$ в уравнение (3) мы использовали имеющийся в литературе подход [11, 12, 26], предложенный для смесей, молекулы компонентов которых имеют различную конформационную жесткость. В рамках такого подхода при расчете комбинаторной энтропии смешения растворов компонентов, молекулы которых существенно отличаются своими размерами, выделяют два предельных случая. Для гибких молекул (полимеров), молекулы которых можно разбить на кинетически независимые сегменты, энтропия смешения описывается уравнением Флори–Хаггинса. Для абсолютно жестких молекул, не изменяющих свою конформацию, энтропия смешения описывается теорией идеального раствора. В промежуточных случаях энтропия смешения принимает некоторые средние значения. В литературе имеются примеры [11, 12] универсальных моделей, которые включают в себя оба указанных выше предельных случая.
Следует отметить, что имеются формальные основания для того, чтобы рассматривать смеси ПАА–агароза с этих позиций. Полиакриламид является термодинамически гибкоцепным полимером, обладающим небольшими значениями сегмента Куна около 2 нм. В то же время агароза, как любой полисахарид, – полужесткоцепной полимер, сегмент Куна которого по разным оценкам составляет 6–15 нм.
Взяв за основу идею о комбинировании двух различных статистических подходов к описанию комбинаторной энтропии в растворах молекул разного размера, мы предлагаем упрощенную эмпирическую модель, в рамках которой комбинаторная энтропия представляет собой аддитивную сумму энтропийного слагаемого для гибкоцепного полимера по теории Флори–Хаггинса и энтропийного слагаемого для идеального раствора, которые берутся с весовыми коэффициентами, отражающими их относительный вклад в общее изменение энтропии.
В рамках этого предположения для вклада$\Delta {{\mu }_{{1,mix}}}$ мы использовали следующее уравнение:
(6)
$\begin{gathered} \Delta {{\mu }_{{1,mix}}} = RT\left[ {\lambda \left( {\ln (1 - \varphi ) + \left( {1 - \frac{1}{p}} \right)\varphi } \right) + } \right. \\ \left. {_{{_{{_{{_{{_{{_{{_{{}}}}}}}}}}}}}} + \,(1 - \lambda )\ln (1 - x) + \chi {{\varphi }^{2}}} \right] \\ \end{gathered} $Здесь χ – параметр Флори–Хаггинса, λ – относительный вклад “гибкоцепной” комбинаторной энтропии в химический потенциал растворителя (0 ≤ λ ≤ 1), x – эффективная мольная доля полимерных цепей в растворе. Нетрудно показать, что для полимера со степенью полимеризации p мольная доля полимера связана с его объемной долей соотношением
По своему смыслу модифицированная модель учитывает увеличение эффективной жесткости цепи в ряду смесей ПАА с агарозой. Модель, описываемая комбинацией уравнений (3)–(7), содержит неопределенные параметры χ, gads, $\varphi _{V}^{{\text{0}}}$, λ. Она была использована для аппроксимации экспериментальных изотерм адсорбции воды на пленках ПАА, агарозы и их смесей. Предварительно величины адсорбции x/m были пересчитаны в значения объемной доли полимера.
На рис. 3 представлены результаты интерполяции экспериментальных изотерм сорбции с помощью уравнений модифицированной модели, учитывающей эффективную жесткость цепи полимера – уравнения (3)–(7). На рисунке видно, что для всех систем наблюдается хорошее соответствие модельных зависимостей экспериментальным значениям во всей области изменения объемной доли полимера и относительного давления пара. В табл. 1 приведены значения параметров модели, полученных в результате интерполяции.
Рис. 3.
Изотермы сорбции воды в координатах объемная доля полимера–относительное давление пара для пленок ПАА (1), агарозы (7) и смесей ПАА : агароза состава 70 : 30 (2), 60 : 40 (3), 50 : 50 (4), 40 : 60 (5) и 30 : 70 (6). Линии на рисунке соответствуют результату интерполяции уравнениями (2), (4)–(6).
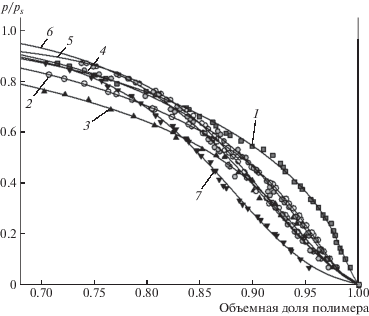
Таблица 1.
Параметры модифицированной модели для описания сорбции воды на пленках ПАА, агарозы и их смесей
| ПАА | Агароза | χ | gads | $\varphi _{V}^{0}$ | λ |
|---|---|---|---|---|---|
| 100 | 0 | 0.77 | 0.748 | 0.087 | 1.000 |
| 70 | 30 | 0.42 | –1.95 | 0.025 | 0.75 |
| 60 | 40 | 0.15 | –1.87 | 0.037 | 0.64 |
| 50 | 50 | 0.13 | –2.05 | 0.051 | 0.39 |
| 40 | 60 | –0.11 | –3.30 | 0.066 | 0 |
| 30 | 70 | –0.06 | –3.16 | 0.074 | 0 |
| 0 | 100 | –0.13 | –4.33 | 0.074 | 0 |
Несмотря на то, что трактовка значений параметров модели носит в значительной мере условный характер, тем не менее установленные тренды согласуются с качественными представлениями об ожидаемых изменениях термодинамических свойств в смесях ПАА–агароза. Из таблицы следует, что параметр Флори–Хаггинса монотонно уменьшается по мере увеличения содержания агарозы в смеси, что может быть связано с образованием более прочных водородных связей молекул воды с гидроксильными группами агарозы по сравнению с амидными группами звеньев ПАА. В этом же ряду значения параметра свободной энергии gads становятся все более отрицательными. Доля неравновесных вакансий $\varphi _{{\text{V}}}^{{\text{0}}}$ существенно уменьшается при добавлении 30% агарозы к ПАА, а затем монотонно увеличивается, достигая практически уровня ПАА для индивидуальной агарозы. Параметр λ, характеризующий относительный вклад конформационной гибкости цепей, равен единице для гибкоцепного ПАА. В смесях с содержанием агарозы 30, 40, 50% этот параметр уменьшается, что коррелирует с повышением эффективной жесткости цепей и указывает на смешанный характер комбинаторной энтропии. Для полужесткоцепной агарозы и для смесей с ее содержанием 60 и 70% параметр λ обращается в нуль.
Существенным плюсом предложенной модифицированной модели является возможность интерполяции изотерм сорбции в область большого относительного давления пара вблизи насыщения (р/рs > 0.95), где экспериментальные измерения технически трудно осуществимы. Эта возможность является критически важной для расчета величин энергии Гиббса смешения ПАА с агарозой.
В соответствии с приведенным выше термодинамическим циклом искомая величина энергии Гиббса смешения полимеров рассчитывается по уравнению
(8)
$\Delta {{G}_{m}} = {{\omega }_{1}}\Delta {{g}_{x}}(1) + {{\omega }_{2}}\Delta {{g}_{x}}(2) + \Delta g{\text{*}} - \Delta {{g}_{x}}(3),$На основании изотерм адсорбции, представленных на рис. 3, были определены концентрационные зависимости химического потенциала воды $\Delta {{\mu }_{1}}$ в системах на основе ПАА, агарозы и их смесей. Расчет проводили по точкам модельных зависимостей в диапазоне объемной доли полимера от 1 (индивидуальный полимер) до 0.4 (относительное давление р/рs = 0.99). Участок при высоких относительных давлениях получали экстраполяцией модельных зависимостей с использованием параметров, приведенных в табл. 1. Для расчета концентрационной зависимости химического потенциала полимера использовали уравнение Гиббса–Дюгема
(9)
$\Delta {{\mu }_{2}} = - \int\limits_{ - \infty }^{\Delta {{\mu }_{1}}} {\frac{{1 - \omega }}{\omega }d(\Delta {{\mu }_{{\text{1}}}})} ,$На основании величин $\Delta {{\mu }_{1}}$ и $\Delta {{\mu }_{2}}$ рассчитывали концентрационную зависимость средней удельной энергии Гиббса смешения раствора (Δgm) по уравнению
Величины Δgx, используемые в уравнении (8), рассчитывали из концентрационной зависимости Δgm как отрезок, отсекаемый на оси ординат касательной, являющейся продолжением прямолинейного участка [28]. Формально эта величина отвечает энергии Гиббса растворения полимера в избытке растворителя. Возможность экстраполяции изотерм адсорбции в область относительного давления, близкого к насыщенному, очень полезна в этом отношении.
В качестве примера на рис. 4 представлены расчетные концентрационные зависимости энергии Гиббса смешения для водных растворов агарозы и одной из смесей ПАА–агароза. Видно, что интерполяция изотерм сорбции модельной зависимостью по уравнениям (3)–(7) позволила надежно локализовать прямолинейный участок на концентрационной зависимости и провести расчет величин Δgx. На основании полученных величин Δgx по уравнению (8) были рассчитаны значения энергии Гиббса смешения ПАА с агарозой в зависимости от соотношения компонентов. Они представлены на рис. 5. Из рисунка следует, что концентрационная зависимость энергии Гиббса смешения ПАА и агарозы является знакопеременной: в области избытка ПАА энергия Гиббса смешения положительна, а в области избытка агарозы – отрицательна. Это означает, что смеси ПАА–агароза, содержащие менее 50 мас. % агарозы термодинамически несовместимые, в то время как смеси, содержащие большее количество агарозы, термодинамически устойчивые.
Рис. 4.
Расчетная зависимость энергии Гиббса смешения водных растворов агарозы (1) и смеси ПАА–агароза в соотношении 40 : 60 (2). Точки на кривых – результат расчетов по уравнениям (9), (10) с использованием интерполированных изотерм сорбции. Штриховые линии показывают прямолинейный участок зависимостей, используемый для расчета величин Δgx.
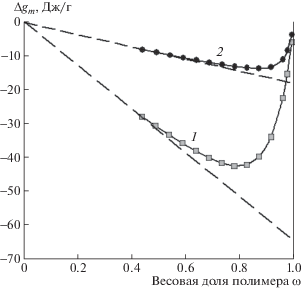
Рис. 5.
Концентрационные зависимости энергии Гиббса ΔGm (1) и энтропии TΔSm смешения ПАА и агарозы (2). Температура 25°С. Линии проведены как визуальный ориентир.
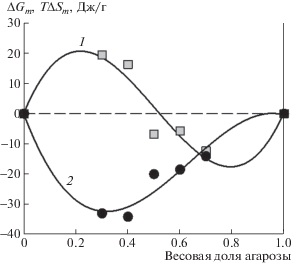
На рис. 5 также представлена концентрационная зависимость энтропии смешения ПАА и агарозы, рассчитанная с использованием значений энтальпии смешения этих полимеров, приведенных на рис. 1. Расчетные значения энтропии смешения отрицательны во всем диапазоне соотношений агарозы и ПАА, что свидетельствует об упорядочении в данной полимерной смеси. Причина упорядочения, очевидно, заключается в энергетически выгодном взаимодействии звеньев агарозы и ПАА, проявляющемся в отрицательных значениях энтальпии смешения, представленных на рис. 1. Наибольшие по абсолютной величине значения энтропии смешения наблюдаются в смесях с избытком ПАА. В этом диапазоне составов тенденция к упорядочению приводит к сегрегированию системы и появлению термодинамической несовместимости компонентов.
Термодинамические данные не раскрывают структурные особенности строения смесей ПАА–агароза. Можно лишь высказать предположения о причинах знакопеременного характера кривых ΔGm. Известно, что макромолекулы агарозы, как и других полужесткоцепных полисахаридов, существуют в спиралевидной конформации, стабилизированной внутримолекулярными водородными связями. Гибкоцепные макромолекулы ПАА характеризуются конформацией гауссового клубка. Логично предположить, что избыток того или иного компонента в смеси предопределяет доминирующий характер конформационного набора. С этой точки зрения смешение агарозы с избытком ПАА должно сопровождаться разрушением спиралевидных конформаций, что термодинамически не выгодно для полужесткоцепных макромолекул. С другой стороны, в смесях на основе избытка агарозы спиралевидные конформации ее макромолекул сохраняются, а гибкие цепи ПАА, по-видимому, распределяются в межспиральном пространстве. Для гибкоцепного полимера это не сопровождается существенным проигрышем в энтропии. Учитывая, что взаимодействие звеньев ПАА и агарозы является энергетически выгодным (см. рис. 1), смеси на основе избытка агарозы термодинамически совместимы.
Указанные термодинамические особенности сказываются на поведении гелей на основе полувзаимопроникающих сеток ПАА и агарозы. На рис. 6 представлены значения степени набухания DN- гидрогелей на основе сшитого ПАА и линейной агарозы в сопоставлении со степенью набухания гелей на основе сшитого ПАА. Приведены зависимости степени набухания от концентрации акриламида в реакционной смеси. Концентрация агарозы была постоянной и составляла 0.57%.
Рис. 6.
Зависимость степени набухания в воде от концентрации акриламида при синтезе для гелей ПАА (1) и DN-гелей на основе сшитого ПАА и линейной агарозы (2). Степень сшивки ПАА равна 1 : 100. Содержание агарозы в реакционной смеси 0.57%. Температура 25°С.
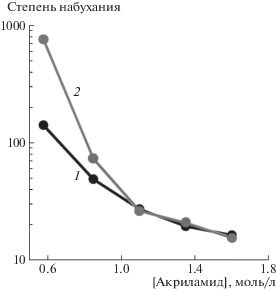
Видно, что по мере уменьшения концентрации акриламида возрастает степень набухания геля на его основе. Такой эффект обычно связывают со снижением степени сетчатости за счет уменьшения числа пространственных узлов, образующихся при синтезе сетки в условиях повышенной концентрации мономера. Обращает на себя внимание то, что введение линейной агарозы значительно увеличивает степень набухания сетки. При минимальной концентрации акриламида – 0.6 моль/л, степень набухания увеличилась почти на порядок. При данной концентрации акриламида концентрация агарозы в полимерной смеси ПАА–агароза около 7%. Такая концентрация отвечает области термодинамической несовместимости ПАА и агарозы (рис. 5). Ввиду того, что сеткообразующие полимеры несовместимы друг с другом, их цепи стараются обособиться в пространстве, что приводит к резкому увеличению степени набухания.
Эффект увеличения степени набухания DN-геля снижается при повышении концентрации акриламида при синтезе и исчезает при достижении концентрации акриламида более 1 моль/л. Вероятно, это связано с влиянием пространственной сетки зацеплений, которая препятствует пространственному разделению полимерных компонентов DN-гелей.
Представляет интерес сопоставить данные изучения термодинамики совместимости ПАА и агарозы с механическими свойствами DN-гелей на основе указанных полимеров. На рис. 7 приведены деформационные кривые сжатия гелей, полученных на основе сшитого ПАА с различным содержанием агарозы.
Рис. 7.
Деформационные кривые DN-гелей на основе ПАА с содержанием агарозы 0 (1), 4.0 (2), 7.8 (3), 14.4 (4), 20.2 (5) и 25.2% (6). Содержание агарозы указано как доля в общей полимерной компоненте геля.
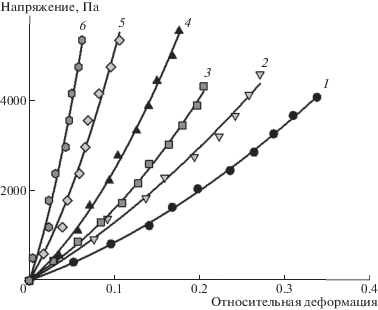
Видно, что зависимости имеют вид плавных кривых, исходящих из начала координат, и их положение зависит от содержания полисахарида. С увеличением содержания агарозы деформация геля уменьшается при фиксированном значении приложенного механического напряжения. По тангенсу угла наклона начального прямолинейного участка каждой деформационной кривой был определен модуль Юнга соответствующего геля. Концентрационная зависимость величины модуля Юнга от процентного содержания агарозы в составе полимерной сетки представлена на рис. 8. Видно, что с повышением содержания агарозы в составе DN-гелей на основе сшитого ПАА их модуль Юнга значительно увеличивается и достигает целевых значений для материалов биомедицинского назначения. Сопоставление с приведенными выше термодинамическими данными показывает, что в диапазоне содержания агарозы до 25% наблюдается термодинамическая несовместимость компонентов, обусловленная большими отрицательными значениями энтропии смешения.
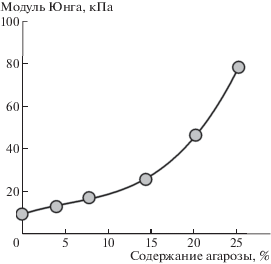
Прямое соотнесение данных, полученных для полимерной смеси с результатами механических испытаний DN-гелей на основе этой смеси требует осторожности, поскольку в составе гидрогеля присутствует третий компонент – вода, которого нет в бинарной полимерной смеси. Тем не менее, определенные заключения предположительного характера могут быть сделаны. Вероятно, что термодинамическая несовместимость агарозы с ПАА в данном диапазоне составов приводит к тому, что макромолекулы агарозы не смешиваются с субцепями сетки ПАА на сегментальном уровне, и физическая сетка агарозы играет роль армирующего наполнителя в химической сетке ПАА. Сегрегация двух полимеров происходит в водной среде, причем оба полимера, и агароза, и ПАА являются гидрофильными и сильно гидратированными. Иными словами, и тот и другой компонент DN-гидрогеля несет на себе сольватные оболочки, которые находятся в равновесии с объемной фазой воды. Таким образом, для геля на основе сегрегированных термодинамически несовместимых гидратированных макромолекул наблюдается улучшение механических характеристик. Данная ситуация подобна улучшению механических свойств наполненных полимерных композитов со слабым межфазным взаимодействием. Для таких композитов повышение модуля связывают со структурированием частиц наполнителя по механизму формирования коагуляционных сеток, пронизывающих объем полимерного материала и, тем самым, оказывающих армирующий эффект [29–31]. Можно предположить, что в геле на основе несовместимых полимеров физическая сетка агарозы играет роль армирующего наполнителя. Полисахарид как компонент с большей жесткостью цепи формирует надмолекулярную структуру из физически связанных спиралевидных макромолекул, пронизывающих объем сетки ПАА, повышая модуль упругости материала.
ЗАКЛЮЧЕНИЕ
Полиакриламид и агароза используются для синтеза гидрогелей с двойной сеткой (DN), в основе которых лежит сочетание химически сшитой сетки синтетического полимера и физической сетки биополимера. Свойства DN-гелей, перспективных для применения в медицине, биотехнологии и биоинженерии, во многом обусловлены взаимодействием образующих их полимерных компонентов. В настоящей работе был проведен термодинамический анализ взаимодействия ПАА и агарозы и определены значения энтальпии, энтропии и энергии Гиббса смешения указанных полимеров во всем диапазоне их соотношений. Для этого методами микрокалориметрии и интервальной изотермической сорбции было исследовано взаимодействие с водой смесей линейного ПАА с агарозой и проведен расчет функций смешения с использованием термодинамических циклов. Для описания и анализа изотерм сорбции воды пленками смесей ПАА–агароза была использована модель, учитывающая полужесткоцепную природу полисахарида. Исходя из условий приготовления смесей, которые определяли предысторию полимеров, полученные термодинамические данные следует отнести к взаимодействию ПАА с агарозой в конформациях неупорядоченных клубков.
Было показано, что энтальпия смешения ПАА с агарозой отрицательна во всем диапазоне составов, при этом ее наибольшее по абсолютной величине значение составляет –6.33 кДж/моль звеньев и приходится на мольное соотношение звеньев ПАА и агарозы, равное 1 : 2. Это свидетельствует об энергетически сильном взаимодействии звеньев полимеров в их смеси, что связано с образованием между ними водородных связей. Вместе с тем было установлено, что термодинамическая совместимость ПАА с агарозой реализуется не во всей области составов, а лишь в смесях с избыточным содержанием агарозы, для которых энергия Гиббса смешения принимает отрицательные значения. Для смесей, содержащих менее 50 мас. % агарозы, энергия Гиббса смешения положительна. Энтропия смешения ПАА с агарозой отрицательна во всем диапазоне соотношений, что свидетельствует о процессах упорядочения в смеси их линейных макромолекул. Наиболее сильное упорядочение, отвечающее наибольшим по абсолютной величине значениям энтропии смешения, наблюдается в смесях с избыточным содержанием ПАА. Это приводит к сегрегации цепей и термодинамической несовместимости ПАА и агарозы в данном диапазоне составов.
Вместе с тем термодинамическая несовместимость ПАА и агарозы не сказывается отрицательным образом на поведении DN-гелей на их основе, в составе полимерной сетки которых ПАА находится в избытке. Напротив, введение агарозы в химическую сетку ПАА приводит одновременно к увеличению степени набухания и к увеличению модуля сжатия DN-гидрогеля. Вероятный механизм “усиливающего” действия агарозы состоит в армирующем действии ее физической сетки, термодинамически несовместимой с химической сеткой DN-гидрогеля.
Работа выполнена при финансовой поддержке Российского научного фонда (код проекта 20-12-00031).
Список литературы
Sahiner N., Demirci S. // J. Appl. Polym. Sci. 2017. V. 134. № 21. P. 44854.
Chen, Q., Zhu L., Huang L., Chen H., Xu K., Tan Y., Wang P., Zheng J. // Macromolecules. 2014. V. 47. № 6. P. 2140.
Xin H., Brown H.R., Naficy S., Spinks G.M. // J. Polym. Sci., Polym. Phys. 2016. V. 54. № 1. P. 53.
Li Z., Su Y., Haq M.A., Xie B., Wang D. // Polymer. 2016. V. 103. P. 146.
Haque Md.A., Kurokawa T., Gong J.P. // Polymer. 2012. V. 53. № 9. P. 1805.
Park S.-B., Lih E., Park K.-S., Joung Y.K., Han D.K. // Progr. Polym. Sci. 2017. V. 68. P. 77.
Chen Q., Chen H., Zhu L., Zheng J. // Macromol. Chem. Phys. 2016. V. 217. № 9. P. 1022.
Tarashi S., Nazockdast H., Sodeifian G. // Polymer. 2020. V. 188. P. 122138.
Terziyan T.V., Safronov A.P., Belous Yu.G. // J. Polym. Sci., Polym. Chem. 2015. V. 57. P. 200.
Adamova L.V., Safronov A.P., Terziyan T.V., Shabadrov P.A., Klyukina A.V. // J. Polym. Sci., Polym. Chem. 2018. V. 60. № 2. P. 190.
Lichtenthaler R.N., Abram D.S., Prausnitz J.M. // Can. J. Chem. 1973. V. 51. P. 3071.
Donohue M.D., Prausnitz J.M. // Can. J. Chem. 1975. V. 53. P. 1586.
Mostafavi F.S., Zaeimb D. // Int. J. Biol. Macromol. 2020. V. 159. P. 1165.
Rani G.U., Konreddy A.K., Mishra S. // Int. J. Biol. Macromol. 2018. V. 117. P. 902.
Hamzavi N., Dewavrin J.-Y., Drozdov A.D., Birgersson E. // J. Polym. Sci., Polym. Phys. 2017. V. 55. P. 444.
Kang Peng, Kaixiang Yang, Yujiao Fan, Akram Yasin, Xiang Hao, Haiyang Yang // Macromol. Chem. Phys. 2017. № 1700170.
Суворова А.И., Тюкова И.С., Сафронов А.П., Адамова Л.В. Высокомолекулярные соединения: Лабораторный практикум: Уч. пособие. Екатеринбург: Изд-во Уральского гос. ун-та, 2006.
Rochas C., Lahaye M. // Carbohydr. Polymers. 1989. V. 10. № 4. P. 289.
Calvet E., Prat H. Microcalorimetrie. Paris: Masson, 1956.
Arnott S., Fulmer A., Scott W.E., Dea I.C.M., Moorhouse R., Rees D.A. // J. Mol. Biol. 1974. V. 90. № 2. P. 269.
Тагер А.А. Физико-химия полимеров. М.: Научный мир, 2007.
Safronov A.P., Adamova L.V. // Polymer. 2002. V. 43. № 9. P. 2653.
Safronov A.P., Adamova L.V. // Polymer Science A. 2002 . V. 44. № 4. P. 408.
Safronov A.P., Adamova L.V., Kurlyandskaya G.V. // J. Polym. Sci. Polym. Chem. 2019. V. 61. № 1. P. 29.
Flory P.J. Principles of Polymer Chemistry. Ithaca: Cornell Univ. Press, 1953.
Смирнова Н.А. Молекулярные теории растворов. Л.: Химия, 1987.
Чалых А.Е., Герасимов В.К., Чертков В.Г. // Высокомолек. соед. Б. 1994. Т. 36. № 12. С. 2077.
Safronov A.P., Suvorova A.I., Tyukova I.S., Smirnova Y.A. // J. Polym. Sci., Polym. Phys. 2007. V. 45. № 18. P. 2603.
Baeza G.P., Genix A.-C., Oberdisse J., Degrandcourt C., Petitjean L., Couty M., Gummel J. // Macromolecules. 2013. № 46. P. 317.
Ai-Jun Zhu, Sternstein S.S. // Compos. Sci. Technol. 2003. № 63. P. 1113.
Jancar J., Douglas J.F., Starr F.W., Kumar S.K., Cassagnau P., Lesser A.J., Sternstein S.S., Buehler M.J. // Polymer. 2010. V. 51. № 15. P. 3321.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия А)