

Высокомолекулярные соединения (серия С), 2019, T. 61, № 1, стр. 165-176
Радикальное замещение дитиокарбонильной группы полиметилметакрилата, полученного полимеризацией с обратимой передачей цепи
М. З. Беканова a, b, Н. К. Неумолотов a, А. Д. Ябланович c, А. В. Плуталова a, Е. В. Черникова a, b, *
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
b Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
c Московский государственный университет имени М.В. Ломоносова. Факультет наук о материлах
119991 Москва, Ленинские горы, 1, стр. 73, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 03.03.2019
После доработки 17.03.2019
Принята к публикации 22.03.2019
Аннотация
Проведено систематическое исследование реакции радикального замещения дитиокарбонильной группы полиметилметакрилата, полученного радикальной полимеризацией с обратимой передачей цепи, при взаимодействии с радикальным азоинициатором в инертном растворителе при 80°С. Показано, что для полимера с дитиобензоатной группой повышение мольного отношения концентраций инициатора и макромолекул с концевой дитиобензоатной группой до 100 эквивалентов способствует быстрому и количественному замещению дитиобензоатной группы на фрагмент инициатора и подавлению побочных реакций обрыва с участием радикальных интермедиатов. Для замещения тритиокарбонатной группы требуются более мягкие условия – двадцатикратный мольный избыток инициатора и непродолжительное время реакции 2–5 ч. Определяющую роль в выборе условий проведения реакции замещения играет стабильность радикальных интермедиатов. Образующийся в ходе реакций замещения агент обратимой передачи цепи – низкомолекулярное соединение, содержащее дитиокарбонильный фрагмент, можно использовать повторно для синтеза ПММА.
ВВЕДЕНИЕ
Радикальная полимеризация с обратимой деактивацией цепи широко используется в последние десятилетия для синтеза макромолекул заданной молекулярной массы, строения и архитектуры [1–5]. Среди известных ее вариантов следует выделить радикальную полимеризацию с обратимой передачей цепи по механизму присоединения–фрагментации (ОПЦ), которая не только толерантна к функциональным группам мономеров, но и позволяет получать полимеры с заданной функциональностью концевых групп [6–12]. Согласно механизму ОПЦ-полимеризации [13–19], концевые α- и ω-группы в макромолекулах, образующихся с использованием эффективного ОПЦ-агента при условии [ОПЦ-агент] : [инициатор] $ \gg $ 1, задаются исключительно химической природой ОПЦ-агента R–S–C(=S)–Z и представляют собой, соответственно, “уходящую” группу R и фрагмент ОПЦ-агента –S–C(=S)–Z, т.е. структуру макромолекулы в общем виде можно представить как R–Pn–S–C(=S)–Z.
ω-Группа, как показали многочисленные исследования, является удобным объектом для дальнейшей модификации, расширяющей потенциальные возможности применения ОПЦ-процесса в макромолекулярном дизайне [5, 9, 10, 20]. Среди известных способов модификации дитиокарбонильной группы, обобщенных в ряде обзоров и статей, наиболее часто на практике используются термолиз дитиокарбонильной группы, приводящий к появлению в макромолекуле концевой кратной связи С=С; взаимодействие с нуклеофилами и/или восстанавливающими агентами, в результате чего образуется тиольная группа SH; реакция с радикальными инициаторами, вследствие которой дитиокарбонильная группа замещается фрагментом инициатора и т.д. [19–37].
Одним из удобных способов удаления дитиокарбонильной группы и ее замещения на другую концевую функциональную группу в полимере является его обработка избытком радикального инициатора в инертном растворителе. В этом случае при распаде инициатора образуются радикалы, присоединяющиеся по связи C=S к макромолекуле, образуя радикальный интермедиат. Фрагментация интермедиата приводит к высвобождению макрорадикала, который в условиях избытка радикального инициатора X–X обрывается на радикалах инициатора, а в присутствии ловушек (Н-доноров) обрывается на них:
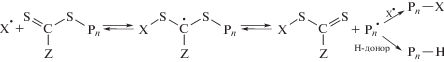
Впервые метод удаления дитиокарбонильной концевой группы в ПММА (дитиобензоатной) и ПС (дитиобензоатной и тритиокарбонатной) с помощью радикального азоинициатора был описан в работе [19]. Образование низкомолекулярного ОПЦ-агента было подтверждено хроматографически. ММР полимера после реакции не изменяется, а по результатам УФ-спектроскопии в полимере перестает регистрироваться поглощение при 510 нм, отвечающее, по мнению авторов, дитиокарбонильной группе. Данная реакция протекает количественно в течение 2.5 ч при 80°С и мольном отношении [инициатор] : [ОПЦ] = 20. При этом в ходе синтеза происходит образование ОПЦ-агента X–SC(=S)Z. Авторами был продемонстрирован двойной цикл регенерации ОПЦ-агента: синтез ПММА под действием ОПЦ-агента X–SC(=S)Z – обработка полимера 20-кратным избытком инициатора X–X – отделение ОПЦ-агента X–SC(=S)Z – синтез ПММА и т.д.
Такую методику использовали для получения телехелика ПММА с концевыми пентафторфенильными сложноэфирными группами –C(CH3)(CN)CH2CH2C(=O)OC6F5 путем взаимодействия ПММА с концевой дитиобензоатной группой и 20-кратного мольного избытка подходящего инициатора [38].
Несмотря на кажущуюся простоту данного эксперимента, оказалось, что 100%-ное замещение концевой дитиокарбонильной группы в полимерах протекает не всегда [21, 24, 28, 33–35]. Более того, даже для ПММА есть свои ограничения. Например, возможно образование побочного продукта с более высокой ММ и при мольном избытке инициатора по отношению к дитиобензоатным группам полимера [39]. Аналогичные результаты были получены и для поли(н-бутилакрилата) с дитиобензоатной концевой группой. Детальный анализ возможных реакций, проведенный в работе [40], показал, что причиной этого могут быть реакции обрыва с участием интермедиатов, макрорадикалов и радикалов инициатора.
Результаты подобных экспериментов для разных полимеров систематизированы в нескольких обзорах [6–11]. Более подробные исследования реакции замещения концевой дитиобензоатной и тритиокарбонатной групп на остаток радикала в полистироле [33, 34] и поли(н-бутилакрилате) [33] продемонстрировали, что для них степень замещения далека от 100% и составляет от 20 до 80% в зависимости от концентрации инициатора. Повысить эффективность реакции замещения можно либо добавкой к азоинициатору пероксида лауроила или пероксида бензоила [33], либо введением Н-донора (например, толуола, изопропанола, трибутилолова, силанов или гипофосфитов) [11, 21, 41]. Последний вариант использовали для ПММА с концевой тритиокарбонатной группой [21], однако его прямая реакция с инициатором без Н-донора в литературе не описана. Несмотря на многочисленные данные о реакции замещения дитиокарбонильной группы в полимерах, бóльшая часть работ посвящена стиролу и алкилакрилатам. Работа [19], в которой анализируется поведение ПММА с дитиобензоатной группой в аналогичной реакции, является единичной, а для ПММА с тритиокарбонатной группой таких данных вообще нет. Ранее мы на примере ряда виниловых полимеров показали, что полное замещение тритиокарбонатной группы требует применения более высокой концентрации радикального инициатора и более длительного времени проведения реакции [28, 42]. Кроме того, на наш взгляд, открытым остается вопрос и о роли побочных процессов в ходе реакции замещения в ПММА с дитиобензоатной группой.
Цель настоящей работы – систематическое исследование влияния мольного соотношения концентраций инициатора и дитиокарбонильных групп в полимере и времени протекания реакции на эффективность радикального замещения в ПММА с двумя типами концевых групп – дитиобензоатной и тритиокарбонатной. Основываясь на результатах наших предыдущих исследований и литературных данных, мы планировали определить оптимальные условия проведения реакции. Как и ожидалось, эти условия оказались различными, и они определяются в первую очередь стабильностью образующихся радикальных интермедиатов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ММА перед применением перегоняли в вакууме. ДАК перекристаллизовывали из метанола. 4-S-дитиобензоат 4-цианопентановой кислоты (ЦКБ) и 2-циано-2-пропилдодецилтритиокарбонат (ЦТК) фирмы “Aldrich” использовали без дополнительной очистки. Растворители очищали по стандартным методикам.
Для синтеза поли(метилметакрилат)дитиобензоата (ПММА-Б) в свежеперегнанном ММА растворяли заданное количество ДАК и ЦКБ. Реакционную смесь заливали в ампулу, дегазировали до остаточного давления ~5 × 10–3 мм рт.ст. и отпаивали. Ампулу помещали в термостат при 80°С и полимеризовали в течение 24 ч, затем охлаждали в жидком азоте и вскрывали; образовавшийся полимер высаживали в 10-кратный избыток холодного метанола и отфильтровывали. Затем полимер растворяли в 10-кратном избытке бензола и сушили лиофильно. По данным ГПХ, Mn = 7.8 × 103, Ð = 1.12.
Синтез поли(метилметакрилат)тритиокарбоната (ПММА-К) осуществляли аналогичным образом, используя в качестве ОПЦ-агента ЦТК. По данным ГПХ, Mn = 7.7 × 103, Ð = 1.25.
Для исследования реакции замещения тиокарбонильной группы цианоизопропильной группой инициатора ДАК готовили раствор ПММА-Б или ПММА-К и ДАК в бензоле. Мольное отношение концентраций инициатора и тиокарбонильных концевых групп полимера варьировали как 20 : 1, 40 : 1 и 100 : 1. Раствор заливали в ампулу, дегазировали, ампулу отпаивали и помещали в термостат при 80°С на 2.5, 5 или 24 ч. По окончании эксперимента ампулу охлаждали, полимер высаживали в 10-кратный избыток холодного гексана и центрифугировали. Осадок полимера растворяли в 10-кратном избытке бензола и сушили лиофильно в вакууме. Полимер растворяли в ТГФ и анализировали методом УФ-спектроскопии.
Маточный раствор декантировали, упаривали на роторе, образовавшийся остаток растворяли в ТГФ и регистрировали УФ-спектр. Затем ТГФ досуха отгоняли. К сухому остатку, полученному в экспериментах с прогреванием в течение 2.5 и 5 ч, добавляли ММА, а к полученному после нагревания в течение 1 суток – ММА, содержащий 10–3 моль/л ДАК. Реакционные смеси заливали в ампулы, дегазировали до остаточного давления ~5 × 10–3 мм рт. ст. путем четырехкратного повторения циклов заморозки–разморозки, запаивали и ампулы помещали в термостат, разогретый до 80°С на 4 ч. Смеси разбавляли бензолом, полимер высушивали лиофильно и анализировали методом ГПХ.
Концентрацию дитиокарбонильных групп в полимерах определяли методом УФ-спектроскопии, используя определенные ранее значения коэффициента молярного поглощения для ЦКБ и ЦТК [43]. Спектры растворов полимеров в ТГФ регистрировали на спектрофотометре “Unico 2804” (США).
Анализ молекулярно-массовых характеристик полимеров проводили методом ГПХ в ДМФА с 0.1 мас. % LiBr при 50°С на хроматографе GPC-120 фирмы “PolymerLabs” с двумя колонками PLgel 5 µm MIXED B (М = (5 × 102)–(1 × 107)), оборудованном дифференциальным рефрактометром. Для калибровки применяли узкодисперсные стандарты ПММА.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Полиметилметакрилат с дитиобензоатной концевой группой
Нагревание ПММА с концевой дитиобензоатной группой с радикальным инициатором в инертном растворителе вызывает цепочку последовательных реакций:
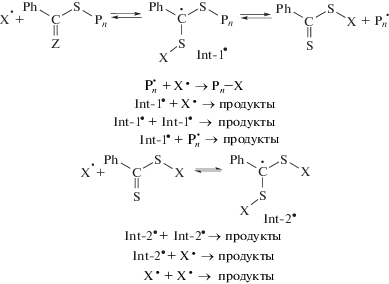
Согласно схеме 1, в результате взаимодействия радикала инициатора X• с полимером образуется радикальный интермедиат Int-1•. Интермедиат может распадаться с отщеплением макрорадикала ${\text{P}}_{n}^{\centerdot }$ и образованием ОПЦ-агента X–SC(=S)Z (Z = Ph). При отношении [инициатор] : [ОПЦ] ${\text{P}}_{n}^{\centerdot }$ $ \gg $ 1 радикалы инициатора способны захватывать макрорадикалы ${\text{P}}_{n}^{\centerdot }$ и интермедиаты Int-1•, а также погибать при взаимодействии друг с другом. При накоплении в системе интермедиатов они могут участвовать в квадратичном и перекрестном обрыве с макрорадикалами. Увеличение концентрации соединения X–SC(=S)Z будет приводить к вовлечению его в аналогичную цепочку реакций присоединения–фрагментации–обрыва, в ходе которых возникает новый интермедиат Int-2•. Квадратичный обрыв макрорадикалов в этих условиях маловероятен.
Мы провели серию экспериментов, в которых ПММА-Б (3.5 × 10–3 моль/л) нагревали с 20, 40 и 100-кратным мольным избытком ДАК в растворе бензола при 80°С в течение 2.5, 5 и 24 ч (τ1/2 = = 90 мин [44]). Следует отметить, что окраска полимера, выделенного после переосаждения, изменялась с розовой на белую, и тем быстрее, чем выше концентрация ДАК. Изменение окраски однозначно указывает на уменьшение концентрации хромофорных дитиобензоатных групп в полимере.
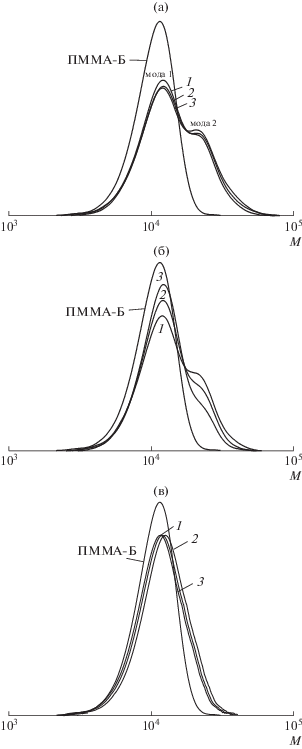
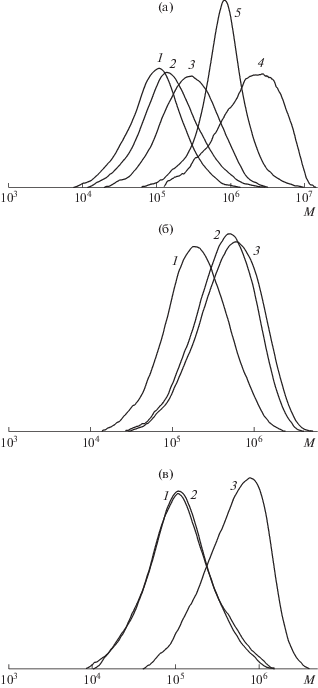
На рис. 1 приведены нормированные к единичной площади кривые ГПХ полимера до и после реакции с ДАК. При [ДАК] : [ПММА-Б] = = 20 : 1 кривые ГПХ полимеров после реакции с ДАК становятся бимодальными. Положение ММ максимума пика моды 1 практически совпадает с ММ максимума пика ПММА-Б; ее небольшое смещение в область высоких ММ обусловлено незначительными потерями при осаждении полимера. Величина ММ максимума пика моды 2 в два раза выше, чем у ПММА-Б. Увеличение продолжительности реакции практически не оказывает влияния на соотношение интенсивностей этих мод. С ростом мольного отношения [ДАК] : : [ПММА-Б] до 40 : 1 картина качественно не изменяется, но заметно меняется соотношение площадей мод: интенсивность пика моды 2 уменьшается, и она дополнительно понижается при увеличении времени реакции. Наконец, при стократном мольном избытке ДАК по сравнению с полимером ММР полимера становится унимодальным. Следует заметить, что используемая система хроматографических колонок не позволяет зарегистрировать низкомолекулярные продукты реакции с М < 500.
Рис. 1.
Кривые ГПХ, нормированные к единичной площади, продуктов реакции ПММА-Б и ДАК после очистки от низкомолекулярных продуктов реакции. Мольное соотношение [ДАК] : [ПММА-Б] = 20 : 1 (а), 40 : 1 (б) и 100 : 1 (в); время реакции 2.5 (1), 5 (2) и 24 ч (3); Т = 80°С.
Очевидно, что методом ГПХ невозможно различить макромолекулы с концевой цианизопропильной группой ПММА-C(CH3)2(CN) и с исходной дитиобензоатной группой, имеющих близкую ММ. Аналогичная проблема возникает и с продуктами обрыва интермедиата Int-1• на радикалах инициатора, ПММА-SC(C(CH3)2(CN))(Ph)S-C(CH3)2(CN). Макромолекулы с более высокой, чем у исходного полимера, ММ могут образовываться только в результате обрыва радикальных интермедиатов друг с другом или с макрорадикалами. Однако напрямую зарегистрировать продукты обрыва радикальных интермедиатов ни ЯМР-спектроскопией, ни спектрометрией MALDI-TOF не представляется возможным [45].
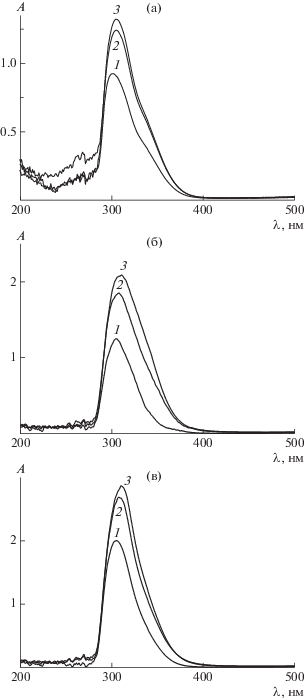
Для анализа содержания дитиобензоатных групп в полимере до и после реакции с ДАК использовали метод УФ-спектроскопии. Предварительная оценка показала, что ПММА-Б содержит практически 100% “живых” цепей с концевой дитиобензоатной группой. После взаимодействия с ДАК количество “живых” полимерных цепей будет уменьшаться, и при полном замещении дитиобензоатной группы на цианизопропильную оно будет равно нулю. УФ-спектры ПММА-Б до и после реакции с ДАК представлены на рис. 2. При соотношении [ДАК] : [ПММА-Б] = 20 : 1 (рис. 2а), интенсивность поглощения, отвечающего дитиобензоатной группе в полимере (λ = 302 – 310 нм, ε = (16.0 ± 0.3) × 103 моль/(л см) [43]), резко уменьшается через 2.5 ч реакции. Затем через 5 ч реакции она понижается еще примерно вдвое и после этого меняется незначительно. Результаты расчета доли “живых” цепей в полимере приведены в табл. 1. Видно, что даже через 1 сутки после начала реакции в полимере регистрируется поглощение остаточных дитиобензоатных групп. При увеличении концентрации инициатора до [ДАК] : [ПММА-Б] = 40 : 1 (рис. 2б) понижается концентрация “живых” цепей в полимере за то же время реакции, и наконец при [ДАК] : [ПММА-Б] = = 100 : 1 (рис. 2в) поглощение дитиобензоатных групп не регистрируется уже через 2.5 ч реакции.
Рис. 2.
УФ-спектры растворов в ТГФ ПММА-Б до (1) и после нагревания с избытком ДАК в бензоле при 80°С в течение 2.5 (2), 5 (3) и 24 ч (4). [ДАК] : [ПММА-Б] = = 20 (а), 40 (б) и 100 (в).
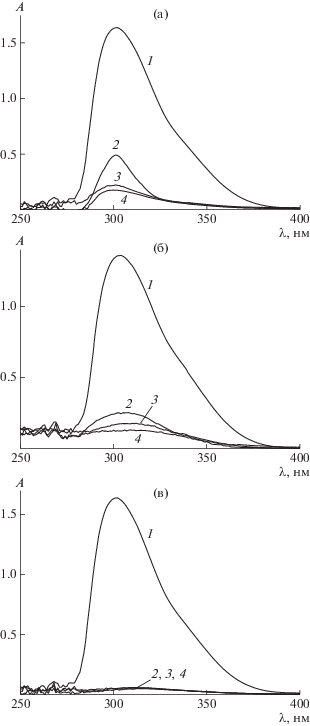
Таблица 1.
Результаты замещения дитиобензоатной группы в ПММА-Б и постполимеризации ММА с регенерированным ОПЦ-агентом
| [ДАК] : [ПММА], мол.% | Время, ч | “Живые” цепи, мол. % | [ДАК] ×103, моль/л | Постполимеризация | |
|---|---|---|---|---|---|
| Mn × 10–3 | Ð | ||||
| 20 | 2.5 | 30.0 | 22* | 65 | 2.0 |
| 5 | 9.1 | 7* | 95 | 2.1 | |
| 24 | 7.9 | 1 | 180 | 2.1 | |
| 40 | 2.5 | 11.7 | 44* | 125 | 2.1 |
| 5 | 5.1 | 14* | 270 | 2.0 | |
| 24 | 3.7 | 1 | 310 | 2.1 | |
| 100 | 2.5 | 0 | 110* | 70 | 2.0 |
| 5 | 0 | 35* | 70 | 2.1 | |
| 24 | 0 | 1 | 340 | 2.1 | |
| − | 24 | − | 1 | 1100 | 2.3 |
| − | 24 | − | 1 | 570 | 1.67 |
Анализируя полученные данные, можно предположить, что повышение концентрации ДАК по отношению к ПММА-Б приводит к подавлению побочных реакций обрыва макрорадикалов на радикальных интермедиатах. Иными словами, радикалы инициатора успевают оборвать радикалы ПММА• до их взаимодействия с интермедиатами Int-1• или Int-2•. Таким образом, чем выше концентрация ДАК, тем вероятнее становится реакция радикалов инициатора с макрорадикалами и интермедиатом Int-1•. С этой точки зрения оптимальным является соотношение [ДАК] : [ПММА-Б] = = 100 : 1 и относительно низкая абсолютная концентрация полимерных цепей, которая предотвращает образование заметной концентрации интермедиатов.
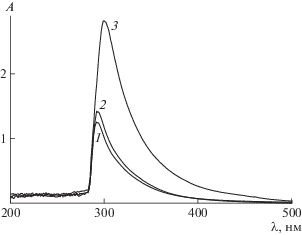
При осаждении полимера, прогретого с ДАК, в метанол маточный раствор приобретает розовую окраску, что может быть вызвано только образованием цианизопропилдитиобензоата (ЦИБ) PhC(=S)S-C(CH3)2(CN), содержащего дитиобензоатную группу. Согласно схеме 1, других продуктов с дитиобензоатной группой и поглощающих при 302–310 нм в системе образовываться не может. Однако нельзя исключать появления других низкомолекулярных соединений – продуктов обрыва с участием радикалов инициатора и/или интермедиата Int-2•, а также остаточного инициатора. После высаживания полимера был собран супернатант, органические растворители были удалены испарением в вакууме, а сухой остаток был растворен в ТГФ и проанализирован методом УФ-спектроскопии (рис. 3).
Рис. 3.
УФ-спектры растворов в ТГФ низкомолекулярных продуктов реакции ПММА-Б с ДАК в бензоле при 80°С в течение 2.5 (1), 5 (2) и 24 ч (3), выделенных из маточного раствора после осаждения полимера. [ДАК] : [ПММА-Б] = 20 (а), 40 (б) и 100 (в).
Наблюдается отчетливая корреляция между результатами, приведенными на рис. 2 и 3: с повышением концентрации ДАК интенсивность сигнала возрастает (т.е. образуется больше ЦИБ). Следовательно, предположение о том, что рост концентрации ДАК приводит к подавлению побочной реакции обрыва макрорадикалов на интермедиате Int-1•, подтвердилось в независимом эксперименте. С увеличением продолжительности реакции интенсивность сигнала также возрастает.
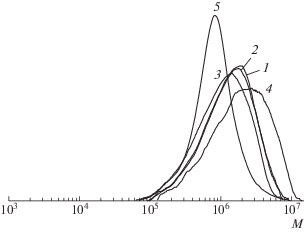
После регистрации УФ-спектров отгоняли ТГФ в вакууме и к сухому остатку добавляли чистый ММА, а к остатку, выделенному через 24 ч – ММА, содержащий 10–3 моль/л ДАК. По грубым оценкам концентрация ЦИБ в этих экспериментах составляет (1.3–2.9) × 10–3 моль/л. Реакционные смеси заливали в ампулы, дегазировали, запаивали и помещали в термостат при 80°С на 4 ч. Полученный полимер выделяли и анализировали методом ГПХ (рис. 4, табл. 1). Все образующиеся полимеры характеризуются широким ММР. В рамках каждой серии экспериментов с сокращением продолжительности реакции замещения, т.е. с ростом концентрации регенерирующегося ЦИБ и уменьшением концентрации распавшегося ДАК, кривые ГПХ продуктов постполимеризации смещаются в область более высоких ММ. При сравнении с ПММА, синтезированным в отсутствие ЦИБ, видно, что ММ полимеров, полученных в описанных выше экспериментах, ниже, а ММР уже. Напротив, ПММА, синтезированный в контрольном эксперименте с эквимолярным количеством ДАК и ЦИБ, характеризуется более узким ММР и более высокой ММ, чем обсуждаемые полимеры.
Рис. 4.
Кривые ГПХ продуктов полимеризации ММА, инициированной ДАК, с участием ЦИБ, выделенного из экспериментов с прогреванием с избытком ДАК в бензоле при 80°С в течение 2.5 (1), 5 (2) и 24 ч (3). [ДАК] : [ПММА-Б] = 20 (а), 40 (б) и 100 (в). Для сравнения на рис. 4а приведены кривые ГПХ для ПММА, полученного с ДАК, 10–3 моль/л (4) и с эквимолярным количеством ДАК и ЦИБ, 10–3 моль/л (5).
Очевидно, что более низкие ММ и относительно широкое ММР полимеров, полученных после регенерации ЦИБ, может быть обусловлено высокой концентрацией остаточного ДАК (табл. 1). Мы воспользовались идеей, описанной в работе [19], и внесли изменения в эксперимент: после осаждения полимера маточный раствор был собран и упарен досуха, после чего сухой остаток был прогрет в шкафу при 95°С в течение 4 ч. Термолиз ОПЦ-агента в этих условиях практически не происходит [46]. В результате мы ожидали, что остаточный ДАК разложится и степень контроля молекулярно-массовых характеристик ПММА увеличится.
Сухие остатки низкомолекулярных продуктов, выделенные в описанных опытах после нагревания ПММА-Б в течение 2.5 ч при мольном соотношении [ДАК] : [ПММА-Б] = 20, 40 и 100 и прогревания при 95°С, были растворены в ТГФ и проанализированы методом УФ-спектроскопии. Как видно на приведенных УФ-спектрах (рис. 5), тенденция не изменяется и с повышением начальной концентрации ДАК растет количество регенерированного ЦИБ.
Рис. 5.
УФ-спектры растворов в ТГФ низкомолекулярных продуктов реакции ПММА-Б с ДАК в бензоле при 80°С в течение 2.5 ч, выделенных из маточного раствора после осаждения полимера и выдержанных при 95°С в течение 4 ч. [ДАК] : [ПММА-Б] = 20 (1), 40 (2) и 100 (3).
Сухие остатки были растворены в 2 мл мономера, содержащего 10–3 моль/л ДАК, смеси разлиты по ампулам, дегазированы и помещены в термостат, разогретый до 80°С на 1 сутки. Полимеры лиофильно высушены из бензола и проанализированы методом ГПХ. Как следует из рис. 6, при использовании 20 и 40-кратного мольного избытка ДАК для замещения дитиобензоатной группы в ПММА-Б и регенерации ЦИБ образуются полимеры близкой ММ (Mn = (830–850) × 103, Ð = 1.95–1.98, кривые 1 и 2), а в случае 100-кратного избытка ДАК ее значение ниже (Mn = 690 × 103, Ð = 1.99, кривая 3). Это согласуется с данными УФ-спектроскопии (рис. 5): концентрация ЦИБ увеличивается с повышением концентрации ДАК, взятого для замещения, что и приводит к уменьшению ММ постполимера. При этом ММ полимеров выше, чем в предыдущих экспериментах, что связано с предварительным разложением ДАК при прогревании сухих остатков и уменьшением его вклада в образование цепей. Однако ММР полимеров шире, чем у контрольного образца, полученного с ЦИБ и ДАК (кривая 5), но уже, чем в случае ПММА, синтезированного под действием той же концентрации ДАК (кривая 4).
Рис. 6.
Кривые ГПХ продуктов постполимеризации ММА под действием ЦИБ, регенерированного из ПММА-Б при 80°С в течение 2.5 ч (1–3). [ДАК] : : [ПММА-Б] = 20 (1), 40 (2) и 100 (3). Для сравнения приведены кривые ГПХ для ПММА, полученного с ДАК, 10–3 моль/л (4) и с эквимолярным количеством ДАК и ЦИБ, 10–3 моль/л (5).
Таким образом, повышение мольного отношения концентраций ДАК и макромолекул с концевой дитиобензоатной группой до 100 эквивалентов способствует быстрому и количественному замещению дитиобензоатной группы на фрагмент инициатора и подавлению побочных реакций.
Полиметилметакрилат с тритиокарбонатной концевой группой
Радикальные интермедиаты, образуемые тритиокарбонатами, существенно менее стабильные, чем интермедиаты, образующиеся с участием дитиобензоатов [15]. Можно предположить, что в аналогичных условиях вероятность побочных реакций обрыва интермедиатов при взаимодействии ПММА-К с ДАК будет ниже, чем в случае ПММА-Б, и в основном будут образовываться ПММА–C(CH3)2(CN) (Pn–X) и несимметричный тритиокарбонат с цианизопропильным и додецильным заместителями X–SC(=S)S–C12H25:
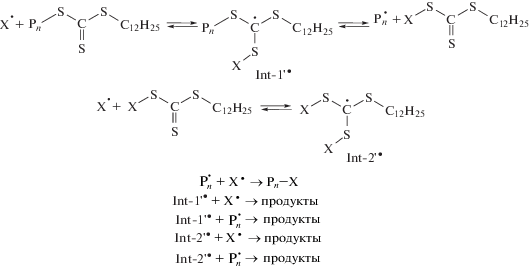
На рис. 7 приведены нормированные к единичной площади кривые ГПХ полимера после взаимодействия с ДАК. Независимо от соотношения [ДАК] : [ПММА-К] и продолжительности реакции кривые ГПХ полимеров остаются унимодальными и практически совпадают с кривой исходного полимера. Таким образом, наше предположение подтверждается – вероятность образования высокомолекулярных побочных продуктов обрыва в случае ПММА с тритиокарбонатной группой оказалась существенно ниже, чем в случае дитиобензоатной.
Рис. 7.
Кривые ГПХ продуктов реакции ПММА-К и ДАК после очистки от низкомолекулярных продуктов реакции. [ДАК] : [ПММА-К] = 20 (а) и 40 (б), время реакции 2.5 (1), 5 (2) и 24 ч (3), Т = 80°С.
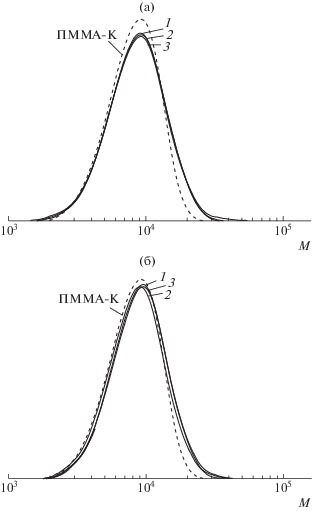
Обесцвечивание полимера наблюдается уже после 2.5 ч реакции с 20-кратным мольным избытком инициатора. На рис. 8 представлены УФ-спектры полимера, очищенного от низкомолекулярных продуктов реакции переосаждением. Для сравнения приведен УФ-спектр исходного ПММА-К, для которого характерен единственный максимум поглощения при длине волны λ = 309 нм (ε = = (10.2 ± 0.9) × 103 моль/(л см)) [43], отвечающий тритиокарбонатной группе. Видно, что 20-кратного мольного избытка ДАК и 2.5 ч реакции достаточно для практически полного замещения тритиокарбонатной группы в ПММА. Результаты расчета доли “живых цепей” в ПММА-К представлены в табл. 2. Из сравнения с данными для ПММА-Б (табл. 1) следует, что скорость замещения тритиокарбонатной группы в ПММА выше, чем дитиобензоатной. Это может быть связано с более высокой скоростью фрагментации тритиокарбонатных интермедиатов по сравнению с дитиобензоатными. Напротив, полимерный дитиобензоат более активен в реакции присоединения, чем аналогичный тритиокарбонат [15, 40, 47].
Рис. 8.
УФ-спектры растворов в ТГФ ПММА-К до (1) и после нагревания с избытком ДАК в бензоле при 80°С в течение 2.5 (2), 5 (3) и 24 ч (4). [ДАК] : : [ПММА-К] = 20 (а) и 40 (б).
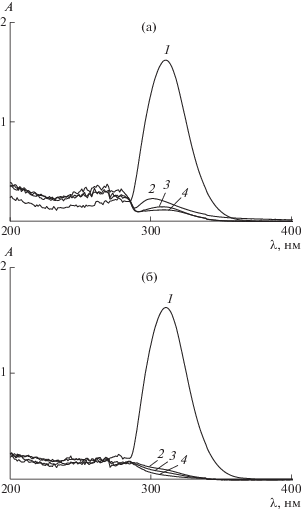
Таблица 2.
Результаты замещения тритиокарбонатной группы в ПММА-К
| [ДАК] : [ПММА-К] | Время, ч | “Живые” цепи, мол. % |
|---|---|---|
| 20 | 2.5 | 12.3 |
| 5.0 | 8.6 | |
| 24.0 | 6.8 | |
| 40 | 2.5 | 5.5 |
| 5.0 | 4.3 | |
| 24.0 | 2.5 |
Аналогичным описанному выше образом был упарен маточный раствор, полученный при осаждении полимера, прогретого с ДАК (20- и 40-кратный мольный избыток), в метанол, и проанализирован сухой остаток. Желтоватая окраска маточного раствора обусловлена наличием S-(2-циан-2-пропил)-S-додецил тритиокарбоната C12H25-SC(=S)S-C(CH3)2(CN), содержащего хромофорную тритиокарбонатную группу.
УФ-спектры растворов низкомолекулярных продуктов до и после нагревания остатка при 95°С в течение 4 ч представлены на рис. 9. Интенсивность поглощения мало изменяется при увеличении продолжительности реакции, что согласуется с приведенными выше данными о практически полном замещении тритиокарбонатной группы уже при 20-кратном мольном избытке инициатора при нагревании в течение 2.5 ч.
Рис. 9.
УФ-спектры растворов в ТГФ низкомолекулярных продуктов реакции ПММА-К с ДАК в бензоле при 80°С в течение 2.5 (1), 5 (2) и 24 ч (3), выделенных из маточного раствора после осаждения полимера (а, б) и дополнительно выдержанных при 95°С в течение 4 ч (в). [ДАК] : [ПММА-К] = 20 (а, в) и 40 (б).
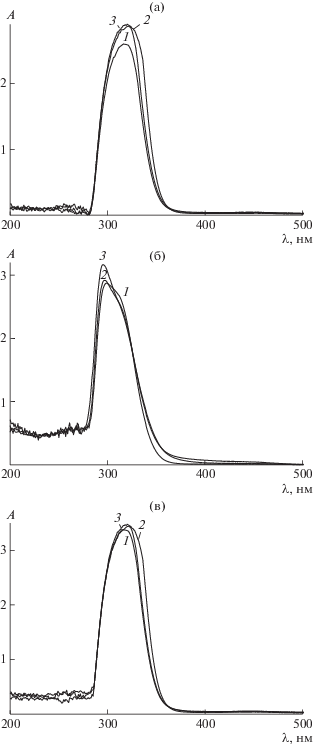
Таким образом, в отличие от дитиобензоата для тритиокарбоната требуются более мягкие условия для реакции замещения. Чтобы проверить возможность регенерации ЦТК в этих условиях, к низкомолекулярному остатку, прогретому 4 ч при 95°С, был добавлен раствор 10-3 моль/л ДАК в 2 мл ММА. Реакционная смесь была заполимеризована при 80°С, а полимер выделен и проанализирован методом ГПХ. Полученный полимер характеризуется широким ММР (рис. 10, кривая 1). Однако его ММ ниже, чем в случае контрольного образца, т.е. ПММА, полученного в присутствии 10–3 моль/л ДАК и в отсутствие ОПЦ-агента.
ЗАКЛЮЧЕНИЕ
В результате систематического исследования реакции радикального замещения на примере ПММА с дитиобензоатной и тритиокарбонатной концевыми группами было показано, что для дитиобензоата подавление побочных реакций обрыва с участием радикальных интермедиатов происходит при использовании стократного мольного избытка инициатора по отношению к дитиобензоатным группам. Для замещения тритиокарбонатной группы достаточно использовать двадцатикратный мольный избыток инициатора. В ходе реакций замещения образуется низкомолекулярный ОПЦ-агент, который можно использовать повторно для синтеза ПММА.
Работа выполнена в рамках государственного задания ИНХС РАН (исследование полимера с дитиобензоатной группой) и государственного задания МГУ по теме “Современные проблемы химии и физико-химии высокомолекулярных соединений” № АААА-А16-116031050014-6 (исследование полимера с тритиокарбонатной группой).
Список литературы
Shipp D.A. // Polym. Rev. 2011. V. 51. № 2. P. 99.
Reversible Deactivation Radical Polymerization: Mechanisms and Synthetic Methodologies/ Ed. by K. Matyjaszewski, H. Gao, B.S. Sumerlin, N.V. Tsarevsky // ACS Symp. Ser. Washington, 2018. V. 1284.
Destarac M. // Polym. Chem. 2018. V. 9. P. 4947.
Klumperman B. // Encyclopedia of Polymer Science and Technology. New York: Wiley, 2015. P. 1.
Vinciguerra D., Tran J., Nicolas J. // Chem. Commun. 2018. V. 54. P. 228.
Moad G., Rizzardo E., Thang S.H. // Polymer. 2008. V. 49. P. 1079.
Moad G., Rizzardo E., Thang S.H. // Aust. J. Chem. 2009. V. 62. P. 1402.
Moad G., Rizzardo E., Thang S.H. // Polym. Int. 2011. V. 60. P. 9.
Moad G., Rizzardo E., Thang S.H. // Aust. J. Chem. 2012. V. 65. P. 985.
Willcock H., O’Reilly R.K. // Polym. Chem. 2010. V. 1. P. 149.
Moad G., Rizzardo E., Thang S.H. // Polym. Int. 2011. V. 60. P. 9.
Chong B., Moad G., Rizzardo E., Skidmore M., Thang S.H. // Aust. J. Chem. 2006. V. 59. P. 755.
Handbook of RAFT Polymerization / Ed. by C. Barner-Kowollik. Weinheim: Wiley-VCH, 2008.
Boyer C., Stenzel M.H., Davis T.P. // J. Polym. Sci., Polym. Chem. 2011. V. 49. P. 551.
Chernikova E.V., Sivtsov E.V. // Polymer Science B. 2017. V. 59. № 2. P. 117.
Moad G. // Macromol. Chem. Phys. 2014. V. 215. P. 9.
Chiefari J., Chong Y.K., Ercole F., Krstina J., Jeffery J., Le T.P.T., Mayadunne R.T.A., Meijs G.F., Moad C.L., Moad G., Rizzardo E., Thang S.H. // Macromolecules. 1998. V. 31. P. 5559.
Barner-Kowollik C., Buback M., Charleux B., Coote M.L., Drache M., Fukuda T., Goto A., Klumperman B., Lowe A.B., McLeary J.B., Moad G., Monteiro M.J., Sanderson R.D., Tonge M.P., Vana P. // J. Polym. Sci., Polym. Chem. 2006. V. 44. P. 5809.
Perrier S., Takolpuckdee P. // J. Polym. Sci., Polym. Chem. 2005. V. 43. P. 5347.
Harvison M.A., Lowe A.B. // Macromol. Rapid Commun. 2011. V. 32. P. 779.
Chong Y.K., Moad G., Rizzardo E., Thang S. // Macromolecules. 2007. V. 40. P. 4446.
Scales C.W., Convertine A.J., McCormick C.L. // Biomacromolecules. 2006. V. 7. P. 1389.
Zelikin A.N., Such G.K., Postma A., Caruso F. // Biomacromolecules. 2007. V. 8. P. 2950.
Moad G., Chong Y.K., Postma A., Rizzardo E., Thang S.H. // Polymer. 2005. V. 46. P. 8458.
Heredia K.L., Grover G.N., Tao L., Maynard H.D. // Macromolecules. 2009. V. 42. P. 2360.
Inglis A.J., Sinnwell S., Davis T.P., Barner-Kowollik C., Stenzel M.H. // Macromolecules. 2008. V. 41. P. 4120.
Sinnwell S., Inglis A.J., Davis T.P., Stenzel M.H., BarnerKowollik C. // Chem. Commun. 2008. V. 44. P. 2052.
Chernikova E.V., Plutalova A.V., Garina E.S., Vishnevetsky D.V. // Polym. Chem. 2016. V. 7. P. 3622.
Spruell J.M., Levy B.A., Sutherland A., Dichtel W.R., Cheng J.Y., Stoddart F.J., Nelson A. // J. Polym. Sci., Polym. Chem. 2009. V. 47. P. 346.
Grover G.N., Alconcel S.N.S., Matsumoto N.M., Maynard H.D. // Macromolecules. 2009. V. 42. P. 7657.
Li M., De P., Gondi S.R., Sumerlin B.S. // J. Polym. Sci., Polym. Chem. 2008. V. 46. P. 5093.
Boyer C., Bulmus V., Davis T.P. // Macromol. Rapid Commun. 2009. V. 30. P. 493.
Postma A., Davis T.P., Evans R.A., Li G., Moad G., O’Shea M.S. // Macromolecules. 2006. V. 36. P. 5293.
Chen M., Moad G., Rizzardo E. // J. Polym. Sci., Polym. Chem. 2009. V. 47. P. 6704.
Postma A., Davis T.P., Moad G., O’Shea M.S. // Macromolecules. 2005. V. 38. P. 5371.
Lima V., Jiang H., Brokken-Zijp J., Schoenmakers P.J., Klumperman B., Linde R.V.D. // J. Polym. Sci., Polym. Chem. 2005. V. 43. P. 959.
Patton D.L., Mullings M., Fulghum T., Advincula R.C. // Macromolecules. 2005. V. 38. P. 8597.
Roth P.J., Wiss K.T., Zentel R., Theato P. // Macromolecules. 2008. V. 41. P. 8513.
Chernikova E.V., Tarasenko A.V., Garina E.S., Golubev V.B. // Polymer Science . A. 2008. V. 50. № 4. P. 353.
Chernikova E.V., Golubev V.B., Filippov A.N., Garina E.S. // Polymer Science C. 2015. V. 57. № 1. P. 94.
Studer A., Amrein S. // Synthesis. 2002. № 7. P. 835.
Vishnevetski D.V., Chernikova E.V., Garina E.S., Sivtsov E.V. // Polymer Science B. 2013. V. 55. № 9–10. P. 515.
Litmanovich E.A., Bekanova M.Z., Shandryuk G.A., Chernikova E.V., Talroze R.V. // Polymer. 2018. V. 142. P. 1.
Polymer Handbook/ Ed. by J. Brandrup, E.H. Immergut, E.A. Grulke. New York: Wiley, 1999.
Moad G. // Macromol. Chem. Phys. 2014. V. 215. P. 9.
Zhou Y., He J., Li C., Hong L., Yang Y. // Macromolecules. 2011. V. 44. P. 8446.
Golubev V.B., Filippov A.N., Chernikova E.V., Coote M.L., Lin C.Y., Gryn’ova G. // Polymer Science C. 2011. V. 53. № 1. P. 14.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)