

Высокомолекулярные соединения (серия С), 2019, T. 61, № 1, стр. 39-49
РУТЕНИЙ-КАРБЕНОВЫЕ КОМПЛЕКСЫ В СИНТЕЗЕ ПОЛИБУТАДИЕНА И ЕГО КРОСС-МЕТАТЕЗИСЕ С ПОЛИНОРБОРНЕНОМ
А. А. Моронцев a, М. Л. Грингольц a, *, М. П. Филатова a, А. С. Перегудов b, Т. Р. Акмалов b, С. М. Масоуд b, С. Н. Осипов b, Ю. И. Денисова a, Я. В. Кудрявцев a
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский пр., 29, Россия
b Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: gringol@ips.ac.ru
Поступила в редакцию 13.04.2019
После доработки 30.04.2019
Принята к публикации 13.05.2019
Аннотация
Изучена активность ряда как известных, так и новых, недавно синтезированных Ru-карбеновых катализаторов, в метатезисной полимеризации 1,5-циклооктадиена и в межцепном кросс-метатезисе полученного полибутадиена с полинорборненом. В качестве катализаторов испытаны Ru-комплексы Граббса первого и второго поколения, Граббса–Ховейды второго поколения и их модифицированные аналоги с несимметричными фторсодержащими лигандами и ненасыщенными имидазолильными фрагментами. Показано, что на модифицированных и немодифицированных катализаторах второго поколения образуется частично кристаллический полибутадиен, в котором блоки с транс-связями С=С в ∼4 раза длиннее блоков, содержащих цис-связи С=С. В присутствии катализаторов с более объемными лигандами длина транс-блоков меньше, а температура плавления полимера ниже. Проведено сравнение активности катализаторов методом ЯМР 1Н in situ мониторинга. Впервые путем кросс-метатезиса между полибутадиеном и полинорборненом синтезированы новые мультиблок-сополимеры норборнена и бутадиена. Наибольшая степень блочности сополимеров достигается на катализаторах Граббса и Граббса–Ховейды второго поколения. Построены ряды активности изученных катализаторов. Показано, что макромолекулярный кросс-метатезис протекает более активно в присутствии катализаторов с менее объемными лигандами.
ВВЕДЕНИЕ
Реакция метатезиса олефинов находит широкое применение в полимерном синтезе – от производства промышленных полинорборнена (Norsorex®), полидициклопентадиена (Telene®, Metton®, Pentam® и Proxima™), полиоктенамера (Vestenamer®), их гидрированных аналогов (Zeonex®, Zeonor®, Arton®) [1–8] до синтеза макромолекул сложной структуры и архитектуры [9–11]. Значительные успехи в макромолекулярном дизайне были достигнуты после открытия Р. Шроком и Р. Граббсом высокоэффективных катализаторов – металлокарбеновых комплексов Мо, Ru, W, Re и т.д. [12–14]. Способность металлокарбеновых комплексов вести “живую” полимеризацию циклоолефинов превратило метатезисную полимеризацию с раскрытием цикла в эффективный инструмент синтеза макромолекул с контролируемой длиной и строением основной цепи, функциональными боковыми заместителями, а также получения блок-, привитых- и других сополимеров сложной архитектуры. Разработаны методы синтеза биодеградируемых, биологически активных, биовозобновляемых, самозаживляемых, супрамолекулярных полимеров, телехеликов, функциональных и монолитных подложек, наночастиц и т.д. [1, 9, 10]. Развиваются подходы к постфункционализации метатезисных полимеров путем модификации двойных связей основной цепи, боковых и концевых групп [11, 15–29]. В последнее десятилетие появились публикации, посвященные реакции кросс-метатезиса между макромолекулами, являющейся по сути реакцией межцепного обмена между полимерами, содержащими двойные связи в основной цепи [30–36]. Ее исследования развиваются в направлении синтеза и изучения свойств новых статистических мультиблок-сополимеров. Помимо возможности получения сополимеров из гомополимеров, синтезированных по различным механизмам, макромолекулярный кросс-метатезис эффективен в синтезе сополимеров мультиблочного строения, которые не могут быть получены из мономеров. Примером являются сополимеры высокоактивного норборнена с циклооктеном и его производными [36–42]. Настоящая работа, в которой изучается кросс-метатезис между полинорборненом и полибутадиеном, является продолжением исследований такого рода. Оба полимера производятся в промышленности и обладают целым рядом интересных свойств, которые могли бы быть объединены в мультиблок-сополимере. Синтез сополимеров норборнен (НБ)–1,4-бутадиен (БД) из мономеров по реакции кросс-метатезиса невозможен, поскольку БД не полимеризуется по схеме метатезиса. Статистические сополимеры НБ–БД, не имеющие тенденции к блочности, могут быть получены по реакции сометатезиса норборнена и 1,5-циклооктадиена [43–47]. Синтез таких сополимеров из полинорборнена (ПНБ) и полибутадиена (ПБД) представляется более перспективным как для получения макромолекул блочного строения, так и с точки зрения доступности промышленных реагентов.
Ранее нами была изучена реакция макромолекулярного кросс-метатезиса на примере ПНБ и полиоктенамера на катализаторах Граббса [36–42]. Было показано, что сначала катализатор взаимодействует с гомополимерами с образованием активных центров реакции – полимерных Ru-карбеновых комплексов, которые затем ведут обмен фрагментами полимерных цепей между гомополимерами по схеме кросс-метатезиса. В результате взаимодействия катализатора с полимерами снижается средняя ММ цепей. Существует ряд факторов, способных препятствовать этому, но наибольший эффект оказывает уменьшение количества катализатора за счет увеличения его активности. В настоящей работе впервые изучена реакция кросс-метатезиса между ПНБ и ПБД под действием ряда известных, а также недавно синтезированных Ru-катализаторов метатезиса. Рутений-карбеновые катализаторы представляют особый интерес для химии высокомолекулярных соединений благодаря способности вести “живую” полимеризацию, толерантности к большинству функциональных групп, стабильности при хранении и в ходе превращений, высокой эффективности, возможности мониторинга активных центров реакции с помощью спектроскопических методов [10, 14]. В качестве катализаторов были выбраны коммерчески доступные Ru-карбеновые катализаторы Граббса первого (G-1)
и второго поколения (G-2)
 ,
,
катализатор Граббса–Ховейды второго поколения (HG)
 ,
,
его аналог с ненасыщенным имидазолильным циклом (uHG)
 ,
,
а также ряд недавно синтезированных Ru-карбеновых комплексов с несимметричными фторсодержащими лигандами – производных G-2 и HG, продемонстрировавших высокую активность в реакции метатезиса [48, 49]. Катализаторы с несимметричными лигандами в последние годы привлекают внимание благодаря способности проявлять стереоселективность в реакции метатезиса олефинов, см. обзор [50] и ссылки в нем, [51–53]. Кроме того, наличие объемной фторсодержащей группы в непосредственной близости к атому рутения способно обеспечить повышенную стабильность комплекса за счет экранирования металлоцентра от негативного воздействия окружающей среды [54]. Активность выбранных катализаторов изучена в синтезе ПБД метатезисной полимеризацией 1,5-циклооктадиена (ЦОД), а также в новой межцепной реакции ПНБ с ПБД.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Методы анализа
Спектры ЯМР 1Н и ЯМР 13С{1H} регистрировали для растворов полимеров и сополимеров в CDCl3 на спектрометре “BrukerAvance 600” с рабочей частотой 600.22 и 150.92 МГц соответственно. Химические сдвиги определяли относительно остаточного сигнала растворителя (7.28 м.д. для ПМР и 77.23 м.д. для ЯМР 13C) и пересчитывали к тетраметилсилану. Спектры ЯМР для кинетических исследований регистрировали для растворов цис,цис-1,5-циклооктадиена в CDCl3 на ЯМР-спектрометре “Bruker MSL-300” c рабочей частотой по протонам 300 МГц. Химические сдвиги фиксировали относительно остаточных сигналов хлороформа.
Калориметрические измерения проводили на дифференциальном сканирующем калориметре DSC823e (“Mettler Toledo”) при скорости изменения температуры 20 град/мин до 200°C в атмосфере аргона (скорость потока газа 70 мл/мин), используя данные второго нагревания. Результаты измерений обрабатывали с помощью сервисной программы STARe, поставляемой в комплекте с прибором. Точность измерения температуры составляла ±0.3°C, энтальпии ±1 Дж/г.
Молекулярно-массовые характеристики определяли методом ГПХ на жидкостном хроматографе “Waters” с рефрактометрическим детектором и системой последовательно соединенных колонок WAT054460 (“Waters”) и G3000HHR (“TosohBiosep”) со сшитым ПС в качестве наполнителя. Элюентом служил ТГФ, скорость подачи 1 мл/мин, температура колонки 25°C, концентрация образца 1 мг/мл, объем пробы 100 мкл. Калибровку прибора выполняли по ПС-стандартам (“PolymerLabs”).
Все операции с соединениями, чувствительными к воздуху и влаге, проводили на стандартной вакуумной установке и линии Шленка в атмосфере аргона с использованием абсолютированных растворителей.
Материалы и методы синтеза
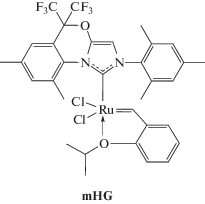
Хлороформ и хлористый метилен абсолютировали кипячением над гидридом кальция. Норборнен (“AcrosOrganics”) и цис,цис-1,5-циклооктадиен (“Sigma–Aldrich”) очищали кипячением над натрием. Абсолютированные растворители и очищенные мономеры перегоняли и хранили в атмосфере аргона. Норборнен использовали в виде 3.15 М раствора в хлороформе, ТГФ (“х. ч.”) перегоняли над щелочью. Метиловый и этиловый спирты (“х. ч.”), ингибитор окисления 2,2′-метилен-бис-(6-трет-бутил-4-метилфенол), катализаторы Граббса первого (PCy3)2Cl2Ru=CHPh (“Sigma–Aldrich”) и второго поколений (“Sigma–Aldrich”) использовали без дополнительной очистки. По методикам, опубликованным в работе [48] и [49], синтезировали катализаторы с несимметричными лигандами G-2a, G-2b, HG-a, HG-b [48] и mHG [49]:
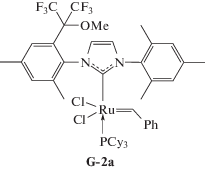
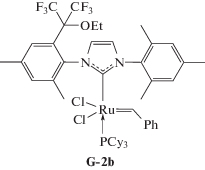
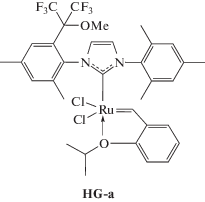
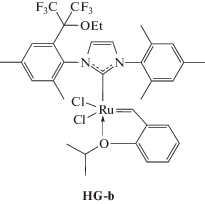
Синтез исходных гомополимеров. Полинорборнен синтезировали метатезисной полимеризацией норборнена, как описано в работе [55]. Выход ПНБ составил 99%, Mw = 343 × 103, Ð = 2.0, Tс = = 39°С, транс С=С – 80%.
Полибутадиен получали следующим образом. В двугорлом реакторе объемом 50 мл, снабженном магнитной мешалкой, в атмосфере аргона готовили раствор 1,5-циклооктадиена (1.60 мл, 1.41 г, 13.0 ммоля) в 1.43 мл хлористого метилена. Раствор трижды дегазировали, заполняли реактор аргоном и добавляли 0.0025 г (0.00294 ммоля) сухого катализатора. Через 5 мин раствор разогревался, начинал пузыриться, и возрастала вязкость. Через 3 ч реакцию останавливали введением 0.15 мл винилэтилового эфира и 0.045 мг ингибитора окисления, через 0.5 ч раствор разбавляли 22 мл хлороформа (“х. ч.”) и полимер осаждали в этаноле. Белый твердый полимер сушили в вакууме до постоянной массы. Получили 1.4 г (99%) ПБД, Mw = 148 × 103, Ð = 1.6, Tс = –96°С, Tпл = = –15 и +5°С, транс С=С – 70%.
Перед реакцией межцепного кросс-метатезиса гомополимеры очищали от следов катализатора колоночной хроматографией на силикагеле (марка Silica 60, 0.04–0.063 мм, MACHEREY-NAGEL GmbH@Co, KG), элюент хлороформ.
Кросс-метатезис между ПНБ и ПБД. В двугорлой колбе объемом 25 мл, оборудованной магнитной мешалкой, в атмосфере аргона растворяли 0.0645 г (0.686 ммоля в расчете на двойные связи) ПНБ и 0.0374 г (0.693 ммоля в расчете на двойные связи) ПБД в 0.82 мл абсолютированного хлороформа в течение 1 суток. Раствор трижды дегазировали, реактор заполняли аргоном и вводили 0.11 мл (0.000514 ммоля) раствора катализатора в хлороформе с концентрацией 0.0045 моль/л. Через 24 ч реакцию останавливали добавлением 0.1 мл винилэтилового эфира, спустя 30 мин вводили 2.4 мг ингибитора окисления и 3.5 мл хлороформа (“х. ч.”). Сополимер высаживали в метанол и сушили в вакууме до постоянной массы. Получили 0.0919 г (90%) белого твердого сополимера. Mw = 165 × 103, Ð = 1.7, LНБ = 30.5, LБД = 25.0.
ЯМР 1Н in situ мониторинг кинетики полимеризации цис,цис-1,5-циклооктадиена. Мономер ЦОД (0.46 ммоля) и 0.66 мл абсолютированного d-хлороформа помещали в ЯМР-ампулу Янга. Смесь трижды дегазировали, используя методику замораживание–вакуумирование–оттаивание. К замороженной смеси добавляли 0.065 мл (5.8 × 10–4 ммоля) отдельно приготовленного раствора катализатора с концентрацией 8.9 ммоль/л и 0.23 мл d-хлороформа (для полного перенесения раствора катализатора в ампулу). Перед съемкой смесь размораживали, перемешивали и немедленно помещали в ЯМР-спектрометр. Расход ЦОД определяли сравнением интегралов сигналов протонов мономера (2.37 м.д.) и полимера (2.08, 2.04 м.д.). Погрешность экспериментов составляла 10–15%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Синтез гомополимеров полинорборнена и полибутадиена
Исходные гомополимеры были получены полимеризацией норборнена и 1,5-циклооктадиена по схеме метатезиса с раскрытием цикла:
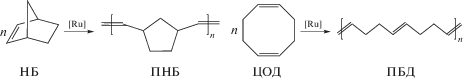 (1)
(1)
В присутствии катализатора Граббса первого поколения (G-1) напряженные молекулы норборнена полимеризуются с большой скоростью, образуя высокомолекулярный полимер. В отличие от норборнена 1,5-циклооктадиен менее активен в полимеризационном метатезисе с раскрытием цикла вследствие низкого напряжения цикла [1]. Изучено влияние типа катализаторов (G-1, G-2 и их модифицированных аналогов) на выход и характеристики получаемого ПБД (табл. 1). Обнаружено, что тип катализатора влияет на соотношение цис/транс двойных связей в ПБД и на его кристалличность. Так, в присутствии катализатора G-1 образуется аморфный ПБД с соотношением цис/транс двойных связей 44:56 (табл. 1, опыт 2), остальные испытанные катализаторы G-2 (опыт 3), катализаторы HG и модифицированные аналоги позволяют получать кристаллический ПБД с соотношением цис/транс двойных связей около 20 : 80 (опыты 4–9). При этом более активные G-2, HG и модифицированные катализаторы требуются в количестве в 6–7 раз меньше, чем G-1.
Таблица 1.
Полимеризация норборнена (опыт 1) и 1,5-циклооктадиена (опыты 2–9) по схеме метатезиса с раскрытием цикла на Ru-катализаторах (T = 25°С, 3 ч)
| Опыт, № | Катали-затор | Мольное отношение [мономер] : : [катализатор] | Выход, % | Mw × 10–3 | Ð | Транс С=С*, % | Lцис | Lтранс | Tс, °C | Tпл, °C |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | G-1 | 780 | 93 | 343 | 2.0 | 80 | н/о | н/о | 39 | – |
| 2 | G-1 | 660 | 85 | 111 | 2.0 | 56 | н/о | н/о | –102 | – |
| 3 | G-2 | 4600 | 94 | 149 | 1.6 | 79 | 1.2 | 4.7 | –92 | 11, 29 |
| 4 | HG | 4230 | 91 | 124 | 1.8 | 78 | 1.3 | 5.7 | –93 | 37, 49 |
| 5 | G-2a | 4180 | 94 | 130 | 1.7 | 79 | 1.2 | 4.7 | н/о | н/о |
| 6 | G-2b | 4970 | 93 | 128 | 1.6 | 79 | 1.2 | 4.4 | –93 | 23 |
| 7 | mHG | 3800 | 94 | 131 | 1.6 | 82 | 1.2 | 5.7 | –88 | 18, 28, 38, 53 |
| 8 | HG-a | 4150 | 94 | 128 | 1.6 | 82 | 1.2 | 5.1 | –92 | 25, 40 |
| 9 | HG-b | 3660 | 94 | 139 | 1.6 | 80 | 1.2 | 4.7 | –89 | 13, 34 |
Отметим, что полученные результаты (табл. 1) не дают представления о сравнительной активности изученных катализаторов: выход, ММ, соотношение цис/транс-двойных связей в полимерных продуктах практически одинаковы для всех катализаторов, кроме G-1.
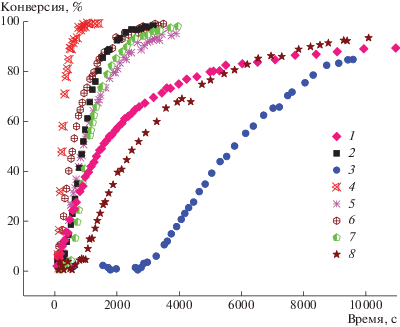
Сравнительная активность катализаторов была изучена путем ЯМР 1H in situ мониторинга степени превращения мономера ЦОД в ходе его полимеризации. Конверсию ЦОД определяли из интегралов сигналов аллильных протонов мономера (2.37 м.д.) и полимера (2.08, 2.04 м.д.). Полученные кинетические кривые представлены на рис. 1. Видно, что катализаторы G-2 и G-2a, G-2b и mHG близки по активности в полимеризации ЦОД, в то время как G-1 и HG-a менее активны. За исключением mHG модифицированные катализаторы в отличие от G-2 и HG содержат двойную связь в имидазолильном кольце. Для выяснения ее роли мы изучили активность ненасыщенных uG-2
Рис. 1.
Кинетические кривые полимеризации цис,цис-1,5-циклооктадиена на Ru-катализаторах G-1 (1), G-2 (2), uG-2 (3), uHG (4), G-2a (5), G-2b (6), mHG (7), HG-a (8). Мольное соотношение ЦОД : катализатор = 850, [ЦОД] = = 0.45–0.47 моль/л, Т = 20°С, CDCl3.
и uHG
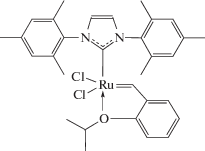
в полимеризации ЦОД. Из рис. 1 следует, что в полимеризации ЦОД наименее активным среди всех катализаторов оказался uG-2, а наиболее активным uHG. Таким образом, в результате модификации фторсодержащими группами активность uHG несколько снижается, а uG-2 значительно повышается.
Интересные результаты получены при исследовании микроструктуры образцов ПБД, синтезированных на Ru-катализаторах (табл. 1, опыты 3–9). В спектре ЯМР 13С полибутадиена хорошо разрешены сигналы диад транс-цис, транс-транс, цис-цис, цис-транс-двойных связей бутадиена (рис. 2). Это позволяет вычислить длину блоков из звеньев, содержащих цис- и транс-двойные связи.
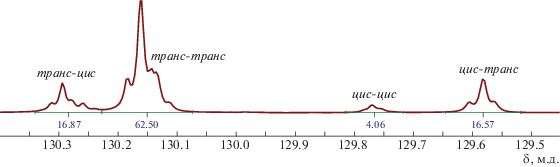
Все испытанные катализаторы формировали практически одинаковые короткие блоки цис-С=С в ПБД, средняя длина которых составляла 1.2–1.3 звена (табл. 1). Средняя длина блоков транс-С=С была в ∼4 раза выше и варьировалась в интервале 4.4–5.7 в зависимости от типа катализатора. Так, в присутствии катализатора G-2 она равна 4.7 звена, а в присутствии HG – 5.7. Для катализаторов с несимметричными фторсодержащими лигандами – производными G-2 (G-2a и G-2b) в случае группы ОМе в лиганде длина транс-блока была такой же, как для G-2 (табл. 1, опыты 3 и 5). Замена группы ОМе на более объемную группу ОЕt (катализатор G-2b) привела к снижению длины транс-блока (табл. 1, опыт 6).
Для модифицированных катализаторов производных HG зависимость длины транс-блока в ПБД от строения лиганда более выражена. Введение в HG группы OMe, наряду с двумя трифторметильными (HG-a), приводит к снижению Lтранс с 5.7 до 5.1 (опыт 8). Замена группы OMe на группу OEt уменьшает Lтранс до 4.7 (опыт 9).
Указанные изменения длины транс-блоков сказываются на термических и кристаллических свойствах получаемых полимеров (табл. 1 и рис. 3). Так, ПБД с наименьшей средней длиной транс-блока в 4.4 звена (катализатор G-2b) имеет один пик плавления при Тпл, близкой к комнатной (табл. 1, опыт 6). С увеличением длины транс-блока до 4.7 для ПБД, синтезированных на различных катализаторах (G-2 и HG-b), появляется второй пик с более высокой Тпл, (табл. 1, опыты 3 и 9; рис. 3). При длине транс-блока до 5.1, значения обеих Тпл несколько возрастают (опыт 8), а на кривой ДСК для ПБД с наибольшей длиной транс-блока, равной 5.7 звеньев, появляется новый эндотермический пик с максимальной Тпл = = 49–53°С (табл. 1, опыт 7).
Рис. 3.
Кривые ДСК полибутадиена, синтезированного на Ru-катализаторах G-2b (1), G-2 (2), HG-b (3), HG-a (4), HG (5) и mHG (6). Стрелками показана температура стеклования.
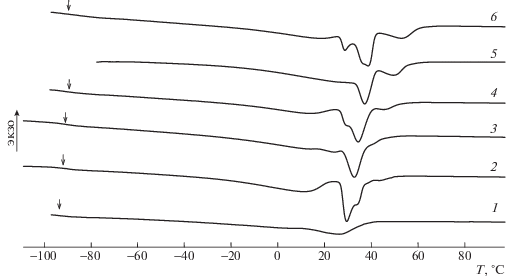
Таким образом, изменяя тип катализатора и структуру лиганда в Ru-катализаторах, можно регулировать кристаллические свойства ПБД.
Макромолекулярный кросс-метатезис между ПНБ и ПБД
Активность Ru-катализаторов была изучена в реакции макромолекулярного кросс-метатезиса между ПНБ и ПБД. Эксперименты проводили в хлороформе, обеспечивающем высокую концентрацию реагентов. К раствору гомополимеров добавляли раствор катализатора, после чего в течение 20–30 мин вязкость реакционной смеси снижалась. Как было показано нами ранее с помощью специальных кинетических исследований, на начальной стадии катализатор взаимодействует с двойными связями гомополимеров, “разрезая” их по механизму олефинового метатезиса с образованием фрагментов полимерных цепей с Ru-карбеном на конце, [Ru]=ПНБ и [Ru]=ПБД [39]:
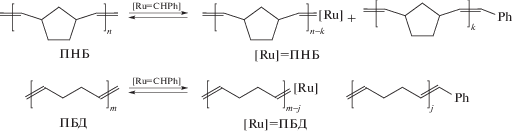 (2a)
(2a)
Такие полимерные карбены ведут цепь превращений, заключающихся в обмене фрагментами макромолекул и образованием сначала диблок-:
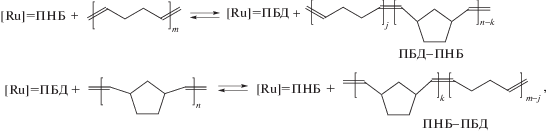 (2b)
(2b)
а затем статистических мультиблок-сополимеров (МБС)
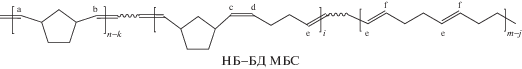 (2c)
(2c)
Об образовании сополимеров судили по появлению сигналов атомов углерода гетеродиад (Сс,d) в спектрах ЯМР 13С (рис. 4). Согласно литературным данным [45, 46], сигналы углеродов в гомо- (цис и транс) и гетеродиадах норборнена и бутадиена (рис. 4) хорошо разрешаются.
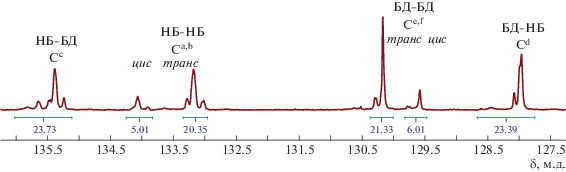
Специально подобранный режим съемки спектров ЯМР 13С позволяет использовать интегральные интенсивности для расчета средней длины блоков в сополимерах по формулам
Были выбраны условия реакции с максимально возможной концентрацией гомополимеров в реакционной смеси (2 моль/л) для подавления внутримолекулярного метатезиса, приводящего к образованию циклоолигомеров, а также мольное соотношение [катализатор] : [гомополимеры] = = 1 : (2500–3000). В указанных условиях был осуществлен кросс-метатезис ПНБ с ПБД в присутствии различных Ru-катализаторов. Из результатов, представленных в табл. 2, видно, что выход сополимеров составляет 84–92%. МБС, синтезированные на модифицированных катализаторах G-2a, G-2b, mHG, HG-a, характеризуются примерно одинаковой ММ, равной (150–165) × 103 (табл. 2, опыты 14–17), на катализаторах G-2, HG и HG-b получены МБС с несколько большей MМ – (190–200) × 103 (опыты 11, 12, 18). Существенно различается средняя длина блоков МБС, полученных на катализаторах Граббса (G-2, HG, uHG) и на модифицированных несимметричными фторсодержащими лигандами. На катализаторах Граббса образуются МБС с меньшей длиной блока, что свидетельствует о более интенсивном обмене сегментами полимерных цепей. Разница в активности изученных катализаторов более наглядна при сравнении степени блочности МБС, определяемой как отношение доли гетеродиад к значению этой величины для полностью случайного сополимера того же состава (рис. 5). Меньшая активность модифицированных катализаторов может быть связана с большим объемом их лигандов, препятствующих взаимодействию катализатора с макромолекулами ПНБ и ПБД. На это также указывает наименьшая активность катализаторов G-2b и HG-b (рис. 5), содержащих самые объемные лиганды с группой OEt.
Таблица 2.
Кросс-метатезис между ПНБ и ПБД под действием различных Ru-карбеновых катализаторов и характеристики синтезированных мультиблок-сополимеров
| Опыт, № | Катализатор | Выход МБС, % | Mw × 10–3 | Ð | LНБ | LБД | φ** | Цис-С=С ПНБ,% | Цис-С=С ПБД, % |
|---|---|---|---|---|---|---|---|---|---|
| 10* | G-1 | 87 | 88 | 1.6 | 23 | 23.0 | 0.09 | 14 | 19 |
| 11 | G-2 | 91 | 200 | 1.7 | 2.3 | 2.3 | 0.87 | 30 | 20 |
| 12 | HG | 92 | 189 | 1.6 | 2.3 | 2.2 | 0.89 | 30 | 18 |
| 13 | uHG | 89 | 102 | 2.0 | 2.3 | 2.3 | 0.87 | 28 | 19 |
| 14 | G-2a | 84 | 149 | 1.7 | 24 | 21.0 | 0.09 | 19 | 19 |
| 15 | G-2b | 86 | 155 | 1.7 | 46 | 40.0 | 0.05 | 18 | 20 |
| 16 | mHG | 90 | 165 | 1.7 | 30 | 25.0 | 0.07 | 19 | 19 |
| 17 | HG-a | 84 | 159 | 1.7 | 32 | 30.0 | 0.06 | 19 | 18 |
| 18 | HG-b | 92 | 199 | 1.5 | 119 | 130.0 | 0.02 | 18 | 19 |
Рис. 5.
Степень блочности мультиблок-сополимеров норборнена и бутадиена, синтезированных на различных Ru-катализаторах.
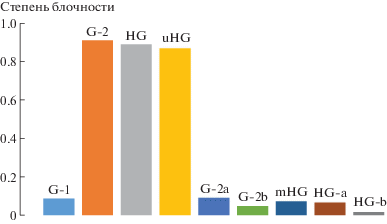
При рассмотрении поведения немодифицированных катализаторов (опыты 10–13) видно, что катализатор G-1 наименее активен в приведенном ряду (кросс-метатезис в его присутствии проводили при низком мольном соотношении G-1 : полимеры = 1 : 440 (опыт 10)). На катализаторах G-2 и HG молекулярная масса получаемых МБС выше, а на uHG – ниже, чем для остальных катализаторов (опыты 11–13). Это может свидетельствовать о различиях в скорости образования Ru-полимерных карбеновых комплексов (схема (2а)), в ходе которого происходит снижение ММ, в то время как собственно межцепной обмен на катализаторах Граббса второго поколения протекает очень эффективно (схема (2б)) [39], и синтезированные на катализаторах G-2, HG и uHG сополимеры по степени блочности близки к полностью случайным (рис. 5).
Необычным является увеличение до 30% доли цис-двойных связей в гомодиадах НБ–НБ на катализаторах Граббса (G-2, HG и uHG) (исходный ПНБ содержал 20% цис-С=С) (опыты 11–13). В кросс-метатезисе ПНБ с полиоктенамерами на катализаторе G-1 этот эффект не проявляется [36–42]. Возможно, катализаторы второго поколения более активно взаимодействуют с транс С=С связями полимерных цепей, что приводит к накоплению цис С=С связей. Таким образом, ряд активности изученных катализаторов в реакции макромолекулярного кросс-метатезиса ПНБ и ПБД выглядит следующим образом:
что существенно отличается от ряда активности катализаторов в метатезисной полимеризации ЦОД с раскрытием цикла:ЗАКЛЮЧЕНИЕ
В работе изучена активность ряда Ru-катализаторов Граббса и их аналогов с несимметричными фторсодержащими лигандами в реакции метатезисной полимеризации 1,5-циклооктадиена с раскрытием цикла и в кросс-метатезисе между полибутадиеном и полинорборненом. Показано, что поведение катализаторов существенно зависит от типа реагентов. В термодинамически выгодной реакции метатезисной полимеризации с раскрытием цикла активность катализаторов различается незначительно. В кросс-метатезисе с участием макромолекул увеличение объема лигандов в Ru-катализаторах приводит к существенному снижению их активности. Обнаружено, что изменением лигандного окружения в катализаторах Граббса можно регулировать термические и кристаллические свойства полибутадиена, получаемого полимеризацией ЦОД. Отмечено изменение стереосостава гомодиад норборнена в синтезируемых мультиблок-сополимерах.
Авторы выражают признательность Г.А. Шандрюку за проведение исследований методом ДСК и А. Шафигулиной – методом ГПХ.
Строение полученных соединений изучено с использованием оборудования Центра исследования строения молекул ИНЭОС РАН и Центра Коллективного пользования ИНХС РАН.
Работа выполнена в рамках Государственного задания ИНХС РАН и при частичной финансовой поддержке Российского фонда фундаментальных исследований (проект 17-03-00596) (Межцепной кросс-метатезис между ПНБ и ПБД).
Список литературы
Ivin K.J., Mol J.C. Olefin Metathesis and Metathesis Polymerization. London: Acad. Press, 1997.
Mol J.C. // J. Mol. Cat. 2004. V. 213. P. 39.
Claverie J.P., Soula R. // Prog. Polym. Sci. 2003. V. 28. P. 619.
Esteruelas M.A., González F., Herrero J., Lucio P., Oliván M., Ruiz-Labrador B. // Polym. Bull. 2007. V. 58. P. 923.
Xu J., Li A., Wang H., Shen Y. // Adv. Mech. Eng. 2016. V. 8. P. 1.
Slugovc C. // Olefin Metathesis: Theory and Practice / Ed. by K.Grela. New Jersey: Wiley, 2014. P. 329.
Erkeçoğlu S., Sezer A.D., Bucak S. // Smart Drug Delivery System / Ed. by A.D. Sezer. InTechOpen, 2016. P. 396.
Yamazaki M. // J. Mol. Cat. A. 2004. V. 213. P. 81.
Handbook of Metathesis / Ed. by R.H. Grubbs. Weinheim: Wiley-VCH, 2003. V. 3. P. 419.
Handbook of Metathesis / Ed. by R.H. Grubbs, E. Khosravi. Weinheim: Wiley-VCH, 2015. V. 3. P. 424.
Chen Y., Abdellatif M.M., Nomura K. // Tetrahedron. 2018. V. 74. P. 619.
Schrock R.R. // Angew. Chem. Int. Ed. 2006. V. 45. P. 3748.
Grubbs R.H. // Angew. Chem. Int. Ed. 2006. V. 45. P. 3760.
Ogba O.M., Warner N.C., O’Leary D.J., Grubbs R.H. // Chem. Soc. Rev. 2018. V. 47. P. 4510.
Boaen N.K., Hillmyer M.A. // Chem. Soc. Rev. 2005. V. 34. P. 267.
Li Z.-L., Sun L., Ma J., Zen Z., Hong J. // Polymer. 2016. V. 84. P. 336.
Zhao Y., Zhang K. // Polym. Chem. 2016. V. 7. P. 4081.
Dong Y., Matson J.B., Edgar K.J. // Biomacromolecules. 2017. V. 18. P. 1661.
Sinclair F., Alkattan M., Prunet J., Shaver M.P. // Polym. Chem. 2017. V. 8. P. 3385.
Morrison S.D., Liskamp R.M.J., Prunet J. // Org. Lett. 2018. V. 20. P. 2253.
Lunn D.J., Discekici E.H., de Alaniz J.R., Gutekunst W.R., Hawker C.J. // J. Polym. Sci., Polym. Chem. 2017. V. 55. P. 2903.
Boyd T.J., Schrock R.R. // Macromolecules. 1999. V. 32. P. 6608.
Trupej N., Novak Z., Knez Ž., Slugovc C., Kovačič S. // J. CO2 Util. 2017. V. 21. P. 336.
Morontsev A.A., Gringolts M.L., Filatova M.P., Finkelshtein E.Sh. // Polymer Science B. 2016. V. 58. № 6. P. 695.
Berron B.J., Payne P.A., Jennings G.K. // Ind. Eng. Chem. Res. 2008. V. 47. P. 7707.
Arend van Hensbergen J., Burford R.P., Lowe A.B. // J. Polym. Sci., Polym. Chem. 2013. V. 51. P. 487.
Zhao Yu., Chen J., Zhu W., Zhang Ke. // Polymer. 2015. V. 74. P. 16.
Morontsev A.A., Zhigarev V.A., Nikiforov R.Yu., Belov N.A., Gringolts M.L., Finkelshtein E.Sh., Yampolskii Yu.P. // Eur. Polym. J. 2018. V. 99. P. 340.
Belov N.A., Gringolts M.L., Morontsev A.A., Starannikova L.E., Yampolskii Yu.P., Finkelshtein E.Sh. // Polymer Science B. 2017. V. 59. № 5. P. 560.
Wagner N.L., Timmer F.J., Arriola D.J., Jueptner G., Landes B.G. // Macromol. Rapid Commun. 2008. V. 29. P. 1438.
Otsuka H., Muta T., Sakada M., Maeda T., Takahara A. // Chem. Commun. 2009. P. 1073.
Maeda T., Kamimura S., Ohishi T., Takahara A., Otsuka H. // Polymer. 2014. V. 55. P. 6245.
Ohishi T., Suyama K., Kamimura S., Sakada M., Imato K., Kawahara S., Takahara A., Otsuka H. // Polymer. 2015. V. 78. P. 145.
Radlauer M.R., Matta M.E., Hillmyer M.A. // Polym. Chem. 2016. V. 7. P. 6269.
Daniele S., Mariconda A., Guerra G., Longo P., Giannino L. // Polymer. 2017. V. 130. P. 143.
Gringolts M.L., Denisova Yu.I., Shandryuk G.A., Krentsel L.B., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // RSC Adv. 2015. V. 5. P. 316.
Denisova Yu.I., Gringolts M.L., Krentsel’ L.B., Shandryuk G.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science. B. 2016. V. 58. № 3. P. 292.
Gringolts M.L., Denisova Y.I., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2019. V. 15. P. 218.
Denisova Yu.I., Gringolts M.L., Peregudov A.S., Krentsel L.B., Litmanovich E.A., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Beilstein J. Org. Chem. 2015. V. 11. P. 1796.
Denisova Y.I., Gringolts M.L., Roenko A.V., Shandryuk G.A., Finkelshtein E.Sh., Kudryavtsev Y.V. // Mendeleev Commun. 2017. V. 27. P. 416.
Shandryuk G.A., Denisova Y.I., Gringolts M.L., Krentsel L.B., Litmanovich A.D., Finkelshtein E.Sh., Kudryavtsev Y.V. // Eur. Polym. J. 2017. V. 86. P. 143.
Denisova Yu.I., Gringolts M.L., Krentsel’ L.B., Shandryuk G.A., Peregudov A.S., Finkelshtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2017. V. 59. № 4. P. 412.
Greene R.M.E., Ivin K.J., Rooney J.J., Kress J., Osborn J.A. // Makromol. Chem. 1988. V. 189. P. 2797.
Stelzer F., Graiman Ch., Hummel K. // Colloid Polym. Sci. 1982. V. 260. P. 829.
Ivin K.J., Lapienisa G., Rooney J.J. // Makromol. Chem. 1982. V. 183. P. 9.
Cetinkaya S., Karabulut S., ImamogˇluY. // Eur. Polym. J. 2005. V. 41. P. 467.
Michel X., Fouquay S., Michaud G., Simon F., Brusson J.-M., Roquefort P., Aubry T., Carpentier J.-F., Guillaume S.M. // Polym. Chem. 2017. V. 8. P. 1177.
Akmalov T.R., Masoud S.M., Petropavlovskikh D.A., Zotov M.A., Nefedov S.E., Osipov S.N. // Mendeleev Commun. 2018. V. 28. P. 609.
Masoud S.M., Akmalov T.R., Palagin K.A., Dolgushin F.M., Nefedov S.E., Osipov S.N. // Eur. J. Org. Chem. 2018. P. 5988.
Paradiso V., Costabile Ch., Grisi F. // Beilstein J. Org. Chem. 2018. V. 14. P. 3122.
Van Veldhuizen J.J., Garber S.B., Kingsbury J.S., Hoveyda A.H. // J. Am. Chem. Soc. 2002. V. 124. P. 4954.
Thomas R.M., Keitz B.K., Champagne T.M., Grubbs R.H. // J. Am. Chem. Soc. 2011. V. 133. P. 7490.
Hartung J., Grubbs R.H. // J. Am. Chem. Soc. 2013. V. 135. P. 10183.
Masoud S.M., Mailyan A.K., Dorcet V., Roisnel T., Dixneuf P.H., Bruneau C., Osipov S.N. // Organometallics. 2015. V. 34. P. 2305.
Morontsev A.A., Denisova Yu.I., Gringolts M.L., Filatova M.P., Shandryuk G.A., Finkel’shtein E.Sh., Kudryavtsev Ya.V. // Polymer Science B. 2018. V. 60. № 5. P. 688.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)