

Высокомолекулярные соединения (серия С), 2019, T. 61, № 1, стр. 4-19
Метатезисная полимеризация в ионных средах
Д. О. Понкратов a, *, А. С. Шаплов a, b, Я. С. Выгодский a
a Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119991 Москва, ул. Вавилова, 28, Россия
b Luxembourg Institute of Science and Technology
L-4362 Esch-sur-Alzette, 5 Avenue des Hauts-Fourneaux, Люксембург
* E-mail: Mattur@mail.ru
Поступила в редакцию 24.01.2019
После доработки 10.04.2019
Принята к публикации 24.04.2019
Аннотация
Обобщены литературные данные о синтезе полимеров метатезисом олефинов и ацетиленов в среде ионных жидкостей – солей, находящихся в жидком состоянии при комнатной или близкой к ней температуре. Обсуждаются особенности и методы оптимизации процесса полимеризации в ионных средах, в том числе и с использованием ионных катализаторов. Демонстрируется возможность многократного использования ионных растворителей и растворенных в них катализаторов. Представлены данные о полимеризации ионных мономеров и перспективы применения ионных полимеров, полученных метатезисной полимеризацией.
ВВЕДЕНИЕ
Ионные жидкости (ИЖ) – это жидкие при температурах ниже 100°С соли, как правило состоящие из объемного органического катиона
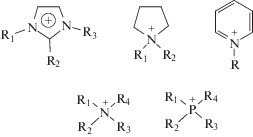
и (не)органического аниона
ИЖ характеризуются крайне низким давлением паров в широком интервале температур, хорошей растворяющей способностью по отношению к разнообразным органическим и неорганическим соединениям, а также каталитическим системам, малой токсичностью, высокой ионной проводимостью, широким окном электрохимической стабильности, хемостойкостью, низкой воспламеняемостью [1–4].
Такое уникальное сочетание свойств делает ИЖ перспективными средами при создании “чистых” химических производств и осуществлении технологических рециклов. В то же время огромное число возможных сочетаний анионов [Br–, Cl–, BF$_{4}^{ - }$, PF$_{6}^{ - }$, (CF3SO2)2N–, (FSO2)2N–, (CN)2N–, CF3SO$_{3}^{ - }$ и т.д.] и катионов (имидазолия, пирролидиния, аммония и т.д.) открывает перспективы для варьирования свойств ИЖ и оптимизации их структуры при использовании в различных физико-химических технологиях. Совершенно иная по сравнению с традиционными органическими растворителями ионная природа ИЖ обусловливает их каталитическую активность, что приводит к повышению селективности многих органических реакций и выхода целевого продукта [5]. В ряде случаев представители этого нового класса растворителей стабилизируют органические катионы и радикалы, активируют катализаторы на основе металлов, обеспечивают высокую скорость процесса, облегчают выделение продуктов реакции и т.д. [1–4]. Более подробную информацию по использованию ИЖ в качестве реакционных сред в органическом и металлоорганическом синтезе можно найти в ряде специализированных обзоров [4–8]. В частности, ИЖ зарекомендовали себя как удобные растворители для метатезиса олефинов [9, 10]. Метатезис можно рассматривать как перераспределение заместителей между парой молекул, содержащих двойную (либо тройную) связь. Реакция метатезиса нашла широкое применение в лабораторной практике и в промышленном органическом синтезе для получения как низкомолекулярных соединений, так и полимеров [11]. В настоящем обзоре будут рассмотрены три типа полимеризации, основанные на реакции метатезиса олефинов и ацетиленов: наиболее распространенная метатезисная полимеризация с раскрытием цикла (ROMP)
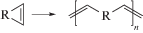 ,
,
полимеризация реакцией обмена ациклических диенов (ADMET)
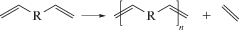
и метатезисная циклополимеризация диинов (MCP)
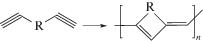
Среди отечественных работ, внесших наибольший вклад в изучение реакций метатезисной полимеризации, следует отметить статьи Е.Ш. Финкельштейна, М.Л. Грингольц, М.В. Бермешева и других авторов [12–17].
КАТАЛИЗАТОРЫ МЕТАТЕЗИСНОЙ ПОЛИМЕРИЗАЦИИ
Важнейшую роль в реакциях метатезиса играет катализатор. В начале 1960-х годов в метатезисной полимеризации с раскрытием цикла применялись каталитические системы, представляющие собой смеси различных соединений, содержащие хлориды переходных металлов, например, WCl6/AlEt2Cl/этанол [18]. В конце 1980-х годов группой R.R. Schrock были разработаны катализаторы на основе молибдена и вольфрама, позволяющие осуществлять “живую” метатезисную полимеризацию с раскрытием цикла для синтеза полимеров с заданной молекулярной массой [19, 20].
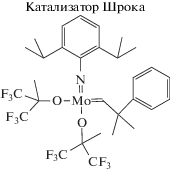
Такие катализаторы показали высокую активность в ROMP-полимеризации широкого ряда замещенных норборненов, норборнадиенов, циклооктадиенов и циклооктатренов с различными функциональными группами [21]. Была обнаружена и повышенная чувствительность катализаторов Шрока к влаге и кислороду воздуха, которая снижает ценность их практического применения.
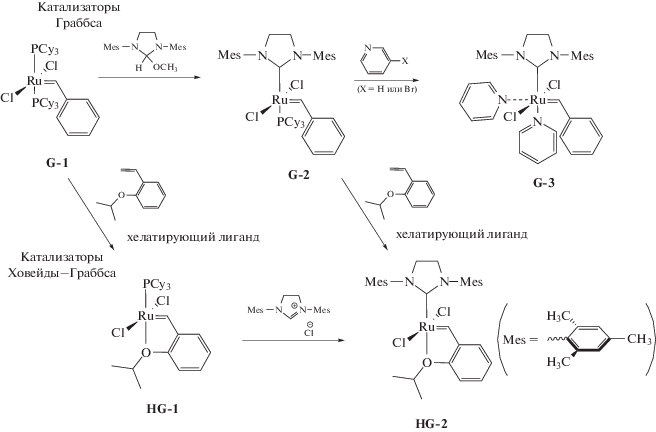
В 1995 г. Граббсом были получены рутениевые катализаторы (G-1), более толерантные к функциональным группам в мономере [22]. Синтезированные позже катализаторы Граббса второго поколения (G-2) с N-гетероциклическим карбеновым лигандом имеют большую активность в реакциях метатезиса, чем G I, при этом они устойчивы к влаге и кислороду воздуха [23, 24]. Третье поколение катализаторов Граббса (G-3) с пиридиновыми лигандами вместо фосфинового отличается повышенной скоростью стадии инициирования роста цепи, что способствует хорошо контролируемой “живой” полимеризации и получению полимеров с заданной ММ и узкой полидисперсностью [25].
В катализаторах Ховейды–Граббса первого и второго поколений (HG-1 и HG-2) бензилиденовые лиганды содержат хелатную орто-изопропоксигруппу, присоединенную к бензольным кольцам. Эти катализаторы дороже катализаторов Граббса и менее активны на стадии инициирования полимеризации, но обладают повышенной стабильностью, что позволяет выделять их из реакционной среды и использовать повторно [26]. Катализаторы Ховейды–Граббса относительно легко синтезируются из катализаторов Граббса соответствующего поколения путем добавления хелатирующего лиганда 1-изопропокси-2-винилбензола.
Катализаторы Граббса и Ховейды–Граббса хорошо растворимы в хлорированных растворителях (хлороформе, дихлорметане и т.д.), а также эфире и ТГФ. Следует отметить, что в ИЖ такие катализаторы растворяются весьма медленно даже при повышенной температуре [27]. Так, при 45°С растворение HG-2 в 1,2-диметил-3-бутил имидазолий гексафторфосфате ([BdmIm]PF6) занимает в среднем 4–5 ч [27]. Для экономии времени катализатор предварительно растворяют в минимальном количестве дихлорметана, добавляют выбранную ИЖ и снова перемешивают до образования раствора, после чего CH2Cl2 удаляют на вакууме [28, 29]. В последнем случае процесс растворения занимает не более 15 мин. Гораздо более быстрым растворением в ионных средах характеризуются ИЖ-модифицированные или ионные катализаторы. Их синтез аналогичен синтезу катализаторов Ховейды–Граббса, но вместо 1-изопропокси-2-винилбензола используется ионный хелатирующий лиганд, т.е. содержащий ИЖ вместо изопропиловой группы или присоединенную к бензольному кольцу [9, 30–43].
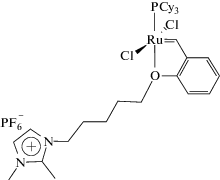
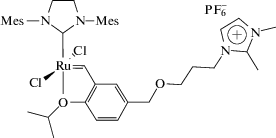
Возможна также модификация катализаторов внедрением ИЖ-модифицированного N-гетероциклического карбенового лиганда [44]
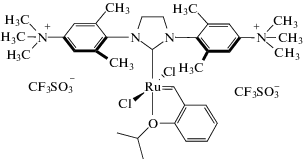 ,
,
заменой хлор-лигандов на ИЖ [36]
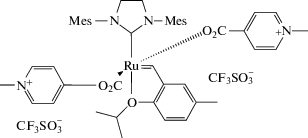 ,
,
синтезом комплексов добавлением солей серебра и ДМФА к HG-2 [45, 46]
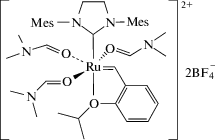
и т.д. Более подробно взаимодействие катализаторов реакций метатезиса с ионными средами рассмотрены в обзорах [9, 10, 34].
Именно те обстоятельства, что ИЖ растворяют многие комплексные соединения металлов [1, 5, 8] в том числе и катализаторы Шрока и Граббса, могут быть использованы в рецикле и ускоряют органические реакции метатезиса [47–50], и послужили основанием для начала изучения их применения в качестве растворителей при синтезе полимеров в реакциях ROPM, ADMET и MCP.
ПОЛИМЕРИЗАЦИЯ В ИОННЫХ СРЕДАХ
Метатезисная полимеризация с раскрытием цикла (ROMP)
Было показано, что в ИЖ по сравнению с молекулярными растворителями метатезисная полимеризация протекает с большей скоростью, с лучшим контролем степени полимеризации и приводит к образованию высокомолекулярных полимеров с количественным выходом [27, 28, 43, 51–55]. В некоторых случаях ИЖ позволяют проводить метатезисную полимеризацию мономеров, не растворимых в традиционно применяемых в реакциях метатезиса органических растворителях [56], или осуществлять сополимеризацию мономеров различной природы, для которых сложно подобрать общий растворитель [57].
Взаимодействие ионной среды с катализатором. Помимо специфической растворяющей способности ИЖ, хорошие результаты полимеризации объясняются также специфическим взаимодействием такой среды с катализатором. При этом во избежание нежелательных побочных реакций, приводящих к пассивации катализатора важен правильный выбор структуры ИЖ. Так, при использовании в качестве реакционной среды наиболее распространенных ИЖ с катионом 1,3-замещенного имидазолия может происходить замещение лигандов катализатора и образование нового комплекса с пониженной каталитической активностью [58]:
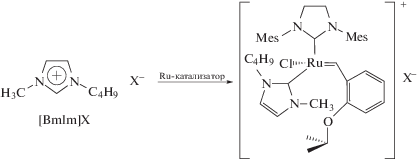
(X– = ${\text{PF}}_{6}^{ - }$, ${\text{BF}}_{4}^{ - }$, CF3${\text{SO}}_{3}^{ - }$, (CF3SO2)2N– и т.д.)
Образованием такого низко реакционного комплекса были объяснены результаты полимеризации экзо,эндо-5-норборнен-2-карбонитрила в [BmIm]PF6
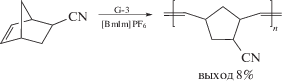 ,
,
при которой полимер либо не удавалось получить вообще, либо при увеличенной до 1 мол. % загрузке катализатора он образовывался с крайне низким (8%) выходом. В дальнейшем вместо ИЖ с незамещенным “кислым” атомом водорода во втором положении имидазолиевого кольца были использованы соли 1,2,3-алкилзамещенного имидазолия
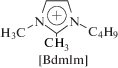
или производные фосфония
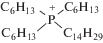
При этом удалось повысить выход полимера до 22 и 85% соответственно [54].
Другим способом защиты катализатора от побочных реакций с 1,3-замещенным имидазолием является добавление к ИЖ таких кислот, как H3PO4 [51] или пинаколфенилбороната [57]. Так, при циклополимерзиации ионного мономера – модифицированного 1,6-гептадиина в среде [BmIm]PF6, разбавленной ТГФ (2:1), добавлением 10% H3PO4 к катализатору G-2 удалось добиться повышения выхода полимера с 60 до 82% [51]:
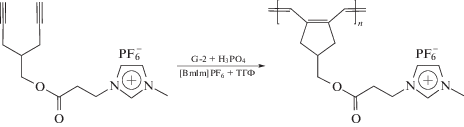
Было показано [54], что при выборе ИЖ важен не только правильный подбор катиона, но и выбор природы аниона, поскольку последний также вносит свой вклад во взаимодействие ионной среды с катализатором. Установлено, что в солях с сильно координированными анионами (Cl–, Br–, NO$_{3}^{ - }$ и т.д.) под действием катализаторов G-3 и HG-2 ROMP-полимеризация метил-(экзо,эндо-5-норборнен-2-карбоксилат)а не протекает:

Напротив, использование в данной реакции ИЖ с анионами BF$_{4}^{ - }$, PF$_{6}^{ - }$, CF3SO$_{3}^{ - }$ и (CF3SO2)2N– способствовало образованию полимеров с высокой молекулярной массой [54].
Растворимость полимеров в ИЖ. Растворимость в ИЖ образующегося полимера также играет огромную роль при осуществлении метатезисной полимеризации в ионной среде. При полимеризации метил (экзо,эндо-5-норборнен-2-карбоксилат)а в ИЖ с катионом [BdmIm] и анионами BF$_{4}^{ - }$, PF$_{6}^{ - }$, CF3SO$_{3}^{ - }$ и (CF3SO2)2N– молекулярная масса Mn образующегося полимера была значительно выше расчетной (до 50 раз) при весьма широком молекулярно-массовом распределении (1.9–4.1). Завышенные ММ наряду с широкой полидисперсностью обусловлены тем, что в указанных ионных растворителях образующийся полимер выпадал в осадок уже через несколько минут после начала реакции, и ROMP протекала в условиях осадительной полимеризации. Несмотря на широкую полидисперсность и невозможность контроля ММ, преимуществом такого процесса является возможность легкого выделения полимера из реакционной массы с высокой степенью чистоты [48]. При переходе к [BdmIm]CF3COO полимеризацию удалось осуществить в растворе. Полученный полимер был выделен с молекулярной массой близкой к расчетной и относительно узким молекулярно-массовым распределением (2.1). Аналогично введение в мономер оксиэтиленовых фрагментов улучшающих растворимость в ионных соединениях, позволило провести метатезисную полимеризацию бис-2-(2-(2-этокси-этокси)этокси)этилового эфира эндо, эндо-норборнен-2,3-дикарбоновой кислоты под действием G-2 в растворе [BmIm]BF4.
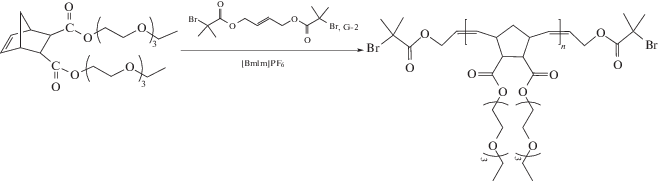
В свою очередь это привело к хорошей контролируемости полимеризации и получению полимера с Ð < 1.5 [29].
Рецикл ионного растворителя и катализатора. В процессе метатезисной полимеризации значительное количество катализатора захватывается растущей полимерной цепью, что, во-первых, приводит к экономическим потерям, во-вторых, продукт сильно загрязняется рутением [47]. Выделение катализаторов хроматографическими методами с целью повторного использования весьма неэкономично и технологически сложно. Благодаря нелетучести ИЖ было предположено, что при осуществлении реакции метатезиса в ионных средах можно не только многократно использовать катализатор, но и его раствор в ИЖ, что значительно улучшит технологичность процесса (рис. 1а). Принимая это во внимание, на примере ROMP-полимеризации ионного производного норборнена была продемонстрирована принципиальная возможность многократного рециклирования системы ИЖ–катализатор [54]:
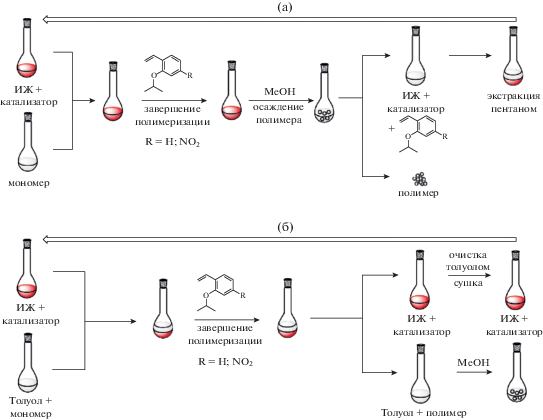
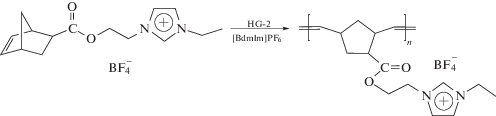
Предложенная схема рецикла состояла в следующем: 1) после завершения реакции в реакционную массу добавляли 2-(2-пропокси)стирол для отделения катализатора (HG-2) от полимерной цепи; 2) далее полимер осаждали метанолом и отфильтровывали; 3) метанол упаривали, а оставшийся раствор катализатора в [BdmIm]PF6 очищали от 2-(2-пропокси)стирола экстракцией пентаном; 4) после удаления следов метанола и пентана из раствора катализатора в ионном растворителе, его использовали в последующем цикле полимеризации (рис. 1а). Установлено, что при повторном использовании раствора катализатора в ИЖ сохраняется высокий выход полимера (91%), однако его молекулярная масса снижается вдвое. При третьем цикле выход полимера уменьшился до 14% [54]. По-видимому, одной из причин снижения выхода полимера с каждым последующим циклом является то, что в самом ионном мономере имеется “кислый” протон у атома С2 имидазольного цикла, который, как было показано выше, способен образовывать низко реакционноспособный комплекс с катализатором HG-2, снижая его активность.
Другой подход к рециклу катализаторов заключается в осуществлении метатезисной полимеризации в двухфазной системе, где одной фазой служит ИЖ, а другая фаза представляет собой несмешивающийся с ИЖ органический растворитель [43, 53, 59–61]. В качестве второй фазы чаще всего применяют дихлорметан или толуол. Основное преимущество данного метода – легкость разделения не растворимого в ИЖ полимера и растворимого катализатора (рис. 1б). При этом смесь ИЖ+катализатор удается многократно использовать в рецикле [44, 59, 60, 62], особенно ИЖ-модифицированные катализаторы с повышенным сродством к ионной среде и практически не растворимых во второй, неионной фазе.
Исследование рецикла катализатора и ИЖ при ROMP-полимеризации норборнена в двухфазной системе [BdmIm]PF6/толуол показало, что катализатор G-1 выдерживает не более одного рецикла, а в случае катализатора G-2 выход полимера уменьшается до 31 и 7% при третьем и четвертом циклах соответственно. Напротив, применение ИЖ-модифицированного катализатора позволило синтезировать полинорборнен в течение пяти циклов с хорошим выходом (97% на четвертом, 64% на пятом цикле) и заданной ММ. В то время как на шестом цикле наблюдалось падение выхода до 27%, на седьмом цикле выход удалось снова повысить до 82% после увеличения времени полимеризации с 30 мин до 2 ч [59].
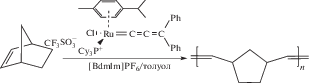
ММ и молекулярно-массовое распределение полимеров при ROMP полимеризации в ионной среде. Следует отметить, что ММ и Ð полимеров, полученных в ИЖ метатезисной полимеризацией варьировались в широком интервале (Mn от 8 × 103 до 1500 × 103, Ð от 1.1 до 4.1). Оба параметра зависели от следующих факторов: выбранной ИЖ и ее взаимодействия с использованным катализатором, типа катализатора и растворимости образующегося полимера в ионной среде. Таким образом, при правильном подборе ионного растворителя и ионного катализатора можно осуществить контролируемую ROMP и синтез полимеров с ММ, близкой к расчетной величине, и узким значением полидисперсности.
Полимеризация реакцией обмена ациклических диенов (ADMET)
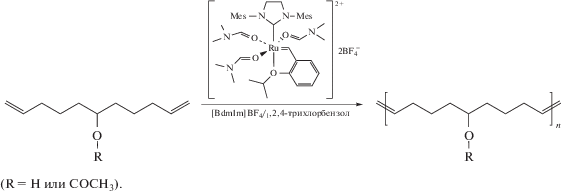
Реакция ADMET протекает по ступенчатому механизму и относится к реакциям поликонденсации. Ввиду этого с целью достижения высокой ММ полимера выделяющийся побочный газообразный продукт этилен необходимо удалять из сферы реакции. Высокая термостойкость и крайне низкое давление паров ИЖ позволяют проводить ADMET реакцию в ионной среде при высоких температурах и/или в вакууме для эффективного удаления этилена и получения высокомолекулярных продуктов. Так, при синтезе триптиценсодержащих полимеров в [BmIm]PF6 наибольшую Мn (11.5 × 103) удалось достигнуть, начиная реакцию при 80°С, а после образования нелетучих олигомеров, выдержкой реакционной массы при 100°С [57]:
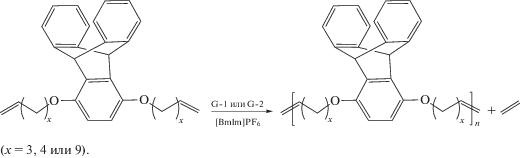
При ADMET в двухфазной системе [BdmIm]BF4/1,2,4-трихлорбензол замена HG-2 на ИЖ-модифицированный катализатор приводит к увеличению Mn с 4.5 × 103 до 35.0 × 103 при полимеризации 6-гидрокси-1,10-ундекандиена и с 10 × 103 до 40 × 103 при полимеризации 6-ацетокси-1,10-ундекандиена. При этом благодаря высокому сродству ионного катализатора к ИЖ, он не загрязняет продукт: содержание катализатора в получаемых полимерах является крайне низким даже в отсутствие дополнительной очистки (0.0012–0.0018%) [46]:
Метатезисная циклополимеризация диинов (MCP)
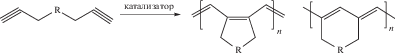
К настоящему времени опубликована лишь одна работа, посвященная метатезисной циклополимеризации ионных диинов в ионной среде [56]. Известно, что при циклополимеризации 1,6-гептадиинов возможно образование как пятичленных, так и шестичленных циклов в основной полимерной цепи:
Разработанные M.R. Buchmeiser модифицированные катализаторы Граббса–Ховейды приводят к стереоселективной полимеризации с образованием исключительно пятичленных звеньев [63, 64]. При проведении MCP в [BdmIm](CF3SO2)2N– показано, что ионная среда не нарушает стереоселективность таких катализаторов и циклополимеризация приводит к образованию полимеров с содержанием пятичленных циклов 95% [56]:
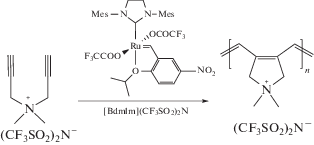
Молекулярная масса полученных полимеров варьировалась в пределах (16.900–21.8) × 103, а полидисперсность составляла 1.01–1.12. Среди других преимуществ осуществления метатезисной циклополимеризации диинов в ионной среде следует отметить тот факт, что ИЖ позволяют проводить метатезисную полимеризацию мономеров, не растворимых в традиционно применяемых в реакциях метатезиса органических растворителях [56].
ПОЛИМЕРНЫЕ АНАЛОГИ ИОННЫХ ЖИДКОСТЕЙ
Отдельно следует остановиться на полимеризации мономеров, самих по себе являющихся ИЖ, т.е. производных имидазолия или аммония с различными анионами. Безусловно, в данном случае также не стоит упускать из виду возможность специфических взаимодействий с катализатором и вызванного этим ускорения или замедления полимеризации [55]. Полимеризация мономеров-ИЖ приводит к образованию полимерных аналогов ионных жидкостей (ПИЖ), обладающих такими уникальными свойствами, как ионная проводимость, специфические диффузионно-адсорбционные свойства к CO2, резкое изменение характеристик при ионном обмене и т.д. [65].
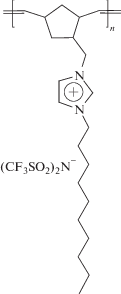
Ряд исследований посвящен ROPM-синтезу сополимеров производных норборнена, содержащих имидазолиевый катион. Полимеры с бис-трифторметансульфонилимидным анионом
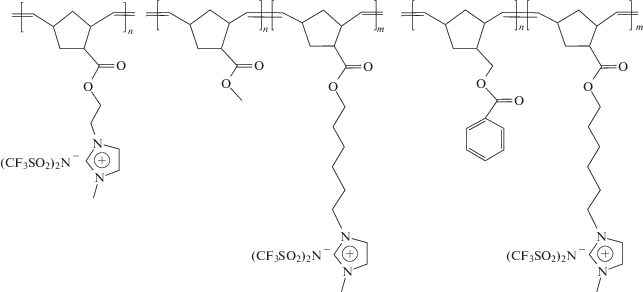 ,
,
характеризующиеся высокой ионной проводимостью и электрохимической стойкостью, предлагается наполнять солью Li(CF3SO2)2N и применять в качестве электролитов литиевых аккумуляторов [27, 66, 67].
Поскольку имидазол токсичен для ряда микроорганизмов, ROMP-полимеризацией на поверхности оксида титана имидазолиевых ПИЖ
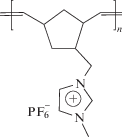 ,
,
можно создать устойчивые антимикробные покрытия [68]. Показано, что в тонких “умных”, или “переключаемых” полимерных покрытиях
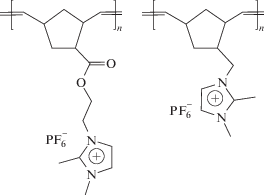
ионным обменом можно легко варьировать свойства поверхности: цвет, проводимость, механические характеристики и смачиваемость [69, 70]. Учитывая способность ряда ПИЖ селективно пропускать углекислый газ, исследуется возможность применения блок-сополимеров
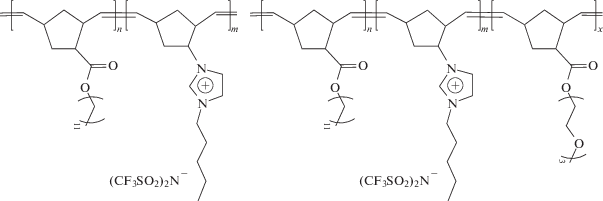
имидазолиевых производных норборненов в качестве газоразделительных мембран [71, 72]. Совмещением в одной макромолекуле TEMPO-фрагментов, способных к обратимым окислительно-восстановительным реакциям, и ион-проводящих звеньев

получали полимер для использования в области энергонезависимой памяти, суперконденсаторов и перезаряжаемых источников тока [73]. Ввиду того, что кроме ковалентных связей, в ионных полимерах имеют место сильные межцепные взаимодействия, на основе ПИЖ
возможно создание самовосстанавливающихся материалов, которые могут “заживлять” различные царапины и трещины [74].
ROMP-cополимеризацией цвиттерионного окосонорборненового сульфобетаина [75]
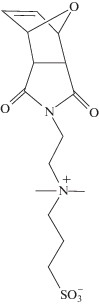
или имидазолиевого циклооктена [76]
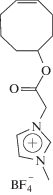
с неионными мономерами получали самособирающиеся полимерные наночастицы. Сополимеризация бифункциональных циклооктенимидазолиевых мономеров
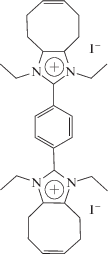
с циклооктеном приводит к образованию макроциклических сшитых анионпроводящих материалов, устойчивых в щелочной среде, которые предлагается использовать в топливных элементах [77].
При ROMP-полимеризации производных циклооктатетраена
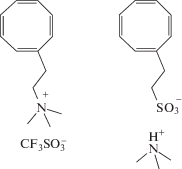
образуются ионные полиацетилены [78, 79]. Сопряженные двойные связи в полимерной цепи обеспечивают электропроводность, а ионные группы в боковых цепях придают полимеру растворимость в органических растворителях. Впрочем, для метатезисного синтеза ионных полиацетиленов чаще используют циклополимеризацию диинов: производные 1,6-гептадиина с имидазолиевыми [45]
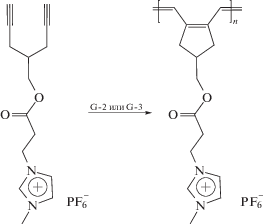 ,
,
триазолиевыми [80]
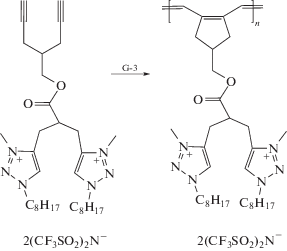 ,
,
пиридиниевыми [81]
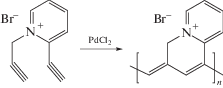 ,
,
аммониевыми [56, 82–84] катионами
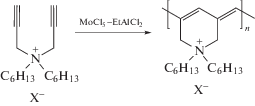
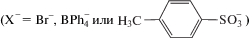
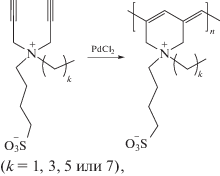
либо содержащими атомы азота для последующей кватернизации [85]
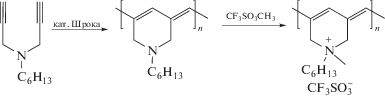
Такие полимеры демонстрируют высокие значения ионной проводимости и находят применение в качестве твердых полиэлектролитов в литиевых аккумуляторах, солнечных батареях, конденсаторах и т.д.
ЗАКЛЮЧЕНИЕ
Таким образом, при использовании ионных сред в метатезисной (со)полимеризации имеют место повышение скорости реакции, достижение количественных конверсий мономеров, управление молекулярно-массовыми характеристиками образующихся полимеров и возможность реализации рецикла дорогостоящего катализатора вместе с растворителем (табл. 1). Однако при этом необходимо учитывать возможность побочных реакций ИЖ с катализатором. Во избежание низкой конверсии реакции следует применять анионы с сильно делокализованным отрицательным зарядом – BF$_{4}^{ - }$, PF$_{6}^{ - }$, (CF3SO2)2N–, (FSO2)2N– и т.д., а при использовании 1,3-замещенного имидазолиевого катиона с “кислым” атомом во втором положении защищать катализатор добавками кислот или выбирать катионы, не содержащие кислых протонов, такие как 1,2-диметил-3-бутил имидазолий, тетраалкиламоний, тетраалкилфосфоний и т.п. При плохой растворимости образующегося полимера в ИЖ реакция становится плохо контролируемой и часто приводит к получению продукта с завышенной молекулярной массой и широким молекулярно-массовым распределением. Повысить эффективность и технологичность реакций метатезиса в ионных средах можно применением ИЖ-модифицированных катализаторов и проведением реакции в двухфазных системах ИЖ/молекулярный растворитель. Полимеризацией ионных мономеров можно получить полимеры с уникальными свойствами, обусловленными их ионной природой: ионной проводимостью, селективной газопроницаемостью и специфической растворимостью.
Таблица 1.
Преимущества применения ИЖ в реакциях полимеризации методами метатезиса
| Преимущества применения ИЖ | Пример | Литература |
|---|---|---|
| Возможность полимеризации и сополимеризации плохо растворимых мономеров | 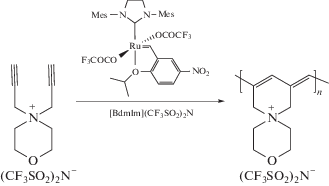 В ИЖ растворимы некоторые мономеры, не растворимые в неионных растворителях, традиционно применяемые для реакций метатезиса; также ИЖ могут выступать в роли общего растворителя для разных мономеров при сополимеризации. |
[56, 57] |
| Увеличение выхода полимера | 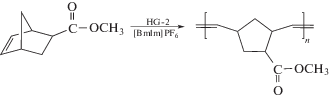 При полимеризации в ИЖ возможно получение полимеров с количественным выходом. |
[27, 28, 43, 51–55] |
| Увеличение скорости реакции | 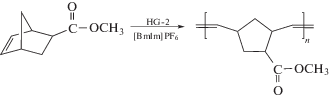 При полимеризации в ИЖ возможно значительное ускорение реакции (полная конверсия мономера в ионной среде достигается за 4 мин, в то время как в органическом растворителе требуется 6–8 ч). |
[27, 28, 43, 51–55] |
| Повышенная ММ | 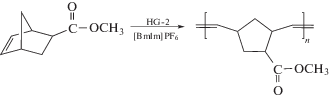 Благодаря протеканию полимеризации на границе фаз ИЖ+мономер/полимер в некоторых случаях образуется чрезвычайно высокомолекулярные полимеры (до 15 × 105). |
[54] |
| Легкость удаления побочного продукта в реакции AMDET | 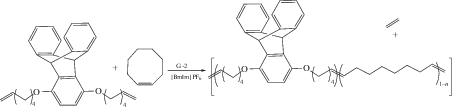 Благодаря низкому давлению паров и нелетучести ИЖ этилен легко удаляется из сферы реакции при нагревании реакционной смеси до 100°С. |
[57] |
| Легкость рецикла катализатора вместе с ИЖ | 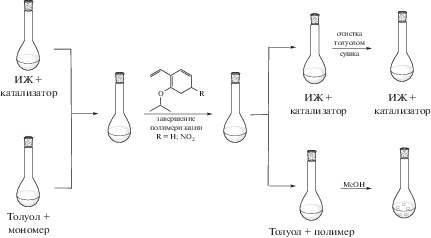 Высокое сродство ИЖ к полярным катализаторам и несмешиваемость с неполярными растворителями открывает возможности легкого выделения и повторного использования каталитических систем. |
[43, 44, 46, 53, 54, 59–62] |
| Сохранение стереоселективной активности катализаторов циклополимеризации | 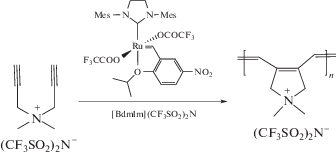 |
[56] |
К сожалению, из огромного количества возможных структур ИЖ с сильно различающимися свойствами на данный момент в большинстве работ, посвященных метатезисной полимеризации, используются лишь соли с катионами 1-бутил-3-метилимидазолия и 1-бутил-2,3-диметилимидазолия. Выбор анионов часто ограничен лишь BF$_{4}^{ - }$, PF$_{6}^{ - }$ и (CF3SO2)2N–. Исходя из этого, можно заключить, что влияние химического строения ИЖ на реакции метатезиса изучено не в полной мере и нуждается в дополнительных исследованиях.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-33-20108).
Список литературы
Ionic Liquids in Synthesis / Ed. by P. Wasserscheid, T. Welton. Weinheim: Wiley, 2007.
Hallett J.P., Welton T. // Chem. Rev. 2011. V. 111. № 5. P. 3508.
Forsyth S.A., Pringle J.M., MacFarlane D.R. // Aust. J. Chem. 2004. V. 57. № 2. P. 113.
Vygodskii Ya.S., Lozinskaya E.I., Shaplov A.S. // Polymer Science C. 2001. V. 43. № 2. P. 236.
Sheldon R. // Chem. Commun. 2001. № 23. P. 2399.
Seddon K.R. // J. Chem. Tech. Biotechnol. 1997. V. 68. № 4. P. 351.
Holbrey J.D., Seddon K.R. // Clean Technol. Environ. Policy. 1999. V. 1. № 4. P. 223.
Welton T. // Chem. Rev. 1999. V. 99, № 8. P. 2071.
Sledz P., Mauduit M., Grela K. // Chem. Soc. Rev. 2008. V. 37. P. 2433.
Thomas P.A., Marvey B.B. // Molecules. 2016. V. 21. P. 184.
Xie M., Han H., Ding L., Shi J. // Polym. Revs. 2009. V. 49. P. 315.
Yampolskii Yu.P., Starannikiva L.E., Belov N.A., Bermeshev M.V., Gringolts M.L., Finkelshtein E.Sh. // J. Membr. Sci. 2014. V. 453. P. 532.
Chapala P.P., Bermeshev M.V., Starannikova L.E., Belov N.A., Ryzhikh V.E., Shantarovich V.P., Lakhtin V.G., Gavrilova N.N., Yampolskii Yu.P., Finkelshtein E.Sh. // Macromolecules. 2015. V. 48. P. 8055.
Finkelshtein E.Sh., Makovetskii K.L., Gringolts M.L., Rogan Yu.V., Golenko T.G., Starannikova L.E., Yampolskii Yu.P., Shantarovich V.P., Suzuki T. // Macromolecules. 2006. V. 39. № 20. P. 7022.
Gringolts M.L., Bermeshev M.V., Yampolskii Yu.P., Starannikiva L.E., Shantarovich V.P., Finkelshtein E.Sh. // Macromolecules. 2010. V. 43. P. 7165.
Alentiev D.A., Dzhaparidze D.M., Gavrilova N.N., Shantarovich V.P., Kiseleva E.V., Topchiy M.A., Asachenko A.F., Gribanov P.S., Nechaev M.S., Legkov S.A., Bondarenko G.N., Bermeshev M.V. // Polymers. 2018. V. 10. P. 1382.
Bermeshev M.V., Chapala P.P. // Progr. Polym. Sci. 2018. V. 84. P. 1.
Truett W.L., Johnson D.R., Robinson I.M., Montague B.A. // J. Am. Chem. Soc. 1960. V. 82. № 9. P. 2337.
Schrock R.R., Feldman J., Cannizzo L.F., Grubbs R.H. // Macromolecules. 1987. V. 20. № 5. P. 1169.
Schrock R.R., Murdzek J.S., Bazan G.C., Robbins J., DiMare M., O’Regan M. // J. Am. Chem. Soc. 1990. V. 112. P. 3875.
Buchmeiser M.R. // Chem. Rev. 2000. V. 100. № 4. P. 1565.
Schwab P., France M.B., Ziller J.W., Grubbs R.H. // Angew. Chem. Int. Ed. 1995. V. 34. № 18. P. 2039.
Nguyen S.T., Grubbs R.H., Ziller J.W. // J. Am. Chem. Soc. 1993. V. 115. № 21. P. 9858.
Handbook of Metathesis: Catalyst Development / Ed. by R.H. Grubbs. Weinheim: Wiley, 2003.
Love J.A., Morgan J.P., Trnka T.M., Grubbs R.H. // Angew. Chem. Int. Ed. Engl. 2002. V. 41. № 21. P. 4035.
Garber S.B., Kingsbury J.S., Gray B.L., Hoveyda A.H. // J. Am. Chem. Soc. 2000. V. 122. № 34. P. 8168.
Vygodskii Y.S., Shaplov A.S., Lozinskaya E.I., Lysenko K.A., Golovanov D.G., Malyshkina I.A., Gavrilova N.D., Buchmeiser M.R. // Macromol. Chem. Phys. 2008. V. 209. P. 40.
Han H., Chen F., Yu J., Dang J., Ma Z., Zhang Y., Xie M. // J. Polym. Sci., Polym. Chem. 2007. V. 45. P. 3986.
Xie M., Kong Y., Han H., Shi J., Ding L., Song C., Zhang Y. // React. Funct. Polym. 2008. V. 68. P. 1601.
Yao Q., Sheets M. // J. Organomet. Chem. 2005. V. 690. P. 3577.
Clavier H., Audic N., Guillemin J.-C., Mauduit M. // J. Organomet. Chem. 2005. V. 690. P. 3585.
Audic N., Clavier H., Mauduit M., Guillemin J.-C. // J. Am. Chem. Soc. 2003. V. 125. P. 9248.
Thurier C., Fischmeister C., Bruneau C., Olivier-Bourbigou H., Dixneuf P.H. // J. Mol. Catal. A. 2007. V. 268. P. 127.
Schmid T.E., Dumas A., Colombel-Rouen S., Crevisy C., Basle O., Mauduit M. // Synlett. 2017. V. 28. P. A.
Clousier N., Filippi A., Borr E., Guibal E., Crevisy C., Caijo F., Mauduit M., Dez I., Gaumont A.-C. // ChemSusChem. 2014. V. 7. P. 1040.
Keraani A., Rabiller-Baudry M., Fischmeister C., Bruneau C. // Catal. Today. 2010. V. 156. P. 268.
Consorti C.S., Aydos G.L.P., Dupont J. // Chem. Commun. 2010. V. 46. P. 9058.
Consorti C.S., Aydos G.L.P., Ebeling G., Dupont J. // Organometallics. 2009. V. 28. P. 4527.
Chen S.-W., Kim J.H., Ryu K.Y., Lee W.-W., Hong J. // Tetrahedron. 2009. V. 65. P. 3397.
Kim J.H., Park B.Y., Chen S.-W., Lee S. // Eur. J. Org. Chem. 2009. № 14. P. 2239.
Wakamatsu H., Saito Y., Masubuchi M., Fujita R. // Synlett. 2008. № 12. P. 1805.
Clavier H., Nolan S.P., Mauduit M. // Organometallics. 2008. V. 27. P. 2287.
Suriboot J., Bazzi H.S., Bergbreiter D.E. // Polymers. 2016. V. 8. P. 140.
Koy M., Altmann H.J., Autenrieth B., Frey W., Buchmeiser M.R. // Beilstein J. Org. Chem. 2015. V. 11. P. 1632.
Autenrieth B., Frey W., Buchmeiser M.R. // Chem. Eur. J. 2012. V. 18. P. 14069.
Zhao J., Wang D., Autenrieth B., Buchmeiser M.R. // Macromol. Rapid Commun. 2015. V. 36. P. 190.
Olefin Metathesis // Metal Catalysed Reactions in Ionic Liquids / Ed. by P.J. Dyson, G.J. Tilmann. Dordrecht: Springer, 2005. Ch. 7. P. 155.
Yao Q., Zhang Y. // Angew. Chem. Int. Ed. 2003. V. 42. № 29. P. 3395.
Audic N., Clavier H., Mauduit M., Guillemin J.-C. // J. Am. Chem. Soc. 2003. V. 125. № 31. P. 9248.
Clavier H., Audic N., Mauduit M., Guillemin J.-C. // Chem. Commun. 2004. № 20. P. 2282.
Song W., Han H., Liao X., Sun R., Wu J., Xie M. // Macromolecules. 2014. V. 47. P. 6181.
Xie M.-R., Ma Z., Han H.-J., Shi J.-X., Wang W.-Z., Li J.-X., Zhang Y.-Q. // Chem. J. Chin. Univ-Chin. 2009. V. 30. № 2. P. 396.
Han H., Liu J., Huang W., Song C., Zhang Y., Xie M. // Acta Polymerica Sinica. 2008. № 5. P. 492.
Vygodskii Y.S., Shaplov A.S., Lozinskaya E.I., Filippov O.A., Shubina E.S., Bandari R., Buchmeiser M.R. // Macromolecules. 2006. V. 39. P. 7821.
Simocko C., Young T.C., Wagener K.B. // Macromolecules. 2015. V. 48. P. 5470.
Vygodskii Ya.S., Shaplov A.S., Lozinskaya E.I., Vlasov P.S., Malyshkina I.A., Gavrilova N.D., Kumar P.S., Buchmeiser M.R. // Macromolecules. 2008. V. 41. № 6. P. 1919.
Simocko C., Yang Y., Swager T.M., Wagener K.B. // ACS Macro Lett. 2013. V. 2. P. 1061.
Daguenet C., Dyson P.J. // Organometallics. 2004. V. 23. P. 6080.
Csihony S., Fischmeister C., Bruneau C., Horvath I.T., Dixneu P.H. // New J. Chem. 2002. V. 26. P. 1667.
Ferraz C.P., Autenrieth B., Frey W., Buchmeiser M.R. // ChemCatChem. 2014. V. 6. P. 191.
Gallagher M.M., Rooney A.D., Rooney J.J. // J. Mol. Catal. A. 2009. V. 303. P. 78.
Ding X., Lv X., Hui B., Chen Z., Xiao M., Guo B., Tang W. // Tetrahedron Lett. 2006. V. 47. P. 2921.
Halbach T.S., Krause J.O., Nuyken O., Buchmaiser M.R. // Macromol. Rapid Commun. 2005. V. 26. P. 784.
Naumov S., Buchmaiser M.R. // Organometallics. 2012. V. 31. P. 847.
Shaplov A.S., Ponkratov D.O., Vygodskii Y.S. // Polymer Science B. 2016. V. 58. № 2. P. 73.
Wang J., He X., Zhu H., Chen D. // RSC Adv. 2015. V. 5. P. 43581.
He X., Wang Z., Zhou W., Jiang X., Han Z., Chen D. // J. Appl. Polym. Sci. 2017. V. 134. P. 44884.
Ye Q., Gao T., Wan F., Yu B., Pei X., Zhou F., Xue Q. // J. Mater. Chem. 2012. V. 22. P. 13123.
Njoroge I., Matson M.W., Jennings G.K.K. // J. Phys. Chem. C. 2017. V. 121. № 37. P. 20323.
Njoroge I., Bout B.W., Matson M.W., Laibinis P.E., Jennings G.K. // ACS Omega. 2018. V. 3. P. 16158.
Wiesenauer E.F., Edwards J.P., Scalfani V.F., Bailey T.S., Gin D.L. // Macromolecules. 2011. V. 44. P. 5075.
Wiesenauer E.F., Nguyen P.T., Newell B.S., Bailey T.S., Noble R.D., Gin D.L. // Soft Matter. 2013. V. 9. P. 7923.
Suga T., Sakata M., Aoki K., Nishide H. // ACS Macro Lett. 2014. V. 3. P. 703.
Cui J., Nie F.-M., Yang J.-X., Pan L., Ma Z., Lia Y.-S. // J. Mater. Chem. A. 2017. V. 5. P. 25220.
Maddikeri R.R., Colak S., Gido S.P., Tew G.N. // Biomacromolecules. 2011. V. 12. P. 3412.
Xie M., Han H., Wang W., He X., Zhang Y. // Macromol. Chem. Phys. 2008. V. 209. P. 544.
You W., Hugar K.M., Coates G.W. // Macromolecules. 2018. V. 51. № 8. P. 3212.
Langsdorf B.L., Zhou X., Adler D.H., Lonergan M.C. // Macromolecules. 1999. V. 32. P. 2796.
Johnston D.H., Gao L., Lonergan M.C. // Macromolecules. 2010. V. 43. P. 2676.
Li H., Wang J., Han H., Wu J., Xie M. // React. Funct. Polym. 2018. V. 127. P. 20.
Gal Y.-S., Lee W.-C., Lee J.-H., Choi S.-K. // Bull. Korean Chem. Soc. 1997. V. 18. № 9 P. 22.
Kang K.-L., Kim S.-H., Cho H.-N., Choi K.-Y., Choi S.-K. // Macromolecules. 1993. V. 26. P. 4539.
Kim S.-H., Choi S.-J., Park J.-W., Cho H.-N., Choi S.-K. // Macromolecules. 1994. V. 27. P. 2339.
Choi D.-C., Kim S.-H., Lee J.-H., Cho H.-N., Choi S.-K. // Macromolecules. 1997. V. 30. P. 176.
Zhang N., Wu R., Li Q., Pakbaz K., Yoon C.O., Wudl F. // Chem. Mater. 1993. V. 5. P. 1598.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)