

Высокомолекулярные соединения (серия С), 2020, T. 62, № 2, стр. 186-199
МОЛЕКУЛЯРНЫЕ КОМПОЗИТЫ НА ОСНОВЕ ПОЛИИМИДОВ
О. Н. Забегаева 1, Д. А. Сапожников 1, Я. С. Выгодский 1, *
1 Институт элементоорганических соединений им. А.Н. Несмеянова Российской академии наук
119991 Москва, ул. Вавилова, 28, Россия
* E-mail: yasvyg@ineos.ac.ru
Поступила в редакцию 29.02.2020
После доработки 18.03.2020
Принята к публикации 06.04.2020
Аннотация
Проанализированы и обобщены результаты исследований в области создания молекулярных композитов на основе полиимидов и ряда полимеризационных мономеров. Показано, что полиимиды активируют, инициируют или выступают в роли агентов передачи цепи при полимеризации лактамов и виниловых мономеров, приводя к образованию ковалентной связи между жестко- и гибкоцепными фрагментами. Продемонстрированы преимущества и перспективность создания поликонденсационно-полимеризационных молекулярных композитов на основе полиимидов для изготовления высокопрочных конструкционных материалов, электроизоляционных пленок и покрытий, мембран, а также применения в роли наноконтейнеров и т.д.
Термин “молекулярные композиты” применим для систем, в которых жесткоцепные макромолекулы диспергированы на молекулярном уровне в матрице гибкоцепных полимеров; при этом жесткоцепные молекулы являются усиливающими агентами. Концепция молекулярных композитов, предложенная в конце 70-х годов [1–3], сразу привлекла внимание исследователей и актуальна по сей день. Особенности формирования молекулярных композитов, их разнообразие, обусловленное только конденсационными или конденсационно-полимеризационными компонентами, и свойства рассмотрены в более ранних работах [1–11]. Очевидные преимущества молекулярных композитов перед традиционными армированными пластиками, побудившие мировое научное сообщество заняться исследованиями в этой области, определяются в первую очередь, отсутствием внутренних дефектов в армирующих элементах (макромолекулах жесткоцепного полимера), характерных для дисперсных материалов, и значительным усиливающим эффектом за счет высокого отношения длины жесткоцепного сегмента к его сечению. При этом можно ожидать проявления синергетического эффекта не только в механических, но и в термических и в ряде других физико-химических свойств.
Становление и развитие концепции молекулярных композитов предопределили интерес к конструированию макромолекул, отличающихся повышенной термодинамической жесткостью. Высокую прочность и жесткость полимерам обеспечивают ковалентные связи в макромолекулах, которые должны характеризоваться высокой степенью выпрямленности, а также наличие карбо- и гетероциклов, лишенных гибких мостиковых групп [12–16].
На первых этапах исследований большое внимание уделялось разработке ароматических полиамидов, свободных от несимметричных фрагментов и “шарнирных” групп, например, на основе поли-п-бензамида и поли-п-фенилентерефталамида и т.д. [17–19]. Следует, однако, отметить, что наличие в макромолекулах амидных групп в значительной степени лимитируется их термической стабильностью. В связи с этим на последующих этапах изучали жесткоцепные полимеры, содержащие только ароматические карбо- и гетероциклы – так называемые жесткоцепные полигетероарилены.
Среди большого числа жесткоцепных полигетероариленов наиболее детально изучены полиимиды [13–16, 20–26]. Молекулярный дизайн ПИ весьма разнообразен и предопределяет термодинамическую гибкость цепей. Простейшие ароматические ПИ со “стержневой” геометрией, характеризующиеся довольно высокой жесткостью макромолекул (сегмент Куна ∼63–175 нм [27]), могут быть получены из пиромеллитового диангидрида или диангидрида дифенил-3,3',4,4'-тетракарбоновой кислоты и карбоциклических (п-фенилендиамина, бензидина и т.д.) или гетероциклических (2,5-бис-(п-аминофенил)пиримидин) диаминов [13]. При этом основная сложность заключалась в том, что многие жесткоцепные ПИ, например содержащие фенильные, дифенильные, нафталиновые и другие группы, растворимы лишь в сильных неорганических кислотах, что ограничивает или вовсе исключает их использование для формирования молекулярных композитов. Сложившаяся проблема стимулировала работы по синтезу органорастворимых жесткоцепных ПИ. Улучшения растворимости ароматических ПИ достигают, в частности, введением в них кардовых и/или перфторалкильных групп [28–34].
Развитие химии ПИ позволило получать жесткоцепные полимеры с набором полезных свойств: высокой прочностью волокон и пленок, высокой термостойкостью и в то же время хорошо растворимых в органических растворителях. Все это определяет целесообразность их использования в формировании молекулярных композитов как перспективных армирующих агентов.
В настоящем обзоре обобщены и проанализированы результаты исследований в области молекулярных композитов на основе ПИ и поликапроамида (ПКА) или виниловых полимеров.
МОЛЕКУЛЯРНЫЕ КОМПОЗИТЫ НА ОСНОВЕ ПОЛИИМИДОВ И ПОЛИКАПРОАМИДА
Существенное препятствие на пути создания молекулярных композитов – термодинамическая несовместимость жестких стержнеобразных макромолекул с гибкоцепной матрицей из-за неудовлетворительной энтропии их смешения, приводящая к разделению фаз и, как следствие, к ухудшению прочностных свойств композиционного материала. Одним из способов подавления фазового разделения и повышения энтропии смешения является подбор близких по химической структуре компонентов, что обеспечивает дополнительное взаимодействие между жесткими и гибкими сегментами (водородные связи, ион-дипольное или диполь-дипольное взаимодействие и т.д.) [35–38]. Алифатические полиамиды благодаря наличию амидных групп, склонных к образованию водородных связей, представляют интерес как гибкоцепной компонент для ПИ.
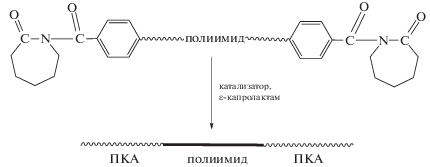
Так, были получены композиционные ПИ-пленки, содержащие ПКА [39–41]. Поскольку основным недостатком ПИ является плохая растворимость, смешение полимеров на молекулярном уровне осуществляли на стадии формирования полиамидокислоты (ПАК). К раствору ПАК в ДМФА добавляли 10–30 мас. % ε-капролактама и подвергали высокотемпературной циклизации. ПКА образовывался при полимеризации ε-капролактама под действием воды, выделяющейся в процессе циклодегидратации ПАК в ПИ. Сформованные таким способом композиционные пленки отличаются высокими деформационно-прочностными характеристиками (зафиксировано увеличение модуля Юнга и прочности на растяжение на 15–40 и 19–47% соответственно), улучшенной термостойкостью, а также незначительным водопоглощением по сравнению с контрольными образцами [39–41]. Полагают, что существенное улучшение термических и механических свойств пленок обусловлено формированием плотной надмолекулярной структуры с высокой степенью кристалличности за счет образования водородных связей между кислородом карбонильной группы имида и амидным водородом ПКА.
Между тем, алифатические полиамиды, в частности ПКА, представляют самостоятельный интерес в производстве пленок, покрытий, волокон, а также конструкционных материалов [42–44].
Исследованы молекулярные композиты на основе смесей коммерчески доступного гибкоцепного ПКА и полимеров с жесткими стержнеобразными макромолекулами, такими как ароматические ПИ, полиамиды и другие [45–49]. Смешение полимеров на молекулярном уровне проводили двумя способами: 1) в общем растворителе (м-крезоле, серной кислоте и других) при концентрации компонентов ниже критической, т.е. в отсутствие фазового разделения; 2) в процессе полимеризации in situ ε-капролактама в присутствии растворенного в расплаве ε-капролактама ПИ или другого жесткоцепного полимера [45–51]. При введении небольшого количества жесткоцепного компонента (менее 5 мас. %) отмечено существенное возрастание модуля Юнга в сформованных пленках. Исследования деформационно-прочностных и термических свойств смесевых пленок, приготовленных из раствора и полимеризацией in situ, кинетики их кристаллизации показали, что при содержании жесткоцепного компонента более 5 мас. % полимеры между собой не смешиваются, и жесткая полимерная составляющая образует отдельную фазу в аморфной области ПКА. Как следствие, из-за снижения роли межмолекулярного взаимодействия между гибкими и жесткими сегментами усиливающий эффект в таких пленках незначительный или вовсе отсутствует [45, 46].
Представляется весьма перспективным целенаправленный поиск новых подобных систем, в том числе синтез блок- и привитых сополимеров, в которых жесткие и гибкие фрагменты связаны более прочными ковалентными связями. Это позволит не только повысить степень совместимости компонентов за счет проявления сильных межмолекулярных взаимодействий, но и создаст возможность для более тонкого регулирования их морфологии и свойств.
Для получения ди- и триблок-сополимеров, в которых одним из блоков является ПКА, необходимо использование полимеров, хорошо растворимых в расплаве ε-капролактама. Кроме того, такие полимеры должны обладать концевыми ациллактамными группами, способными активировать анионную полимеризацию ε-капролактама, либо группами, легко превращающимися в ациллактамные при взаимодействии с ε-капролактамом [52].
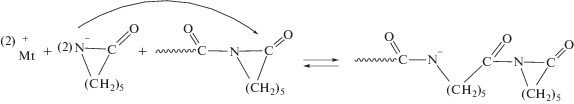
Согласно представленному механизму анионной полимеризации ε-капролактама, рост цепи начинается с нуклеофильного присоединения аниона лактама, образовавшегося при диссоциации катализатора (как правило, лактаматные соли щелочных и щелочноземельных металлов), к эндоциклическому карбонильному атому углерода концевой ациллактамной группы растущего центра с последующим раскрытием лактамного цикла. Далее происходит протонный обмен между образовавшимся полимерным N-анионом и молекулой ε-капролактама. Источник анионов лактама принято считать катализатором, а источник центров роста цепи – активатором полимеризации.
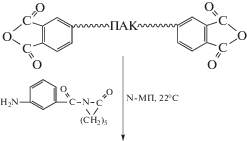
Так, взаимодействием N,N'-терефталоил-ди-ε-капролактама с концевыми аминогруппами предварительно синтезированного в избытке анилинфлуорена ПИ получен полиимидный активатор АПК. Растворителем выступал расплав ε-капролактама; в этих условиях концевые аминогруппы ПИ превращались в активирующие полимеризацию ε-капролактама ациллактамные группы, а затем под действием катализатора полимеризации происходила прививка поликапроамидных цепей по концевым группам ПИ [53]:
В результате были синтезированы сополимеры типа АВА, содержащие гибкоцепные блоки А (ПКА) и жесткоцепные блоки В (ПИ). ε-Капролактам здесь выполнял двоякую функцию: на первой стадии он выступал в роли растворителя, на второй – сомономера. Существенным недостатком данного метода является образование наряду с блок-сополимером гомополикапроамида как побочного продукта.
При раздельном осуществлении процесса получения блок-сополимеров полимеризацию ε-капролактама проводили в присутствии выделенного полиимидного активатора. Последний был получен реакцией аминобензоил-ε-капролактама с предварительно синтезированной ПАК, содержащей концевые ангидридные группы [54].
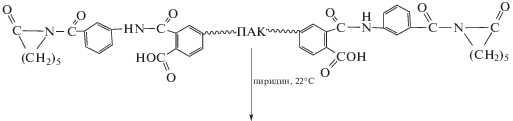
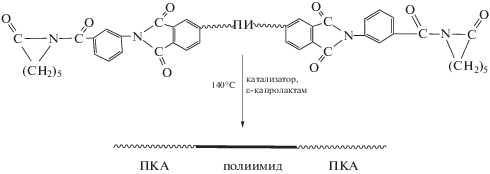
Эффективным активатором полимеризации ε-капролактама оказался ПИ с концевыми сложноэфирными группами [55, 56].
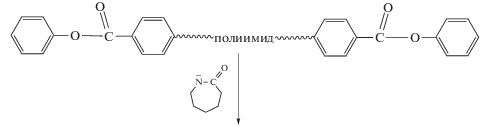
На примере модельного соединения фенилбензоата было установлено, что сложноэфирная группа активирует полимеризацию циклического амида, приводя к формированию поликапроамида с высокой конверсией мономера [57].
Отмечают, что синтез таким путем тройных блок-сополимеров протекает с высоким выходом без образования гомополимеров. При содержании ПИ всего 5 мас. % сополимеры по механическим показателям в 2–3 раза превосходят гомо-ПКА. Заметно улучшается термо- и влагостойкость [53, 58–60].
Исследования структуры, механических и теплофизических свойств сополимеров с привлечением методов рентгеновской дифракции в больших и малых углах рассеивания и ДСК показали, что в тройных сополимерах амидные и имидные компоненты смешиваются на молекулярном уровне без выделения жесткоцепного сегмента в аморфную фазу сополимера. В этом отношении они отличаются от смесевых пленок, приготовленных из раствора или полимеризацией in situ, компоненты которых образуют отдельные фазы или сегментируют после термообработки [45, 46, 61, 62].
Особый интерес представляет получение молекулярных композитов на основе привитых сополимеров. В таких сополимерах активирующие АПК группы расположены не на концах, а вдоль цепи макромолекулы активатора [56, 57, 63].
Одним из первых сополимер гребнеобразного типа был получен анионной полимеризацией ε-капролактама с ПИ, предварительно функционализированным алкилимидными группами [63]. Основная сложность заключалась в трудоемком многостадийном синтезе диамина с привитым N-ациллактамом – 3,5-бис-(4-аминофенокси)бензоил-ε-капролактама. Было также обнаружено [56, 57], что ациллактамные группы при поликонденсации диамина с диангидридом подвержены побочным реакциям.
Предложен альтернативный подход [56, 57], в котором для прививки звеньев ε-капролактама использовали ПИ с боковыми фенилэфирными группами, которые, как было показано [56, 57], являются активаторами косвенного действия, т.е. при взаимодействии с лактамом образуют алкил-имидные группы. ПИ с количественным выходом получали высокотемпературной одностадийной поликонденсацией фенил 3,5-диаминобензоата с различными диаминами и диангидридами.
Все привитые сополимеры характеризуются более высокими деформационно-прочностными и термическими свойствами по сравнению с гомо-ПКА. Так, прочность и модуль упругости при растяжении возрастали в 2–3 раза, а по ударной вязкости с надрезом сополимеры превосходили не только гомо-ПКА (в 2–5 раз), но и соответствующие блок-сополимеры. Существенно улучшались термо- и влагостойкость. Превосходство в механических свойствах привитых сополимеров перед блочными объясняют лучшим распределением жестких и гибких сегментов и наличием сильных межмолекулярных взаимодействий, на что указывают более высокие Tс привитых сополимеров [56, 57, 63].
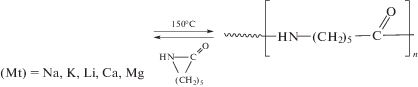
Выше рассмотрено использование двух типов макромолекулярных активаторов, в которых функциональные группы, активирующие полимеризацию лактамов, являются концевыми или представляли собой заместители у основной цепи макромолекул. Известен и третий тип макромолекулярных активаторов, в которых группы, способные активировать полимеризацию лактамов, являются фрагментами основной полимерной цепи [64]. Так, при исследовании АПК в присутствии ароматического полиэфиримида в экструдере при температуре выше температуры плавления ПКА [65] было показано, что полигетероарилен выступает в роли активатора процесса за счет собственных пятичленных имидных циклов, приводя к образованию привитого сополимера. Ориентировочная проверка возможности участия имидных групп ПИ в in situ АПК к такому результату не привела [66].
Было высказано предположение, что ключевым фактором, препятствующим раскрытию имидного цикла ПИ в условиях АПК, является природа катализатора, а именно природа противоиона, который, как считалось ранее, не принимает участия в реакции инициирования [67]. Предложен новый катализатор АПК на основе реактива Гриньяра – магнийбром-ε-капролактам и описан механизм, в котором противоион координирует с имидными карбонилами молекулы активатора, в результате имидные группы поляризуются и атака аниона лактама облегчается [68].
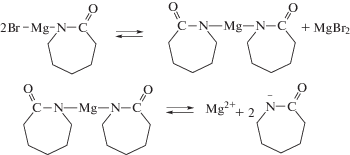
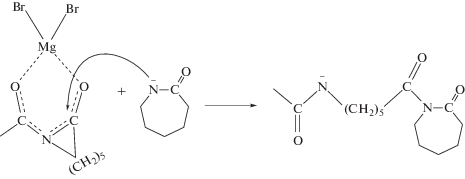
В упомянутой выше статье также использовали катализатор на основе реактива Гриньяра – фенилмагнийбромид, но он, однако, оказался неэффективным [66]. По-видимому, это обусловлено тем, что был использован ПИ относительно невысокой молекулярной массы (6 × 103), при этом повышается концентрация концевых ангидридных групп в полимеризационной системе, которые легко могут вступать во взаимодействие с катализатором полимеризации, дезактивируя его [52, 67]:
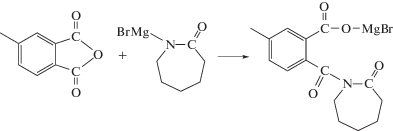
В развитии данного направления исследований было установлено, что ароматические ПИ под действием магнийбром-ε-капролактама действительно активируют полимеризацию ε-капролактама, приводя к формированию привитых сополимеров гребнеобразного типа. При этом на первой стадии при взаимодействии с анионом лактама имеет место раскрытие имидных циклов с образованием ациллактамных группировок, расположенных вдоль цепи полигетероарилена.
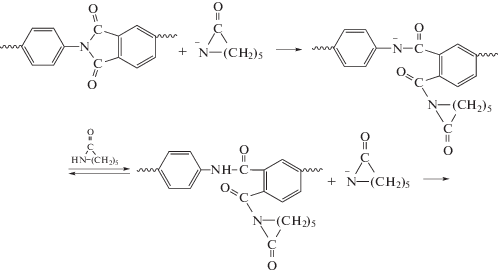
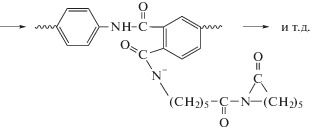
На последующих стадиях наблюдается рост цепи ПКА [69, 70]. Контрольные опыты АПК, катализируемой фенилмагнийбромидом, а также таким известным катализатором, как натриевая соль ε-капролактама, показали, что полимеризация с ароматическими имидными активаторами в первом случае действительно имеет место, но занимает довольно много времени, а во втором случае не протекает вовсе [71, 72].
При изучении кинетики АПК было установлено, что строение ПИ, в частности используемого диамина, существенно влияет на скорость полимеризации: чем выше электроноакцепторные характеристики диамина, тем полимеризация протекает с большой скоростью и приводит к образованию полимера с высокой конверсией мономера [69–71]. Установлено, что привитые сополимеры в противоположность линейным имеют сшитую структуру и набухают в традиционных для ПКА растворителях. Сшивки имеют имидную природу и возникают за счет взаимодействия концевых ациллактамных групп растущих макромолекул с амидными группами других полиамидных макромолекул [69]. Ниже показан механизм сшивания макромолекул при АПК в присутствии ПИ.
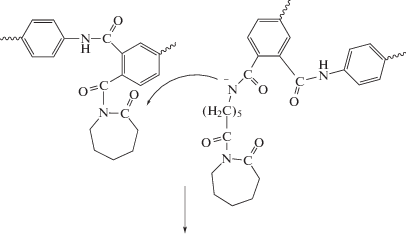
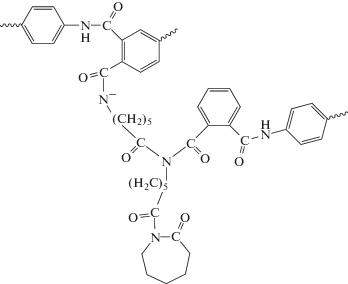
Варьированием природы ПИ возможен контроль таких свойств ПКА, как количество гель-фракции, степень кристалличности, твердость. Это позволяет получать сополимеры с определенным комплексом деформационно-прочностных, теплофизических, и других свойств, выгодно отличающихся от свойств немодифицированного ПКА [70, 73]. Сополимеры характеризуются весьма высокой ударной вязкостью по Изоду, в разы превышающей немодифицированный ПКА. Наибольшего значения ударной вязкости (Ак ∼ ∼ 50 кДж/м2) удалось достичь при использовании ПИ на основе анилинфлуорена и диангидрида дифенилоксидтетракарбоновой кислоты [69, 70, 74, 75]. По данному параметру сополимеры превосходят образцы, синтезированные не только с монофункциональными активаторами (Ак = 4.3–6.9 кДж/м2), но и с полифункциональными активаторами, например, N-акрилоил-капролактамом (Ак = 34 кДж/м2) [76]. Наблюдаемое улучшение механических характеристик сополимеров объясняют формированием прочного структурного полиимидного каркаса, в котором чем жестче макромолекулы ПИ, тем сильнее выражен армирующий эффект [73].
При модификации ПКА полиимидами существенно улучшаются трибологические характеристики. Наилучшие результаты были достигнуты с фторированными ПИ [69, 70]. Так, в два раза снижается коэффициент трения, на 20–30 градусов понижается температура фрикционного контакта, существенно уменьшается износ образца. Низкому и более стабильному коэффициенту трения благоприятствует сшитая структура сополимеров при сохранении кристалличности, а также фторалкильные группы ПИ.
МОЛЕКУЛЯРНЫЕ КОМПОЗИТЫ ПОЛИИМИДОВ И ВИНИЛОВЫХ ПОЛИМЕРОВ
Помимо задачи упрочнения полимерной матрицы молекулярные композиты также успешно используют и при создании функциональных материалов самого разного назначения. Это продемонстрировано на ряде систем, включающих ПИ и ковалентно связанные с ними макромолекулы на основе винильных мономеров.
Необходимость создания материалов с низкой диэлектрической константой k, используемых в микроэлектронике, обусловила значительный интерес к созданию пористых ПИ-пленок, включая материалы с нанопорами [77].
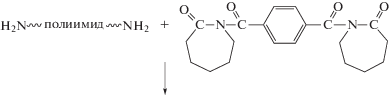
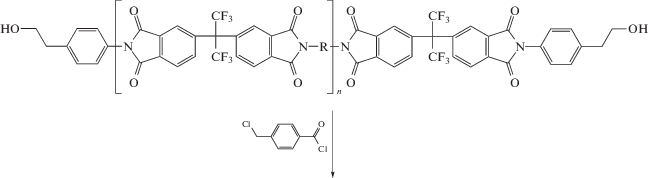
С этой целью рядом авторов модифицированы пленки ПИ на стадии соответствующей ПАК. Ниже приведена схема синтеза молекулярных композитов и пленок ПИ.
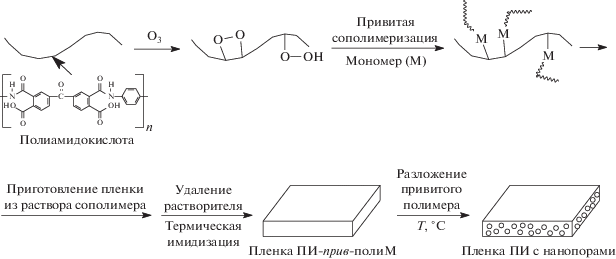
Таким способом ПАК указанного на схеме строения модифицировали прививкой ММА [78] или полиэтиленгликольметакрилата [79] полимеризацией с переносом атома, а также термической привитой сополимеризацией метакрилового мономера, содержащего полиэдральный олигомерный силсесквиоксан [80, 81]. Согласно представленной выше схеме, дальнейшие преобразования данного привитого сополимера позволяют получить полиимидную пленку, содержащую не только нанопоры, но и фрагменты полиэдрального олигомерного силсесквиоксана, значительно понижающего ее k. Так, k со значением, приближающимся к 2.2, достигается прививкой 23.5 мол. % мономера, содержащего полиэдральный олигомерный силсесквиоксан.
По аналогичной схеме протекает прививка акриловой кислоты на макромолекулы ПИ на основе диангидрида 2,2-бис-(3,4-дикарбоксифенил)-1,1,1,3,3,3-гексафторпропана и 4,4′-бис-(4-аминофенокси)дифенилсульфона [82]. ПИ указанного строения растворим в N-метилпирролидоне (N-МП), что позволяет производить прививку непосредственно на ПИ, а не на соответствующую ПАК. Озонированием данного ПИ в растворе N-МП с дальнейшей термической привитой сополимеризацией акриловой кислоты и N-винилпирролидона [83], а также в растворе ДМФА привитой сополимеризацией с переносом атома полиэтиленгликольметакрилата [84] синтезированы соответствующие молекулярные композиты. Их формирование подтверждено различными методами: ИК- и ЯМР-спектроскопией, элементным анализом, ТГА, ДСК и ГПХ. Из растворов сополимеров, содержащих ПИ и поли(полиэтиленгликоль)метакрилат, с помощью инверсии фаз в водной среде, приготовлены мембраны для микрофильтрации [84]. Последующей термической деструкцией акрилатного фрагмента и варьированием его содержания были получены пленки ПИ c равномерно распределенными узкодисперсными нанопорами и характеризующиеся k = 2 [85].
При использовании в синтезе ПИ макромономера с уже привитым поли-α-метилстиролом был также получен сополимер, использующийся при формировании нанопористой пленки [86].
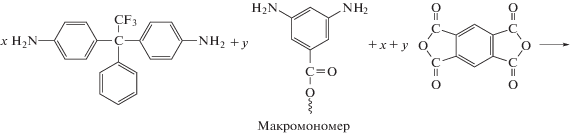
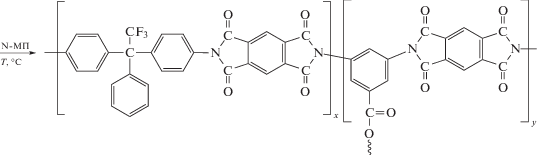
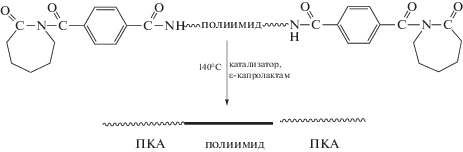
Описан синтез привитых сополимеров на основе ПИ с боковыми функциональными группами, выступающими в роли инициатора полимеризации виниловых мономеров [87]. Согласно представленной ниже схеме, на полиимидный макроинициатор методом контролируемой полимеризации привиты фрагменты ПС и полипентафторстирола (ПФС).
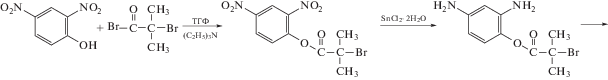
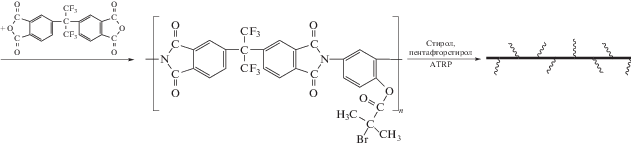
ПИ-прив-ПФС обладает значительно более высокой термостойкостью (до 470°С) и механическими свойствами по сравнению с ПФС. Кроме того, такой сополимер имеет очень низкую диэлектрическую постоянную (k ∼ 2.1), что делает его весьма перспективным для использования в микроэлектронике.
А.В. Якиманский с сотрудниками получили ПИ с боковыми алкилбромидными фрагментами путем полимераналогичных превращений ПИ с группами ОН [88]. Такие ПИ были использованы в контролируемой привитой (со)полимеризации различных акрилатов [88–94] и стирола [89, 95]. Изучены закономерности образования боковых ветвлений, их влияние на свойства сополимеров и поведение в растворах. Отмечено значительное повышение жесткости полимерной цепи (на 1 порядок) привитого сополимера по сравнению с исходным ПИ [95].
Гидролизом поли-трет-бутилметакрилатных фрагментов таких полимерных щеток получены сополимеры с боковыми цепями полиметакриловой кислоты [96, 97] или ее блочными дифильными ветвлениями [94]. Изучены особенности взаимодействия ионов редкоземельных и переходных металлов с некоторыми из сополимеров [98], которые могут найти применение при изготовлении нанореакторов, наноконтейнеров и т.д. [99].
Показана перспективность мембран на основе таких систем в разделении, например, смеси воды и изопропанола [93]. Производительность мембраны при этом легко регулируется плотностью прививки боковых цепей.
Перспективными для мембранных технологий и других приложений являются сополимеры сложной архитектуры, представляющие собой поли(ε-капролактон)–блок–ПИ–блок–поли(ε-капролактон) с привитыми к ПИ макромолекулами ПММА [100].
Прививка к ПИ боковых цепей поли-N,N-диметиламиноэтилметакрилата приводит к образованию материалов, чувствительных к pH и температуре [89, 101]. Такой сополимер можно применять в качестве стабилизатора и нанореактора при синтезе наночастиц Ag [92].
Схожий подход, заключающийся в прививке полиэтиленгликольметакрилата к активным центрам ПИ, использовали для создания полностью твердотельного полимерного электролита c удовлетворительными механическими характеристиками пленок и высокой проводимостью, равной 6.5 × 10–6 С/см при 25°С [102].
Методом радикальной полимеризации с переносом атома таких мономеров, как ММА [103–108], метакрилсодержащего полиэдрального олигомерного силсесквиоксана [109–113], 2-метил-2-адамантилметакрилата [114, 115] и 2-гидроксиэтилметакрилата [116] синтезирован ряд блок-сополимеров типа АВА. Полученный авторами фторированный полиимид с концевыми хлорметильными группами выступал в роли макроинициатора полимеризации.
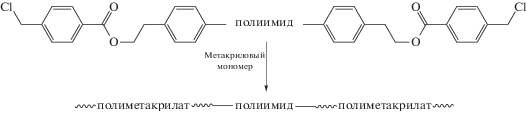
Разработанный метод позволяет в широком диапазоне варьировать длину блоков и свойства конечных материалов. Селективным разложением полиакрилатных блоков, протекающим до начала разложения ПИ, получены перспективные для микроэлектроники пленки с размером пор 50–100 нм и k = 2.36 [104, 105] и газоразделительные/сорбционные мембраны [114, 115, 117]. Пленки из ПММА–блок–ПИ–блок–ПММА характеризуются высокими газобарьерными [106] и гидрофобными свойствами [107, 108], а также низким показателем преломления [106]. Наличие в структуре сополимера фрагментов полиэдрального олигомерного силсесквиоксана приводит к повышению гидрофобности и улучшению газоразделительных свойств по сравнению с немодифицированным ПИ [109–113].
Нами обнаружено, что некоторые кардовые и/или фторсодержащие ПИ, как и другие жесткоцепные конденсационные полимеры, растворимы в ряде ненасыщенных мономеров: (мет)акрилатах, фтор(мет)акрилатах, N-винил-2-пирролидоне и т.д. Свободнорадикальной (со)полимеризацией in situ ММА [118–123], этилакрилата [124, 125], фтор(мет)акрилатов [122, 126, 127], ММА c дифункциональными сомономерами [128, 129], стирола [130], стирола с ММА [130], N-винил-2-пирролидона [131, 132] в присутствии ПИ (4–85 мас. %) получены молекулярные композиты разного состава. Было установлено, что продуктами термо- или фотополимеризации мономеров являются сополимеры, значительно отличающиеся по свойствам от соответствующих гомополимеров и их смесей [118–132]. Методами ДСК и ЭПР установлен механизм образования сополимеров, заключающийся во взаимодействии свободных радикалов реакционной системы с макромолекулами ПИ согласно представленной ниже схеме [120, 124].
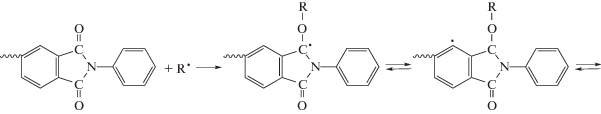
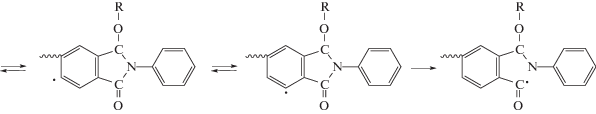
При этом, как правило, карбоцепной гомополимер не образуется даже при содержании ПИ 4 мас. %. Синтезированные сополимеры проявляют отличную от гомополимеров растворимость, а также возросшие показатели тепло-, термостойкости и прочностных свойств пленок [118–132]. Полуалифатические ПИ, не содержащие кардовые или перфторизопропилиденовые группы, также растворяются в ненасыщенных мономерах и участвуют в образовании сополимеров с новыми свойствами [133].
Полученные сополимеры перспективны для применения в микроэлектронике и оптике, в частности в роли защитных покрытий [132].
Таким образом, в обзоре обобщены результаты исследований синтеза, свойств и практического использования молекулярных композитов на основе ПИ и поликапроамида, а также ПИ и виниловых полимеров. Показаны преимущества ковалентного взаимодействия гибкой полимерной компоненты и более жесткого ПИ.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-29-17035 мк), а также Министерства науки и высшего образования Российской Федерации.
Список литературы
Pawlikowski G.T., Dutta D., Weiss R.A. // Annu. Rev. Mater. Sci. 1991. V. 21. P. 159.
Helmimiak T.E., Arnold F.E., Benner C.L. // Am. Chem. Soc., Polym. Prepr. 1975. V. 16. P. 659.
Takayanagi M. // Pure Appl. Chem. 1983. V. 55. № 5. P. 819.
Schartel B., Wendorff J.H. // Polym. Eng. Sci. 1999. V. 39. № 1. P. 128.
Kotomin S.V. // J. Thermoplastic Comp. Mater. 2013. V. 26. № 1. P. 91.
Hwang W.F., Wiff D.R., Benner C.L., Helminiak T.E. // J. Macromol. Sci. B. 1983. V. 22. № 2. P. 231.
Dadmun M.D., Han C.C. // MRS Symp. Proceedings. 1993. V. 305. P. 171.
Cowie J.M.G., Nakata S., Adams G.W. // Macromol. Symp. 1996. V. 112. № 1. P. 207.
Wiley Encyclopedia of Composites / Ed. by L. Nicolais, A. Borzacchiello, M. Lee Stuart. New York: Wiley, 2012.
Polymer Blends Handbook / Ed. by L. Utracki, C. Wilkie. Dordrech: Springer, 2014.
Painter P.C., Tang W.L., Graf J.F., Thomson B., Coleman M.M. // Macromolecules. 1991. V. 24. № 13. P. 3929.
Schazitel B., Wendorff J.H. // Polym. Eng. Sci. 1999. V. 39. № 1. P. 128.
Шифрина З.Б., Русанов А.Л. // Успехи химии. 1996. Т. 65. № 7. С. 648.
High Performance Polymers-Polyimides Based: From Chemistry to Applications / Ed. by M. Abadie. London: IntechOpen, 2012.
Sroog C. // Progr. Polym. Sci. 1991. V. 16. № 4. P. 561.
Liaw D.-J., Wang K.-L., Huang Y.-C., Lee K.-R., Lai J.-Y., Ha C.-S. // Progr. Polym. Sci. 2012. V. 37. № 7. P. 907.
Dobb M.G., McIntyre J.E. // Adv. Polym. Sci. 1984. V. 60. P. 61.
Encyclopedia of Polymer Science and Engineering / Ed. by H.F. Mark, N.M. Bikales, C.G. Overberger, G. Menges. New York: Wiley-Interscience, 1988.
Developments in Orient Polymers / Ed. by A.J. Owen. London; New York: Appl. Sci. Publ., 1987.
Frank F.C. // Proc. Roy Soc. London. A. 1970. V. 319. P. 127.
Fracture Behaviour of Polymers / Ed. by A.J. Kinlock, R.J. Yong. London; New York: Appl. Sci. Publ., 1983.
Read R.T. // Polym. Paint Colour J. 1988. V. 178. P. 664.
Sazanov Y.N. // Russ. J. Appl. Chem. 2001. V. 74. № 8. P. 1253.
Choi M.-C., Kim Y., Ha C.-S. // Progr. Polym. Sci. 2008. V. 33. № 6. P. 581.
Ni H.-J., Liu J.-G., Wang Z.-H., Yang S.-Y. // J. Industr. Eng. Chem. 2015. V. 28. P. 16.
Sanaeepur H., Ebadi Amooghin A., Bandehali S., Moghadassi A., Matsuura T., Van der Bruggen B. // Progr. Polym. Sci. 2019. V. 91. P. 80.
Pat. 4599396A USA. 1986.
Vinogradova S.V., Vasnev V.A., Vygodskii Ya.S. // Russ. Chem. Rev. 1996. V. 65. № 3. P. 249.
Vinogradova S.V., Vygodskii Ya.S. // Russ. Chem. Rev.1973. V. 42. № 7. P. 1225.
Виноградова С.В., Чурочкина Н.A., Выгодский Я.С., Жданова Г.В., Коршак В.В. // Высокомолек. соед. A. 1971. Т. 13. № 5. С. 1146.
Belomoina N.M., Bruma M., Damaceanu M.D., Mikitaev A.K., Kumykov R.M., Rusanov A.L. // Polymer Science B. 2010. V. 52. № 3–4. P. 227.
Korshak V.V., Vinogradova S.V., Vygodskii Y.S. // J. Macromol. Sci. C. 1974. V. 11. № 1. P. 45.
Dhara M.G., Banerjee S. // Progr. Polym. Sci. 2010. V. 35. № 8. P. 1022.
Zhuang Y., Seong J.G., Lee Y.M. // Progr. Polym. Sci. 2019. V. 92. P. 35.
Ballauff M. // J. Polym. Sci., Polym. Phys. 1987. V. 25. № 4. P. 739.
Tang W.-L., Colman M.M., Painter P.C. // Macromol. Symp. 1994. V. 84. P. 315.
Fukai T., Yang J.C., Kyu T., Cheng S.Z.D., Lee S.K., Hsu S.I.C., Harris F.W. // Polymer. 1992. V. 33. № 19. P. 3621.
Schartel B., Wendorff J.H. // Polym. Eng. Sci. 1999. V. 39. № 1. P. 128.
Niyogi S., Maiti S., Adhikari B. // Polym. Degrad. Stab. 2000. V. 68. P. 459.
Niyogi S., Maiti S., Adhikari B. // Eur. Polym. J. 2001. V. 37. P. 2079.
Niyogi S., Maiti S., Adhikari B. // Polym. Eng. Sci. 2002. V. 42. P. 336.
Ageyeva T., Sibikin I., Karger-Kocsis J. // Polymers. 2018. V. 10. P. 357.
Sibikin I., Karger-Kocsis J. // Adv. Ind. Eng. Polym. Res. 2018. V. 1. P. 48.
Russo S. Casazza E. // Encyclopedia Polym. Sci., A Comprehensive Reference. Elsevier Science. 2012. V. 4. P. 331.
Fu Q., Zhang Q., Peng Y., Du R. // Polym. Int. 2000. V. 49. P. 539.
Fu Q., Livengood B., Shen C.-C., Lid F., Harris F.W., Cheng S.Z.D., Hsiao B.S., Yeh F. // Macromol. Chem. Phys. 1998. V. 199. P. 1107.
Lin J., Xi S., Wu H., Li S. // Eur. Polym. J. 1997. V. 33. № 10–12. P. 1601.
Nakata S., Groeninckx G. // Polymer. 1996. V. 37. № 23. P. 5269.
Takayanagi M., Ogata T., Morikawa M., Kai T. // Macromol. Sci., Phys. 1980. V. I7. № 4. P. 591.
Chuah H.H., Kyu T., Helminiak T.E. // Polymer. 1987. V. 28. № 12. P. 2130.
Chuah H.H., Kyu T., Helminiak T.E. // Polymer. 1989. V. 30. № 9. P. 1591.
Фрунзе Т.М., Курашев В.В., Котельников В.А., Волкова Т.В. // Успехи химии. 1979. Т. 48. № 10. С. 1856.
Carriere F.G., Sekiguchi H., Surin N.N., Kotelnikov V.A., Vygodskii Ya.S. // Polym. Bull. 1995. V. 35. P. 441.
Ding H., Harris F.W. // Pure. Appl. Chem. 1995. V. 67. № 12. P. 1997.
Pae Y., Harris F.W. // J. Polym. Sci., Polym. Phys. 2000. V. 38. 4247.
Pae Y. // J. Appl. Polym. Sci. 2006. V. 99. P. 300.
Pae Y. // J. Appl. Polym. Sci. 2006. V. 99. P. 309.
Pae Y., Harris F.W. // J. Polym. Sci., Polym. Chem. 2000. V. 38. 4247.
Harris F.W., Livengood B.P., Ding H., Lin F.-L., Cheng S.Z.D. // Thermochim. Acta. 1996. V. 272. P. 157.
Harris F.W., Pae Y. // Abstrs 46Int. SAMPE Symp. and Exhibition “A Materials and Processes Odyssey”. Book 1. California, USA, 2001. V. 46. P. 546.
Fu Q., Livengood B., Shen C.-C., Lin F., Li W., Harris F.W., Cheng S.Z.D., Hsiao B.S. // J. Polym. Res. 1997. V. 4. № 1. P. 1.
Neverov V.M., Chvalun S.N., Blackwell J., Harris F.W., Cheng S.Z.D. // Polymer Science A. 2000. V. 42. № 3. P. 299.
Nanotechnology. ACS Symp. Ser. // Ed. by H. Ding, F.W. Harris. Washington: Am. Chem. Soc., 1996.
Pat. 1150725. Great Britain. 1969.
Van Buskirk B., Akkapeddi M.K. // Polym. Prepr., Am. Chem. Soc. Div. Polym. Chem. 1988. V. 29. № 1. P. 557.
Ding H., Harris F.W. // Pure Appl. Chem. 1995. V. 67. № 12. P. 1997.
Vygodskii Y.S., Volkova T.V., Batalova T.L., Sapozhnikov D.A., Chekulaeva L.A. // Polymer Science A. 2003. V. 45. № 2. P. 85.
Котельников В.А., Курашев В.В., Данилевская Л.Б., Конова И.О., Гавриленко В.В., Чекулаева Л.А., Гарбузова И.А., Персиц И.Е. // Высокомолек. cоед. А. 1992. Т. 34. № 1. С. 69.
Vygodskii Ya.S., Volkova T.V., Batalova T.L., Zabegaeva O.N., Belavtseva E.M., Sakharova A.A., Gasanov R.G., Sapozhnikov D.A. // High Performance Polymers. 2009. V. 21. P. 579.
Volkova T.V., Vygodskii Ya.S., Zabegaeva O.N., Zubavichus Y.V., Il’ina M.N., Krasnov A.P., Afonicheva O.V., Lozinskaya E.I., Garbuzova I.A., Shaplov A.S. // J. App. Polym. Sci. 2009. V. 114. P. 577.
Vygodskii Y.S., Volkova T.V., Batalova T.L., Sapozhnikov D.A., Nikiforova G.G., Chekulaeva L.A., Lomonosov A.M., Filatova A.G. // Polymer Science A. 2005. V. 47. № 7. P. 645.
Pae Y. // J. Appl. Polym. Sci. 2006. V. 99. P. 292.
Zabegaeva O.N., Chen G., Fang X., Aliev T.M., Vygodskii Y.S. // Polymer Science B. 2020. V. 62. № 2. P. 94.
Volkova T.V., Pashkova O.N., Il’ina M.N., Belavtseva E.M., Vygodskii Ya.S. // Bull. Russ. Acad. Sci. Phys. 2008. V. 72. № 11. P. 1490.
Vygodskii Y.S., Volkova T.V., Pashkova O.N., Batalova T.L., Dubovik I.I., Chekulaeva L.A., Garbuzova I.A. // Polymer Science A. 2006. V. 48. № 6. P. 557.
Курашев В.В., Котельников В.А., Шлейфман Р.Б., Цуцуран С.В., Аскадский А.А., Васильев В.Г., Казанцева В.В., Бычко К.А. // Высокомолек. cоед. А. 1991. Т. 33. № 9. С. 1956.
Handbook of Low and High Dielectric Constant Materials and Their Application: Materials and Processing // Ed. by H.S. Nalwa. San Diego: Acad. Press, 1999.
Fu G.D., Zong B.Y., Kang E.T., Neoh K.G., Lin C.C., Liaw D.J. // Ind. Eng. Chem. Res. 2004. V. 43. № 21. P. 6723.
Chen B.Y., Wang W., Yu W., Yuan Z., Kang E.T., Neoh K.G., Krauter B., Greiner A. // Adv. Func. Mater. 2004. V. 14. № 5. P. 471.
Chen Y., Chen L., Nie H., Kang E.T. // J. Appl. Polym. Sci. 2006. V. 99. № 5. P. 2226.
Chen Y., Kang E.T. // Mater. Lett. 2004. V. 58. № 29. P. 3716.
Wang W.C., Vora R.H., Kang E.T., Neoh K.G., Ong Ch.K., Chen L.F. // Adw. Mater. 2004. V. 16. № 1. P. 54.
Wang W.C., Vora R.H., Kang E.T., Neoh K.G., Liaw D.J. // Ind. Eng. Chem. Res. 2003. V. 42. № 4. P. 784.
Chen Y., Chen L., Nie H., Kang E.T., Vora R.H. // Mater. Chem. Phys. 2005. V. 94. № 2–3. P. 195.
Chen Y.W., Wang W.C., Yu W.H., Kang E.T., Neoh K.G., Vora R.H., Ong C.K. // J. Mater. Chem. 2004. V. 14. № 9. P. 1406.
Hedrick J.L., Di Pietro R., Plummer C.J.G., Hilborn J., Jerome R. // Polymer. 1996. V. 37. № 23. P. 5229.
Fu G.D., Kang E.T., Neoh K.G., Lin C.C., Liaw D.J. // Macromolecules. 2005. V. 38. № 18. P. 7593.
Meleshko T.K., Il’gach D.M., Bogorad N.N., Kukarkina N.V., Vlasova E.N., Dobrodumov A.V., Malakhova I.I., Gorshkov N.I., Krasikov V.D., Yakimanskii A.V. // Polymer Science B. 2010. V. 52. № 9–10. P. 589.
Yakimanskii A.V., Meleshko T.K., Il’gach D.M., Bogorad N.N., Vlasova E.N., Anan’eva T.D. // Russ. Chem. Bull. 2012. V. 61. № 5. P. 999.
Filippov A.P., Belyaeva E.V., Krasova A.S., Simonova M.A., Tarabukina E.B., Meleshko T.K., Il’gach D.M., Bogorad N.N., Yakimansky A.V. // Polymer Science A. 2014. V. 56. № 1. P. 1.
Meleshko T.K., Il’gach D.M., Bogorad N.N., Kukarkina N.V., Yakimanskii A.V. // Polymer Science B. 2014. V. 56. № 2. P. 118.
Meleshko T.K., Ivanova A.S., Kashina A.V., Ivanov I.V., Nekrasova T.N., Zakharova N.V., Filippov A.P., Yakimansky A.V. // Polymer Science B. 2017. V. 59. № 6. P. 674.
Meleshko T.K., Pulyalina A.Yu., Tyan N.S., Polotskaya G.A., Ivanov I.V., Kukarkina N.V., Toikka A.M., Yakimansky A.V. // Polymer Science B. 2017. V. 59. № 2. P. 183.
Meleshko T.K., Ivanov I.V., Kashina A.V., Bogorad N.N., Simonova M.A., Zakharova N.V., Filippov A.P., Yakimansky A.V. // Polymer Science B. 2018. V. 60. № 1. P. 35.
Filippov A.P., Belyaeva E.V., Meleshko T.K., Yakimansky A.V. // J. Polym. Sci., Polym. Phys. 2014. V. 52. № 23. P. 1539.
Pautov V.D., Nekrasova T.N., Anan’eva T.D., Meleshko T.K., Il’gach D.M., Yakimansky A.V. // Polymer Science A. 2013. V. 55. № 9. P. 526.
Yakimansky A.V., Meleshko T.K., Ilgach D.M., Bauman M.A., Anan’eva T.D., Klapshina L.G., Lermontova S.A., Balalaeva I.V., Douglas W.E. // J. Polym. Sci., Polym. Chem. 2013. V. 51. № 20. P. 4267.
Pautov V.D., Nekrasova T.N., Anan’eva T.D., Meleshko T.K., Ivanov I.V., Yakimansky A.V. // J. Polym. Res. 2018. V. 25. № 1. P. 1.
Klimova S.A., Inozemtseva O.A., German S.V., Gorin D.A., Ilgach D.M., Meleshko T.K., Yakimansky A.V. // Protect. Metals Phys. Chem. Surf. 2015. V. 51. № 3. P. 396.
Kashina A.V., Meleshko T.K., Bogorad N.N., Bezrukova M.A., Yakimanskii A.V. // Polymer Science C. 2019. V. 61. № 1. P. 174.
Filippov A.P., Belyaeva E.V., Zakharova N.V., Sasina A.S., Ilgach D.M., Meleshko T.K., Yakimansky A.V. // Colloid Polym. Sci. 2015. V. 293. № 2. P. 555.
Higa M., Yaguch K., Kitani R. // Electrochim. Acta. 2010. V. 55. № 4. P. 1380.
Miyata S., Nagai K. // Chem. Lett. 2007. V. 36. № 9. P. 1114.
Miyata S., Shirokura H., Nagai K. // Kobunshi Ronbunshu. 2009. V. 66. № 5. P. 170.
Miyata S., Yoshida K., Shirokura H., Kashio M., Nagai K. // Polym. Int. 2009. V. 58. № 10. P. 1148.
Sato S., Ichikawa M., Ose T., Miyata, Takahashi Y., Kanehashi S., Matsumoto H., Nagai K. // Polym. Int. 2013. V. 62. № 9. P. 1377.
Kanehashi S., Koyama Y., Ando S., Konishi S., Shindo R., Miyata S., Sato S., Miyakoshi T., Nagai K. // Polym. Int. 2014. V. 63. № 3. P. 435.
Yoshida A., Nagai K. // Abstrs248 ACS National Meeting & Exposition. San Francisco, USA, 2014, PMSE-394.
Suzuki T., Nagai K. // Abstrs 248 ACS National Meeting & Exposition. San Francisco, USA, 2014, PMSE-377.
Suzuki T., Yoshida A., Ando S., Nagai K. // Polym. Int. 2015. V. 64. № 9. P. 1209.
Yoshida A., Nagai K. // Abstrs250 ACS National Meeting & Exposition. Boston, USA, 2015, PMSE-332.
Yoshida A., Nagai K. // Abstrs252 ACS National Meeting & Exposition. Philadelphia, USA, 2016, PMSE-490.
Taniguchi N., Nagai K. // Abstrs252 ACS National Meeting & Exposition. Philadelphia, USA, 2016, PMSE-464.
Ando S., Koyama Y., Miyata S., Sato S., Kanehashi S., Nagai K. // Polym. Int. 2014. V. 63. № 9. P. 1634.
Ando S., Yoshida A., Nagai K. // Polym. Eng. Sci. 2016. V. 56. № 10. P. 1191.
Sasago Y., Nagai K. // Abstrs252 ACS National Meeting & Exposition. Philadelphia, USA, 2016, PMSE-449.
Ando S., Yoshida A., Nagai K. // J. Appl. Polym. Sci. 2015. V. 132. № 27. P. 42208.
Vygodskii Ya.S., Sakharova A.A., Matieva A.M. // Polymer Science B. 1998. V. 40. № 7–8. P. 282.
Vygodskii Ya.S., Volkova T.V., Sakharova A.A., Matieva A.M. // Polymer Science B. 2001. V. 43. № 3–4. P. 90.
Vygodskii Ya.S., Matieva A.M., Sakharova A.A., Sapozhnikov D.A., Volkova T.V. // High Performance Polymers. 2001. V. 13. P. 317.
Abadie M.J.M., Voytekunas V.Yu., Matieva A.M., Vygodskii Ya.S. // Polymer Science B. 2003. V. 45. № 3–4. P. 67.
Vygodskii Ya.S., Sakharova A.A., Matieva A.M. // High Performance Polymers. 1999. V. 11. P. 379.
Vygodskii Ya.S., Volkova T.V., Sakharova A.A., Sapozhnikov D.A., Matieva A.M. // Polymer Science A. 2002. V. 44. № 12. P. 1249.
Sapozhnikov D.A., Volkova T.V., Sakharova A.A., Gasanov R.G., Voytekunas V.Yu., Abadie M.J.M., Sanchez J.-Y., Vygodskii Ya.S. // Int. J. Polym. Sci. 2009. Art. ID 527046.
Sapozhnikov D.A., Volkova T.V., Sakharova A.A., Gasanov R.G., Voytekunas V.Yu., Abadie M.J.M., Sanchez J.-Y., Vygodskii Ya.S. // Polymer Science B. 2009. V. 51. № 1–2. P. 1.
Vygodskii Ya.S., Sakharova A.A., Matieva A.M. // Polym. Prepr. 1998. V. 39. № 2. P. 819.
Vygodskii Ya.S., Sakharova A.A., Matieva A.M. // Polymer Science B. 2000. V. 42. № 3–4. P. 62.
Vygodskii Ya.S., Volkova T.V., Sakharova A.A., Sapozhnikov D.A., Nikiforova G.G., Buzin M.I. // Polymer Science A. 2004. V. 46. № 7. P. 681.
Vygodskii Ya.S., Volkova T.V., Sakharova A.A., Sapozhnikov D.A., Nikiforova G.G., Matieva A.M. // Polymer Science A. 2006. V. 48. № 7. P. 683.
Vygodskii Ya.S., Matieva A.M., Volkova T.V., Sakharova A.A., Sapozhnikov D.A. // Polymer Science A. 2004. V. 46. № 4. P. 352.
Sapozhnikov D.A., Baiminov B.A., Vygodskii Ya.S. // Dokl. Chem. 2016. V. 468. № 2. P. 169.
Vygodskii Ya.S., Sapozhnikov D.A., Bayminov B.A., Semjonov S.L., Kosolapov A.F., Plastinin E.A. // Progr. Org. Coat. 2016. V. 99. P. 210.
Sapozhnikov D.A., Popova N.A., Vygodskii Ya.S. // Polymer Science B. 2013. V. 55. № 9–10. P. 541.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)