

Высокомолекулярные соединения (серия С), 2021, T. 63, № 2, стр. 253-270
СОВМЕСТНОЕ ИСПОЛЬЗОВАНИЕ АТОМНО-СИЛОВОЙ МИКРОСКОПИИ И МОЛЕКУЛЯРНОЙ ДИНАМИКИ ПРИ ИССЛЕДОВАНИИ БИОПОЛИМЕРНЫХ СИСТЕМ
М. А. Смирнов a, *, Д. А. Толмачев a, А. Д. Глова a, М. П. Соколова a, П. В. Гейдт b, Н. В. Лукашева a, С. В. Люлин a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
b Новосибирский государственный университет
630090 Новосибирск, ул. Пирогова, 2, Россия
* E-mail: Smirnov_Michael@mail.ru
Поступила в редакцию 05.04.2021
После доработки 27.05.2021
Принята к публикации 11.06.2021
Аннотация
Проведен анализ имеющихся в литературе публикаций, показывающих перспективность сочетания компьютерного моделирования с атомно-силовой микроскопией при исследовании полимерных биообъектов и материалов различного назначения. Описаны проблемы, возникающие при совместном использовании этих методов. Представлены три основных направления, в рамках которых атомно-силовая микроскопия применяется совместно с компьютерным моделированием: исследование топографии поверхности полимерных материалов; измерение силовой кривой взаимодействия атомно-силового зонда с полимерными структурами; определение энергии межмолекулярных взаимодействий.
ВВЕДЕНИЕ
Направленное регулирование характеристик современных полимерных материалов и многокомпонентных полимерных систем, используемых в качестве сенсоров, мембран, а также материалов и систем биомедицинского назначения, требует детального понимания их структуры и свойств на различных масштабах размеров. Исследование свойств на нанометровом уровне позволяет устанавливать молекулярные механизмы, отвечающие за изменение свойств многокомпонентных систем при вариации химического состава компонентов.
С момента своего создания в 1986 г. атомно-силовая микроскопия превратилась в незаменимый и чрезвычайно информативный метод исследования материалов и молекулярных систем на наномасштабном уровне. АСМ применяется для анализа широкого спектра функциональных материалов [1–13] и биообъектов [14–17]. Более чем 30-летняя история АСМ и ее методология изложены в ряде обзоров, например [18–22]. Являясь одним из немногих методов, обладающих нанометровым разрешением, АСМ позволяет визуализовать полимерные материалы на молекулярном уровне [23]. Показано, что данный метод представляет интерес при исследовании конформации отдельных полимерных цепей [24], молекулярной подвижности полимерных щеток [25], кристаллизации макромолекул [26] и свойств поверхности полимерных материалов [27]. Разработка новых методик в области АСМ, таких как анализ физических свойств поверхности на наномасштабе или молекулярная силовая спектроскопия [28], исследование вязкоупругих или пластических свойств поверхности полимерных материалов, открывает дополнительные возможности изучения физических свойств полимерных молекул и материалов на наноуровне. Вместе с тем метод АСМ дает информацию только о топографии и механических свойствах поверхности, в то время как молекулярная структура и механизм отклика материала на механическое воздействие атомно-силового зонда остаются неизвестными. Таким образом, актуальным является объединение АСМ с методами, позволяющими дополнять данные о физических свойствах поверхности [29]. Обзоры по таким комбинированным подходам в настоящее время только начинают появляться в литературе. Отмечается, что комбинация АСМ и флуоресцентной микроскопии высокого разрешения оказывается востребованной при исследовании биологических объектов [30]. Сопоставление результатов измерений вязкоупругих характеристик клеточных ядер методом АСМ с результатами моделирования деформации ядра методом конечных элементов позволило обнаружить протяженные пространственные неоднородности в структуре этих объектов [31]. Было показано, что комбинирование АСМ и облучения исследуемой поверхности ИК-излучением определенной длины волны дает возможность характеризовать микроструктуру полимерных смесей в каждой точке взаимодействия зонда и поверхности [32].
В последнее десятилетие моделирование методом молекулярной динамики (МД) широко применяется для исследования структуры полимерных материалов и молекулярных механизмов функционирования биологических объектов [33–61 ]. Атомистическое моделирование позволяет получать информацию об объектах на атомарном уровне и обладает большим потенциалом как метод прогнозирования свойств полимерных систем [62–76]. Возможность достаточно простого варьирования состава и типа учитываемых взаимодействий компонентов изучаемых систем делает МД уникальным инструментом, незаменимым для расширения границ понимания молекулярных процессов, особенно в биологических системах [37, 60, 77–83]. Последние достижения в области вычислительной техники расширили доступные для МД временные границы моделирования вплоть до микросекундных масштабов [81, 82, 84–87]. Разработка новых подходов, направленных на увеличение статистического ансамбля в моделировании [88–91], позволила исследовать относительно медленные процессы, такие как свертывание пептидов и их агрегация [92–94] или, например, формирование органоминеральных комплексов [41, 95, 96]. Вместе с разработкой более точных силовых полей это дает возможность расширить круг задач, решаемых методами МД, и в ряде случаев добиться количественного согласия с экспериментальными данными [38]. В то же время сложность и многокомпонентность моделируемых систем обычно требует использования большого набора модельных параметров, что диктует необходимость проверки (валидации) применяемых моделей и протоколов моделирования путем сопоставления результатов, полученных в моделировании и в эксперименте [86, 97–102]. Важно отметить, что характерные размеры периодических ячеек, рассматриваемых в моделировании, составляют ~10 нм [60] и по порядку величины совпадают с размерами острия зондов, применяемых в АСМ. В связи с этим открывается возможность прямого сопоставления результатов моделирования с характеристиками объектов, которые могут быть получены методом АСМ.
Таким образом, можно ожидать кооперативный эффект от комбинации АСМ и моделирования методом молекулярной динамики в области науки о полимерах. Основной проблемой на данном пути оставалось несоответствие времен, доступных для воспроизведения в моделировании, и временных интервалов в эксперименте [103]. Однако произошедшее за последние годы значительное увеличение мощности вычислительных ресурсов и соответственно возможностей компьютерного моделирования позволяет ожидать многообещающих перспектив в расширении границ физики и химии полимеров за счет более тесного сочетания методов АСМ и МД.
Целью данного обзора является обобщение имеющихся в литературе работ, в которых продемонстрирован кооперативный эффект объединения АСМ и МД с акцентом на исследования в области биополимерных систем. Важно отметить, что опубликованный в 2001 г. обзор работ по объединению методов АСМ и моделирования был посвящен в первую очередь неравновесной динамике [104]. За последние 20 лет оба рассматриваемых метода претерпели значительное развитие, и их совместное использование требует нового обзора. Анализ литературы показывает, что можно выделить три направления, в которых АСМ и МД применяются совместно: экспериментальное и теоретическое исследование топологии поверхности полимерных материалов; моделирование и измерение сил, возникающих при взаимодействия атомно-силового зонда с поверхностью; описание молекулярных взаимодействий компонентов сложных молекулярных систем.
МЕТОДЫ ИССЛЕДОВАНИЯ
Атомно-силовая микроскопия
Атомно-силовая микроскопия – это один из методов семейства зондовых микроскопий, который наряду со сканирующей туннельной микроскопией позволяет разрешать наноразмерную и субмолекулярную структуру в различных материалах и молекулярных объектах от гладких полимерных пленок до белков [105, 106]. В области материаловедения АСМ широко применяется для исследования топографии, т.е. карты высот поверхности объектов. Метод дает возможность определить характерные размеры (длина, диаметр), форму объектов и перепад высот на различных участках [107]. Конструкционно АСМ состоит из пьезосканера (трубки, которая изменяет длину и наклон для перемещения лежащего на ней образца), АСМ-зонда (кантилевера), который выполнен в виде балки/консоли с перпендикулярно ориентированной пирамидой, имеющей острое окончание с радиусом скругления острия 1–20 нм. Острота зонда во многом определяет и ограничивает разрешение сканирования в АСМ. На консоль падает луч лазера, который отражается в 4-секционный фотодетектор (рис. 1). Сигнал с фотодетектора попадает в блок обратной связи, который направляет его на рабочую станцию и посылает управляющий сигнал на пьезосканер. Современные атомно-силовые микроскопы позволяют измерять силы с точностью до нескольких пН [106, 108] за времена длительностью в несколько микросекунд.
Рис. 1.
Схема работы атомно-силового микроскопа. Цветные рисунки можно посмотреть в электронной версии.
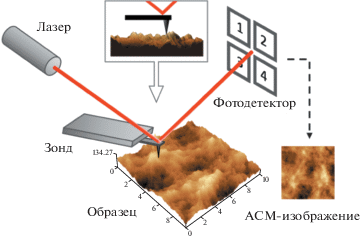
Измерения на атомно-силовом микроскопе выполняются посредством разных методик: контактной, полуконтактной и бесконтактной. Наибольшей популярностью обладают полуконтактные методики, когда зонд простукивает поверхность с резонансной частотой балки зонда. Поскольку размеры этой балки малы (длина – порядка сотни микрометров, ширина – несколько десятков микрометров, а толщина около 5 мкм), резонансная частота колебаний составляет сотни килогерц. Полуконтактный режим оптимален для исследования полимерных образцов, так как позволяет проводить быстрое сканирование (что бывает полезно для визуализации динамических процессов стеклования полимеров) и в то же время заметным образом не деформирует образцы во время простукивания с небольшой силой нажима зондом. Для сканирования участка рельефа поверхности образца и определения механических характеристик широко используются контактный, полуконтактный и бесконтактный методы, а для измерения локальных характеристик в режиме спектроскопии сил взаимодействия зонд–образец (сил притяжения при разрыве связей в макромолекуле и сил отталкивания при продавливании упругого тела) применяется измерение силовых кривых, т.е. зависимости F(Z).
Благодаря развитию компьютерных методик обработки сигнала с большим массивом данных и улучшению инструментов нанодиагностики стало возможным определение механических характеристик в режиме измерения свойств, в том числе силовых кривых, в каждой точке скана (рис. 2) – так называемое количественное наномеханическое картирование. Преимущество такого подхода состоит в том, что при одном акте удара зондом о поверхность материала можно зафиксировать несколько характеристических участков силовой кривой, определяющихся силами взаимодействия между зондом и образцом. Разные участки силовой кривой могут дать информацию уже не только о топографии, но и о модуле упругости материала поверхности, об адгезии к ней зонда, энергии диссипации (потери энергии за один удар в полуконтактном режиме) и других характеристиках, позволяя за один скан получить множество карт свойств на одной измеренной области.
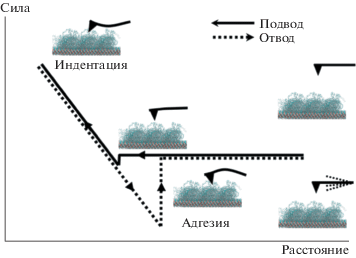
При этом для исследования свойств не плотно упакованных материалов или мягких полимеров, в том числе биополимеров, используется не только картирование, но также анализ упругого взаимодействия в конкретно выбранных локациях на поле сканирования [106]. Иначе говоря, проводится измерение силы взаимодействия зонд–образец F в зависимости от расстояния Z между зондом и образцом. Выбор данной методики связан также с тем, что картирование не видится подходящим методом для исследования силы связи между боковыми цепочками полимеров и биомолекул, так как оно позволяет измерить упругие свойства лишь по вертикальной оси Z, но не по латеральным осям X и Y.
Изучение отклика системы на внешнее механическое воздействие в макросистемах контролируется динамометром. В роли динамометра в атомно-силовом микроскопе выступает юстированная система из четырехсекционного фотодетектора и балки АСМ-зонда с известной жесткостью. При получении зависимости F(Z) в режиме индентации зонд медленно подводится к поверхности, прикасается к ней, углубляется в нее и затем отводится вверх от поверхности. Стоит отметить, что для получения упругих характеристик глубина индентации должна составлять не более 2 нм [109], в противном случае будет исследован текучий режим взаимодействия и измерен модуль пластичности или прочности материала. При отходе от поверхности зонд испытывает дополнительные силы притяжения к ней – силы адгезии. Последние обусловлены слабыми связями между атомами зонда и образца, возникшими в момент соприкосновения. Таким образом, по форме кривой F(Z) можно восстановить участки, отвечающие за прогиб, неупругую деформацию и клейкость, а также количественно охарактеризовать эти величины для конкретного образца в конкретной локации поверхности.
Второй наиболее популярный вид механического АСМ-эксперимента – растягивание одиночной молекулы, которое, как и в случае макроразмерных материалов, предполагает измерение увеличения длины структуры при прикладывании к ней известной силы. Одним из недостатков данного подхода, как будет указано ниже, является невозможность определения точного местоположения “слабой” области, где происходит наиболее сильное растяжение. Вместо этого определяется усредненное значение удлинения всей полимерной цепочки.
Длительность АСМ-эксперимента составляет десятки минут и даже часы, и часто требует нетривиальной подготовки образца и зондов. Тем не менее, АСМ активно используется как диагностический экспериментальный метод исследования морфологии и структуры полимеров и биомолекул.
Компьютерное моделирование
В зависимости от степени огрубления используемой модели методы компьютерного моделирования можно условно разделить на несколько групп в соответствии с их пространственно-временным масштабом (рис. 3).
Рис. 3.
Схематичное представление пространственно-временных масштабов, доступных различным методам компьютерного моделирования в сравнении с таковыми в случае АСМ.
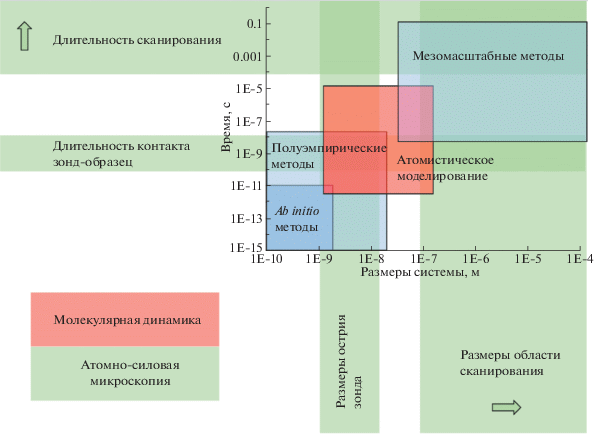
Квантово-химические методы моделирования основаны на решении уравнения Шредингера и учитывают корпускулярно-волновой дуализм моделируемых соединений. Аналитическое решение уравнения Шредингера возможно только для тривиальных случаев, таких как, например, атом водорода. Для расчета более сложных соединений, содержащих большое количество взаимодействующих друг с другом электронов, вводят различные допущения. Использование квантово-химических методов расчета в полимерной науке подробно обсуждено в обзорах R.A. Friesner [110] и F. Ruipérez [111]. Важно отметить, что такие методы ограничены с точки зрения доступных размеров моделируемых систем несколькими сотнями атомов. Тем не менее, доступные масштабы данного подхода позволяют исследовать взаимодействие двух небольших объектов (например, частей зонда и исследуемого образца) с учетом квантово-химических эффектов.
Более грубым подходом к атомистическому моделированию молекулярных систем является метод молекулярной динамики, основанный на численном решении уравнений Ньютона, описывающих движение частиц, взаимодействующих друг с другом. Обычно такие частицы представляют отдельные атомы. Взаимодействие между ними задается с помощью потенциалов и параметров, выбранных на основе эмпирических данных и квантово-химических расчетов. Такой тип огрубления позволяет на микросекундном временном масштабе рассматривать системы с размером порядка 10 нм, причем ячейка моделирования может содержать сотни тысяч атомов. Данный подход дает возможность учитывать особенности химического строения моделируемых систем и непосредственно исследовать протекающие в них процессы и их структуру на молекулярном уровне. Масштабы атомистического моделирования граничат с возможностями АСМ (рис. 3), что с учетом ее разрешения делает метод молекулярной динамики перспективным для их совместного использования.
В последние десятилетия метод молекулярной динамики становится все более популярным, что в числе прочего стимулирует его развитие [34, 50, 52]. С одной стороны, развиваются подходы, позволяющие учесть квантово-химические эффекты в атомистической модели исследуемых объектов [112, 113], что повышает точность моделирования, лишь частично ограничивая пространственно-временные масштабы. С другой стороны, развивается набор методик, расширяющих возможности атомистического моделирования. Важно выделить новые подходы, основанные на расчете свободной энергии системы, например метадинамику и зонтичную выборку, дающие возможность получать информацию о взаимодействии молекулярных объектов друг с другом (например, об адсорбции полимеров на мембрану [48]), а также обеспечивать расширение статистического ансамбля в случае присутствия в системе сильных взаимодействий, препятствующих перестройкам в моделируемой системе [41, 89, 114]. Разработанный в 2002 г. метод метадинамики [115], заключающийся в сканировании энергетического ландшафта системы, на данный момент активно используется для улучшения выборки при моделировании [94]. Моделирование методом зонтичной выборки, основанным на подходе, разработанном G.M. Torrie и J.P. Valleau в 1977 г. [116], заключается в расчете свободной энергии системы как функции расстояния между молекулами. С помощью этих подходов можно напрямую оценить энергию взаимодействия молекулярных объектов, что крайне важно при исследовании таких процессов, как, например, адгезия полимеров на поверхности или образование полимерных агрегатов. Кроме того, важным направлением развития атомистического моделирования является метод неравновесной МД [117], в рамках которого к объектам в системе прикладывается внешнее воздействие. Такой подход позволяет исследовать, в частности, движение зонда АСМ по поверхности материала [118, 119] или разворачивание белков [120, 121]. Тем не менее, поскольку в процессе движения частиц под действием приложенного поля система может не успевать достигнуть равновесного состояния, исследования изменения свободной энергии методом неравновесной МД следует проводить с осторожностью, как было показано в недавней работе [122].
Таким образом, методы атомистического моделирования могут быть использованы для прямого сравнения с АСМ. Вместе с тем, пространственно-временные масштабы данного теоретического подхода все еще сильно ограничены, что существенно затрудняет прямое сравнение структур, полученных в моделировании, и структур, визуализированных с помощью АСМ. В связи с этим могут быть привлечены еще более грубые модели исследуемых систем в рамках мезоскопических методов моделирования, в которых структурным элементом уже является фрагмент из нескольких атомов, например повторяющееся звено или сегмент Куна [123]. Огрубление дает возможность получить информацию о структуре на масштабах, доступных для АСМ. Более того, времена, доступные для мезоскопических методов, также соизмеримы с временами, характерными для сканирования АСМ. Однако использование такой грубой модели зачастую приводит к потере информации о влиянии химической структуры полимеров на процессы, происходящие в исследуемой системе. Следовательно, выбор уровня описания следует проводить с учетом особенностей рассматриваемых объектов.
ТОПОГРАФИЯ ПОВЕРХНОСТИ
Классическим и наиболее распространенным применением метода АСМ является исследование топографических особенностей материалов. АСМ позволяет получать данные о морфологии поверхности на наномасштабном уровне и детектировать на ней субмолекулярные неоднородности, размеры которых близки к размерам ячеек моделирования (~10 нм).
Из-за различия пространственных масштабов, доступных АСМ и атомистическому моделированию, результаты, получаемые этими методами, могут не всегда совпадать количественно. Такое несоответствие продемонстрировано в работе [124], в которой с помощью АСМ и МД исследовано влияние температуры высушивания на структуру поверхности пленок полианилина, нанесенных на наноструктурированную поверхность оксида титана. Предсказанное с помощью МД изменение структуры поверхности было только качественно подтверждено данными АСМ. Оба метода показали, что шероховатость поверхности увеличивается с повышением температуры высушивания. Количественные различия между теоретическими и экспериментальными результатами связаны с тем, что АСМ описывает систему на микромасштабном уровне, в то время как МД дает информацию на наномасштабном уровне.
Другое ограничение совместного использования АСМ и МД было обнаружено S. Drechsler с соавторами при исследовании набора из четырех Y-образных тетрамеров ароматических амидов с различными функциональными возможностями, способных к агрегации в результате нековалентных взаимодействий [125]. Основные механизмы взаимодействия (агрегации) Y-образных амидных олигомеров и структура образующихся супрамолекулярных цепей были исследованы с помощью атомистического компьютерного моделирования методом МД и экспериментальных методов: АСМ, УФ-спектроскопии, реологии, малоуглового рассеяния рентгеновских лучей и растровой электронной микроскопии. Экспериментальные данные показали образование самоорганизующихся структур, форма которых зависит от природы подложки, на которую были нанесены образцы. В моделировании в отличие от АСМ влияние подложки может быть исключено, но, к сожалению, масштабы времени и длины, доступные для атомистической МД, не позволяют детально изучить динамику агрегирования и конечные самоорганизованные структуры. По этой причине в работе [125] с точки зрения значений энергии взаимодействия и структурных параметров изучались только ранние стадии агрегации (образование димеров).
A.R. Bizzarri с соавторами в работе, посвященной характеристике топологических и проводящих свойств белков, адсорбированных на металлических электродах, наблюдали количественное различие между результатами методов МД и АСМ [126]. В этой работе был исследован модифицированный пластоцианин (белок, содержащий медь) в водной среде, ковалентно связанный с поверхностью Au (111) через дисульфидный мостик. Было показано, что совместное использование АСМ и МД открывает новые возможности для более глубокого понимания влияния закрепления белка на металлической поверхности на его структуру и топологические свойства (высоту и ориентацию на подложке). Было обнаружено, что распределения молекул по ориентациям на подложке, полученные с помощью моделирования, заметно уже, чем найденные с помощью АСМ. Одной из причин такого различия может быть давление, оказываемое острием зонда на белок в ходе эксперимента. Другой причиной может быть то, что МД-моделирование проводилось для одной молекулы белка, в то время как данные АСМ относятся к образцу, содержащему большое количество молекул в различных начальных конфигурациях.
Несмотря на неполное (в некоторых случаях) совпадение результатов АСМ и МД, близость разрешения, достигаемого обсуждаемыми методами, делает естественным их сочетание. Объединение возможностей этих методов позволяет получить дополнительную информацию и лучше понять молекулярные процессы, происходящие в исследуемых системах. Атомистическое моделирование помогает выявить структуру и процессы на молекулярном уровне и понять природу объектов, обнаруженных при АСМ-сканировании. Наиболее распространенные работы, в рамках которых используется комплексный подход, включающий методы АСМ и МД-моделирования, посвящены структурным исследованиям на многомасштабном уровне. Комбинация методов, описывающих структуру материала на разных пространственных масштабах, делает такой подход к исследованию естественным и позволяет достичь более полного понимания структуры материала, чем использование каждого из методов по отдельности. Так, в недавнем исследовании пектинового матрикса клеточной стенки [127] P.M. Pieczywek с соавторами использовали МД-моделирование для определения структуры агрегатов, наблюдаемых с помощью АСМ. В МД-моделировании авторы получили распределение по размеру молекул, входящих в состав агрегатов. Подбор комбинации таких распределений к высотному профилю агрегата, полученному методом АСМ, позволил сделать предположение о молекулярном составе агрегатов. Такой же подход применили A.S. Garay с соавторами [128] при исследовании агрегации сополимеров на основе мономеров тимина. Сравнение диаметров агрегатов, предсказанных в результате моделирования, и агрегатов, обнаруженных с помощью АСМ, позволило сделать вывод о том, что отдельные макромолекулы объединены в пучки из двух полимерных цепей. Сочетание методов помогло описать исследуемый объект на многомасштабном уровне и понять молекулярные механизмы образования агрегатов.
Комбинированное исследование методами МД и АСМ дало согласующиеся между собой данные о структуре и динамике сборки геллановой камеди в зависимости от pH раствора и взаимодействия с глинистыми минералами [129]. Моделирование позволило преодолеть ограничения АСМ-визуализации, которая требует удаления растворителя. Полученные данные проливают свет на механизмы взаимодействия, а также помогают контролировать динамические свойства и стабилизировать углеводные гидрогели при помощи глинистых минералов. В свою очередь S. Jeong с соавторами использовали комбинацию АСМ и МД, чтобы продемонстрировать возможность нековалентной адсорбции одноцепочечной ДНК на поверхности так называемых, графеновых квантовых точек [130]. Ими была изучена зависимость адсорбции полимера от степени окисления графеновых квантовых точек. Для этого были исследованы нековалентные биополимерные ассоциации на четырех типах субстратов графеновых квантовых точек в широком диапазоне уровней окисления. Зависимость комплексообразования одноцепочечной ДНК с графеновыми квантовыми точками от уровня окисления графеновых квантовых точек подтверждалась как AСM, так и MД, а также результатами, полученными с помощью теории функционала плотности. Использование МД-моделирования позволило определить энергии комплексообразования. J. Włoch с соавторами исследовали взаимосвязь между шероховатостью поверхности политетрафторэтилена и ее смачиваемостью на наномасштабном уровне [131]. Три серии шероховатых поверхностей из политетрафторэтилена были охарактеризованы путем моделирования АСМ в рамках МД (МД-АСМ). Результаты моделирования структуры политетрафторэтилена и взаимодействия его поверхности с водой позволили связать значения краевого угла смачивания водой с плотностью политетрафторэтилена и со среднеквадратичной шероховатостью поверхности, рассчитанной в МД-АСМ. Данные МД-моделирования продемонстрировали почти такое же поведение угла смачивания в зависимости от шероховатости поверхности, как и эксперимент.
Возможность многомасштабного исследования и определения молекулярных механизмов формирования сложных структур наномасштабного уровня делает комбинацию методов АСМ и атомистического моделирования мощным инструментом для исследования сложнейших биологических систем, особенно при изучении агрегации пептидов. Так, обсуждаемый в обзоре экспериментально-теоретический подход был применен в исследованиях молекулярных механизмов, вызывающих болезнь Альцгеймера [132–134]. Болезнь Альцгеймера представляет собой заболевание, связанное с ошибкой в сворачивании белка, которое характеризуется накоплением пептида β-амилоида (Aβ) в виде сенильных бляшек, прогрессирующей нейродегенерацией и потерей памяти. L. Connelly с соавторами использовали МД-моделирование вместе с АСМ для исследования структуры мембранных пор, образованными β-амилоидами [132]. При этом АСМ служил для подтверждения результатов компьютерного моделирования и получения информации о наноуровневой структуре мембраны. Компьютерное моделирование определило участки в пептиде, которые могут отвечать за ингибирование или индукцию проницаемости пор. Выявленные участки могут стать важной основой для исследований по разработке соответствующих лекарств. Недавно M. Tian с соавторами удалось исследовать процессы самосборки амилоидных протофибрилл β-амилоида в полиморфные многонитевые “зрелые” фибриллы, что необходимо для понимания природы амилоидоза β-амилоида [133]. Анализ изображений АСМ помог идентифицировать самосборку двух, трех и четырех отдельных одноцепочечных протофибрилл в зрелые фибриллы, в то время как МД-моделирование выявило лестничный механизм упаковки фибрилл. С помощью теоретических и экспериментальных методов R. Rajput с соавторами продемонстрировали новый подход к разработке потенциальных антиамилоидных молекул, которые могут быть полезны при разработке эффективных лекарств от болезни Альцгеймера [134].
I.A. Bhat с соавторами применили комбинацию методов АСМ и МД для изучения смесей сурфактантов и гемоглобина [135]. АСМ и другими экспериментальными методами показано образование комплекса сурфактант–гемоглобин и охарактеризовано поведение гемоглобина при взаимодействии с сурфактантом. Моделирование дополнительно подтвердило образование комплекса и позволило определить основные взаимодействия, ответственные за его образование.
B. Cai с соавторами использовали комбинацию АСМ и методов МД для исследования амфифильных пептоидных олигомеров, которые самоорганизуются в различные биомиметические наноструктуры [136]. АСМ показала прямую зависимость морфологии формирующейся структуры и кинетики роста неорганических материалов от пептоидных последовательностей. Моделирование методом МД продемонстрировало такую же зависимость, что дополняет экспериментальные данные, позволяя установить взаимодействия, ответственные за формирование структур.
Развитие АСМ и, в частности, увеличение разрешения делает возможным прямое сравнение структур, изучаемых методами АСМ и компьютерного моделирования. Так, Y. Oda с соавторами с помощью АСМ проследили траекторию одиночной полимерной цепи на твердой поверхности [137]. Высокое пространственное разрешение АСМ позволило исследовать конформацию полимерной цепи. В то же время МД предоставила информацию о конформационных переходах адсорбированных цепей. В другой работе прямое сравнение результатов, полученных методами АСМ и МД, было проведено в рамках исследования синтетических четырехкомпонентных липидных монослоев, созданных для легочного сурфактанта, не содержащего белков [138]. В этой работе J. Liekkinen с соавторами, используя МД-моделирование и визуализацию с помощью AСM, обнаружили наличие латеральных неоднородностей в монослоях липидов, которые могут играть решающую роль в функционировании сурфактанта легких. МД-моделирование и метод АСМ были применены для изучения диффузии и структуры мембраны.
Использование крупнозернистых моделей, в которых один движущийся центр соответствует группе атомов, позволяет добиться такого же масштаба при мезоскопическом моделировании, что и в АСМ. В недавней работе [139] комбинация АСМ и мезоскопического моделирования внесла значительный вклад в понимание структуры полиэлектролитной щетки в присутствии многовалентной соли. Авторами впервые были визуализованы боковые неоднородности на поверхности полиэлектролитных щеток, сколлапсированных в растворах, содержащих многовалентную соль; существование таких неоднородностей было предсказано ранее в теоретической работе [140]. Интерпретация и дальнейший анализ результатов АСМ были подтверждены компьютерным моделированием с крупнозернистыми моделями. Моделирование в этом случае позволяет легко изменять параметры системы, что помогает понять природу исследуемого эффекта. G. Onaran с соавторами применили аналогичный подход к изучению морфологии расплава симметричных триблочных сополимеров. С помощью крупнозернистой модели авторы построили фазовую диаграмму и показали, что изменение молекулярной массы блоков и силы межмолекулярного взаимодействия между ними могут служить для контроля морфологии [141]. Мезоскопическое моделирование позволило авторам использовать АСМ-изображения для проверки параметров моделирования. Это дало возможность сделать выводы, относящиеся к конкретным химическим соединениям, несмотря на грубые модели, задействованные при их моделировании. Комбинация моделирования и AСM также была успешно применена в работе [142]. Это исследование сфокусировано на изучении смесей линейного и сильно разветвленного поли(ариленэфирсульфонов), а именно, на морфологии и механическом поведении смесей в зависимости от расстояния между точками ветвления в разветвленном поли(арилен-эфирсульфоновом) компоненте. Распределения плотности, найденные в моделировании, и фазовые изображения, полученные в рамках АСМ, продемонстрировали схожие наномасштабные топографии, которые свидетельствуют о совместимости всех смесей и отсутствии разделения фаз. Моделирование дополнительно объяснило микрофазовое поведение, смешиваемость и влияние расстояния между точками ветвления в разветвленном поли(ариленэфирсульфоне) на морфологию материала. Совместные экспериментальные исследования и компьютерное моделирование показали, что понимание влияния степени разветвления на межмолекулярные взаимодействия сильно разветвленных полимеров и их линейных аналогов имеет решающее значение для получения конечных смесей полимеров с настраиваемыми механическими свойствами и улучшенными механическими характеристиками.
Таким образом, сочетание методов АСМ и компьютерного моделирования является мощным инструментом для исследования структуры широкого круга объектов, в то время как АСМ дает прямую информацию о структуре материала на наномасштабном уровне, атомистическое моделирование выявляет структуру на молекулярном уровне, описывая молекулярные механизмы ее формирования [129–134, 136, 137, 143–145]. Более того, атомистическое моделирование позволяет интерпретировать результаты АСМ. С помощью компьютерного моделирования можно выдвигать гипотезы о структуре и составе наноразмерных неоднородностей, обнаруживаемых с помощью АСМ [127, 128]. Прогресс в увеличении разрешения АСМ, с одной стороны, и повышение размера ячейки моделирования, с другой, позволяют проводить прямое сравнение структур, полученных разными методами. Это дает возможность обосновать параметры моделирования с помощью АСМ и исследовать методом компьютерного моделирования объекты, наблюдаемые в эксперименте. Такой комплексный подход позволяет объединить положительные стороны теоретических и экспериментальных методов и компенсировать их недостатки. Однако важно отметить, что методы АСМ и МД могут приводить к разным результатам по ряду причин: использование упрощенных моделей в моделировании, наличие специфических взаимодействий между зондом АСМ и исследуемой поверхностью, а также различие в масштабах измерения, доступных рассматриваемым методам [124–126]. Комбинация АСМ и моделирования должна применяться с пониманием специфики исследуемых систем и обязательным обоснованием результатов, полученных данными методами.
НАНОМЕХАНИЧЕСКИЕ ХАРАКТЕРИСТИКИ
Обзор современной литературы указывает на перспективность проведения взаимодополняющих исследований методами компьютерного моделирования и АСМ, а также при анализе механических характеристик полимерных материалов на наномасштабном уровне [146, 147]. Сложная иерархическая структура полимерных материалов, состоящая из кристаллической и аморфной фаз, является одним из факторов, определяющих их механические, транспортные, электрические и другие свойства. Изучение надмолекулярной структуры требует высокого пространственного разрешения, порядка 1–10 нм, что делает атомно-силовую микроскопию одним из наиболее эффективных экспериментальных методов в данной области. Контроль отклонения зонда по оси, перпендикулярной поверхности образца, позволяет определять усилие, приложенное к образцу по закону Гука (F = kd, где где k – жесткость кантилевера, d – отклонение кантилевера), тогда как деформацию образца δ можно рассчитать по отклонению кантилевера и смещению пьезоэлемента. Анализируя взаимосвязь между F и δ с помощью теоретических моделей механики контакта зонда с поверхностью можно количественно охарактеризовать наномеханические свойства материалов, такие как модуль упругости и энергия адгезии на нанометровом масштабе [148]. Однако до сих пор трудно выявить прямую корреляцию между структурой на масштабах цепей полимеров и механическими параметрами, измеренными с помощью атомно-силового микроскопа. Эффективным методом предсказания поведения молекул, в частности, механических свойств различных типов полимерных материалов на наномасштабном уровне служит МД.
Количественное сравнение данных о механике взаимодействия зонда с поверхностью, полученных методами АСМ и МД, встречает ряд трудностей. Так, в работе [149], в которой проанализированы наномеханические свойства полиэтилена высокой плотности, отмечено, что количественное сравнение модулей, найденных методом МД и в эксперименте, сопряжено с двумя проблемами. Первая проблема – правильный выбор теоретической модели контактного взаимодействия кантилевера атомно-силового микроскопа с исследуемой поверхностью. Вторая проблема связана с разной скоростью движения кантилевера в моделировании и в АСМ (расхождение на семь–восемь порядков) [150, 151]. Вместе с тем в результате исследования было обнаружено, что экспериментальные кривые сила–деформация и кривые, полученные в моделировании, сходны по форме. Кроме того, рассчитанные значения модулей и гистограммы их распределения, соответствующие разным методам, также близки [149]. Однако авторы обращают внимание на то, что “реальные” материалы в отличие от “модельных” имеют более неоднородную упаковку цепей в кристаллитах, которые могут быть ориентированными, иметь дефекты структуры, и т.д., что в общем случае затрудняет сопоставление эксперимента и теории.
C целью понимания механизмов деформации поликарбоната и аналогичных ему полимеров в процессе наноиндентирования методом АСМ D. Ikeshima с коллегами провели мезоскопическое моделирование наноиндентирования методом МД [152]. Авторам удалось сконструировать большую систему объемом 25 000 нм3, содержащую 500 цепей поликарбоната со степенью полимеризации 100 и провести моделирование процесса вдавливания наконечника кантилевера в полимер. Для исследования механизма деформации авторы количественно оценивали потенциальную энергию, связанную с различными режимами, такими как растяжение связи, изгиб и кручение. Было обнаружено, что локальная нагрузка при вдавливании вызывает деформацию поверхности поликарбоната вблизи острия кантилевера. При этом необратимая локальная деформация полимерной цепи приводит к возникновению текучести полимера. Сравнение силовых кривых, полученных в эксперименте по АСМ, с расчетными силовыми кривыми показало их идентичность. Кроме того, значения предела текучести, определенные при моделировании, качественно согласовывались с результатами эксперимента. Вместе с тем отличие в скорости движения зонда в моделировании (50 м/с) и эксперименте (500 нм/с) приводит к завышению предела текучести в моделировании по сравнению с экспериментом.
В работе [153] исследовано взаимодействие между зондом из аморфного диоксида кремния и подложкой из аморфного полиметилметакрилата в условиях, подходящих для АСМ в полуконтактном режиме. Для фиксации динамики зонда авторы использовали метод моделирования динамического контакта. В работе показано, как топография поверхности и подповерхностные фазы влияют на локальные свойства, определяемые из кривых сила–смещение. Характеристики, полученные из динамического АСМ (энергия диссипации и модуль Юнга), зависят не только от подложки и скорости деформации, но и от степени вдавливания и топографии локальной поверхности. Кроме того, они отражают локальные изменения свойств поверхности. Форма и амплитуда кривых сила–смещение, а также соответствующие значения энергии диссипации, предсказанные моделированием и являющиеся следствием вязкоупругого поведения аморфного полимера, хорошо согласовывались с экспериментальными данными. Анализ карты свойств композитной системы (образец с нанотрубкой около поверхности) показывал соответствующие различия в свойствах вблизи и вдали от углеродной нанотрубки, что позволило предположить пространственную локализацию приповерхностного упрочнения.
Такой же подход был применен для моделирования взаимодействия платинового зонда с плоской поверхностью из того же металла в условиях, соответствующих полуконтактной моде АСМ [154]. В работе изучалось, как скорость нагрузки во время контакта и номинальное расстояние между наконечником и подложкой влияют на взаимодействие между зондом и поверхностью. При анализе кривых сила–смещение авторы обнаружили, что при использовании зондов на нанометровом масштабе точные значения могут быть получены только для узкого диапазона вдавливаний, достаточно больших для обеспечения эластичности и достаточно малых, чтобы избежать пластической деформации. Несмотря на то, что в данной работе моделирование не было сопоставлено с экспериментом, оно дает полезную информацию для оптимизации условий АСМ исследования в полуконтактном режиме.
Стоит отметить, что в качестве зондового метода исследования упругих характеристик материалов разными авторами активно применяются не только традиционно популярные АСМ подходы: 1) упругая индентация поверхности острым зондом [147, 155] (по моделям механики контактного взаимодействия, например, Герца, Дерягина–Мюллера–Топорова, Джонсона–Кендала–Робертса и т.д.); 2) неупругая индентация объекта острым зондом с протыканием/ломкой поверхности [150]; 3) линейное растяжение молекулы/структуры с напряжением вдоль цепочки углеродного остова [151, 156]; 4) линейное растяжение молекулы/структуры с разрывом боковых нековалентных связей в скрученной глобуле [146]; 5) отрыв молекул АСМ-зондом от гладкой подложки, к которой она прикреплена π–π-связями [156], но и продвинутые активные режимы экспериментов: 6) растяжение структуры при латеральном соскальзывании зонда в поперечном направлении относительно движения зонда [157]. Отметим также, что вместо острого зонда авторами также применяются специальные сферы микрометрового диаметра для прогиба поверхности [155, 157] или модифицированные/декорированные зонды [119]. Перечисленные выше зондовые эксперименты предоставляют возможность изучения различных упругих характеристик макромолекул и в будущем также могут быть успешно использованы совместно с методами компьютерного моделирования.
Отдельной областью исследований, в которой активно применяется сочетание АСМ и МД, является нанолитография. В работе [158] продемонстрировано, что моделирование методом МД является мощным инструментом для исследования процесса наномеханической обработки, который не может быть полностью охарактеризован экспериментально. МД позволила наглядно объяснить микромасштабные механизмы резки на атомистическом масштабе. Параметрические исследования, проведенные с помощью атомистической модели и АСМ, предоставили большой набор данных, на основе которых авторы разработали руководство для нанолитографии методом АСМ с целью получения наножидкостных устройств для медицинских приложений [159]. Однако исследования в этой области посвящены преимущественно обработке неорганических материалов (см., также работы [160, 161]), что выходит за рамки настоящего обзора.
В литературе встречаются упоминания о значительном влиянии окружающей среды на результаты как моделирования, так и АСМ. Проведение исследований в вакууме [162], воде [163], полярных и неполярных жидкостях и растворах (хлороформе, толуоле и т.д. [119]) иногда дает разные результаты в обсуждаемых подходах. Такое расхождение требует проводить коррекцию параметров компьютерных моделей в каждом конкретном случае для достижения сходных результатов в атомистическом моделировании и АСМ-эксперименте. Стоит также заметить, что в некоторых работах авторами были рассмотрены разные среды в АСМ-эксперименте и в компьютерной модели, что затрудняет возможность не только оценки эффекта одновременного использования двух подходов исследования, но и собственно сравнения результатов [163]. Интересно, что в некоторых изученных комбинациях исследуемая молекула–окружение молекулы среды приводили к ускоренному разрушению структур при воздействии зондом [162, 163]. Вместе с тем, часть авторов отмечала, что ими не замечено влияния среды на прочность исследуемых структур [119]. Говоря про прочие методические сложности в каждом из методов и трудности их сравнения/сопряжения, ряд авторов отмечает, что применяемые для моделирования свойств полимеров программы не всегда адекватно отражают свойства материалов при низких температурах [146, 151], а также то, что АСМ-эксперимент ограничен узким диапазоном прикладываемых сил нажима в индентационном эксперименте, в то время как моделирование свободнее в выборе диапазона прикладываемых сил [146, 162, 163].
МОЛЕКУЛЯРНЫЕ ВЗАИМОДЕЙСТВИЯ
Ряд других важных исследований, сочетающих методы АСМ и МД, посвящен оценке молекулярных взаимодействий, поскольку такая комбинация экспериментальных и теоретических подходов может позволить однозначно описать детали этих взаимодействий, а также получить представление об их механизмах и роли в самых разных полимерных системах. Ниже мы приведем характерные примеры таких исследований.
С помощью АСМ и МД наблюдали за комплексами белков Mre11 и Rad50 при их взаимодействии с ДНК-субстратами [164]. Методом АСМ были обнаружены временные взаимодействия между субстратами ДНК и спиральными участками белка Rad50, а также манипуляция и транслокация субстратов ДНК рассматриваемыми белковыми комплексами. Аналогичный режим взаимодействия Rad50 с ДНК наблюдался в моделировании методом МД, что подтвердило экспериментальные результаты. С учетом экспериментальных и теоретических данных была предложена модель воздействия белковых комплексов на ДНК. P. Soto с соавторами [165] использовали АСМ и МД для изучения взаимодействия рецепторов P2X4 и 5-HT3A. Эксперимент показал образование комплексов P2X4/5-HT3A со стехиометрией 1 : 1 в системах с и без адено-зинтрифосфата. Компьютерное моделирование, проведенное после молекулярного докинга рецепторов, установило, что общая энергия взаимодействия рецептора 5-HT3A с рецептором P2X4 в рассмотренных конфигурациях практически одинаковая. Также было обнаружено, что взаимодействие рецептора 5-HT3A с P2X4 может снизить сродство молекул аденозинтрифосфата к рецептору P2X4. Комбинация методов АСМ и МД позволила сделать вывод о том, что стехиометрическая ассоциация, а не накопление или неспецифическая кластеризация, ответственна за аллостерическую регуляцию комплексов P2X4/5-HT3A. В свою очередь Z. Yang с соавторами [166] изучили взаимодействие между амилоидными фибриллами пептида Aβ и графеном, сочетая МД-моделирование с АСМ-экспериментом. Моделирование позволило выявить возможность проникновения графена в амилоидные фибриллы и их последующего разрушения, что объяснялось наличием π–π-стэкинга между поверхностью наночастиц и ароматическими остатками пептида Aβ. Затем с помощью АСМ было установлено, что фибриллы были разрезаны на короткие кусочки после добавления наночастиц оксида графена, что подтверждает эффективность нанолистов на основе графена как ингибитора фибрилляции пептида Aβ. Исследования [164–166] продемонстрировали, что эксперименты с помощью АСМ и моделирование методом МД могут успешно использоваться, чтобы выявить общие аспекты взаимодействий в различных биологических структурах.
Еще одной важной задачей, которую можно решить в рамках комбинирования АСМ и МД, является анализ адгезионных свойств полимерных материалов. В частности, эти методы были применены для изучения устойчивости мембран из полимерных смесей на основе полисульфона и поливинилпирролидона к загрязнению природными органическими веществами [167]. Эксперименты методом АСМ показали уменьшение силы адсорбции с увеличением массовой доли поливинилпирролидона в системе и позволили предположить механизм засорения рассматриваемых мембран. Основываясь на моделировании плотности и межфазных сил с помощью МД, авторы проверили предложенный механизм на молекулярном уровне и объяснили обнаруженные улучшения противообрастающих характеристик. Таким образом, экспериментально и теоретически было показано, что добавление поливинилпирролидона может играть важную роль в предотвращении загрязнения мембраны, вызванного природными органическими веществами.
X. Zhang с соавторами [168] провели измерение силы адгезии между кремний-связывающим пептидом SB7 и поверхностью стекла на уровне отдельной молекулы с использованием АСМ и МД. Методом АСМ было показано, что пептид SB7 имеет значительно более высокую силу адгезии к поверхности, чем другие адгезивные пептиды. МД-моделирование позволило установить, что адгезию пептидов регулируют электростатические взаимодействия между положительно заряженными остатками аргинина и отрицательно заряженной поверхностью. Таким образом, АСМ и МД-моделирование продемонстрировали, что пептид SB7 является многообещающей связывающейся меткой для иммобилизации белков на поверхности диоксида кремния.
Комбинация МД и АСМ была также задействована для оценки работы сил адгезии между париленом C и нержавеющей сталью 316L в системах с силансодержащими молекулами и без них [169]. Методы МД и АСМ привели к качественно похожим данным для работы сил адгезии в зависимости от наличия силансодержащих молекул, хотя из-за упрощений в рассмотренных моделях абсолютные значения работы в моделировании были намного меньше экспериментальных. В частности, было показано, что добавление силансодержащих молекул может немного снизить работу сил адгезии. С помощью моделирования методом МД было также установлено, что адгезия между исследуемыми материалами определяется ван-дер-ваальсовыми взаимодействиями. Таким образом, было обнаружено, что силансодержащие молекулы могут служить для регулирования адгезионных свойств стентов с лекарственным покрытием.
В работе [170] методами АСМ и МД проведена оценка адгезионных свойств акриловых полимеров, чувствительных к давлению, по отношению к электродам на основе диоксида кремния. С одной стороны, измерения адгезионных сил между полимерами и поверхностью электрода с помощью АСМ показали, что увеличение доли 2-этилгексилакрилата в процессе сополимеризации может улучшить характеристики адгезии акриловых полимеров. С другой стороны, моделирование методом МД позволило авторам подтвердить этот результат, а также дать его объяснение. А именно, было сделано предположение, что более высокая адгезия может быть связана с наличием водородных связей между полимером и поверхностью в системе с 2-этилгексилакрилатом.
В работе [171] проведено сравнение значений свободной энергии адсорбции полипептида на поверхность аморфного SiO2, измеренных различными экспериментальными методами и методами компьютерного моделирования, и описаны возможные проблемы, связанные с использованием каждого из методов. В результате сделан вывод о том, что наиболее точным методом измерения свободной энергии является компьютерное моделирование в рамках комплексной методики на основе метода обмена реплик и метадинамики [172]. Однако данный подход крайне требователен к вычислительным ресурсам, и результаты моделирования сильно зависят от выбранного силового поля. Важно отметить, что экспериментальные методы исследования также имеют свои ограничения. Так, для АСМ это – невозможность точного определения положения присоединения кантилевера к адсорбированной молекуле, что приводит к чрезвычайно широкому распределению получаемых расстояний, на которых происходит отрыв молекулы от поверхности. Совместное использование АСМ и МД в некоторых случаях дает возможность обойти ограничения, присущие данным методам по отдельности. В работе [173] оно позволило напрямую измерить энергию гидрофобных взаимодействий отдельной молекулы пептида с модельной поверхностью. В то время как результаты АСМ дополнили и подтвердили результаты моделирования, с помощью моделирования был детально описан процесс растяжения макромолекулы, и отрыв ее от поверхности в процессе проведения эксперимента. Таким образом, методы АСМ и МД не только подтверждали друг друга, но и помогали выявить основные причины наблюдаемых изменений адгезионных свойств различных полимерных систем.
Следует также рассмотреть исследования растяжения полимеров, выполненные методами АСМ и МД. В частности, R.B. Best с соавторами [174] проследили за разворачиванием химерных белковых конструкций на основе барназы и титина I27 с помощью АСМ-экспериментов и неравновесного МД-моделирования. Было обнаружено, что наличие барназы может уменьшить высоту и количество пиков на АСМ-зависимости силы от расстояния по сравнению с высотой и количеством пиков на кривой для октамера титина I27. Моделирование растяжения барназы методом МД показало, что ее разворачивание сопровождается наличием пиков с небольшой величиной силы на кривых растяжения, в то время как концы барназы являются начальной точкой для его разворачивания. Другими словами, было обнаружено, что белок барназа, вероятно, снижает устойчивость структуры к действию приложенной силы.
Методами АСМ и неравновесной МД был выполнен также анализ разворачивания и сворачивания белка с анкириновыми повторами NI6C [175]. Эксперимент и моделирование привели к практически одинаковым кривым растяжения. В результате исследований был предложен путь векторного сворачивания повторяющихся белков. Кроме того, Y. Song с соавторами [176] объединили методы МД и АСМ для изучения процесса сворачивания цепи полиэтиленоксида в монокристалл, полученный из разбавленного раствора или из расплава. Неравновесное моделирование методом МД показало, что кривые растяжения различаются для кристаллов ПЭО с регулярными и нерегулярными складками. Это различие может отражать разницу во внутримолекулярных силах, действующих при растяжении кристаллов. Эксперимент методом АСМ также выявил различия в кривых растяжения, наблюдаемые в моделировании. Был сделан вывод о том, что способ сворачивания цепей ПЭО может зависеть от подхода к получению их кристаллов. Таким образом, отслеживание молекулярных взаимодействий и конформаций с помощью комбинации методов АСМ и МД позволило значительно улучшить понимание процесса упорядочения полимеров [174–176].
Эксперименты с использованием АСМ ранее комбинировались либо с равновесным [164–170, 177–180], либо с неравновесным [166, 174–176, 181–185] моделированием методом МД. При этом было показано, что неравновесная МД с помощью метода зонтичной выборки позволяет рассчитывать профили свободной энергии в зависимости от расстояния между взаимодействующими молекулярными объектами и, таким образом, дополнять АСМ-изображения описанием термодинамических аспектов молекулярных взаимодействий [166, 182–185]. Еще более редко используемый подход к расширению выборки в МД-моделировании – метадинамика также оказалась чрезвычайно полезной для дополнения АСМ-экспериментов данными о ландшафте поверхности свободной энергии [186]. В свою очередь экспериментальные работы [187–192] показали, что свободная энергия и ее профиль могут быть определены методом АСМ. Таким образом, сравнительный анализ данных о свободной энергии, полученных с помощью АСМ и МД, может существенно обогатить знания о молекулярных взаимодействиях в сложных полимерных системах.
В заключение отметим, что совместный экспериментальный и теоретический подход, сочетающий измерения методом АСМ и моделирование методом МД, может служить ценным инструментом для улучшения понимания молекулярных взаимодействий в большом количестве природных и синтетических макромолекулярных систем. Данный подход позволяет подробно описать взаимодействия, оценить адгезионные свойства и проследить реакцию полимеров на внешние воздействия. Характеристики, измеряемые методами АСМ и МД, не всегда совпадают, что затрудняет их прямое сравнение. Тем не менее, АСМ и МД можно успешно сочетать, чтобы они не только подтверждали, но и дополняли друг друга. Несомненно, это может привести к новым интересным результатам. Более того, существуют возможности для дальнейших разработок, направленных на прямое сравнение результатов рассматриваемых методов. Например, может быть перспективным объединение АСМ и МД для анализа термодинамики взаимодействий на основе данных о свободной энергии.
ЗАКЛЮЧЕНИЕ
Комбинация экспериментальных измерений методами АСМ и компьютерного моделирования методом МД успешно применяется для всестороннего изучения морфологии, механических свойств и особенностей взаимодействий, присущих самым разнообразным природным и синтетическим полимерным системам. АСМ и МД могут быть использованы как в рамках прямого сравнения измеряемых характеристик, так и для подтверждения, интерпретации и дополнения результатов, полученных экспериментально и в моделировании. Совместное применение указанных методов, с одной стороны, дает неоспоримые преимущества в исследовании полимерных систем, но, с другой стороны, сопровождается рядом затруднений. В первую очередь это связано с различными пространственными и временными масштабами, доступными для обсуждаемых методов.
Разрешение при построении топографических карт в атомистическом моделировании и эксперименте могут различаться на порядок, что может приводить к расхождению результатов, полученных данными методами, или их различной интерпретации. С другой стороны, разные масштабы делают привлекательным использование комплексного экспериментально-теоретического подхода для многомасштабного исследования структуры и свойств материала. Важно отметить, что чувствительность смещения зонда перпендикулярно поверхности образца позволяет детектировать разницу в высотах на поверхности материала в диапазоне единиц и долей нанометров, что является доступным для исследования методом атомистического компьютерного моделирования. Огрубление теоретических моделей, рост вычислительных возможностей и развитие методов АСМ позволяют получить информацию о структуре материала на сопоставимом пространственном масштабе, что, несомненно, делает еще более перспективным совмещение указанных методик.
Различие между экспериментом и моделированием в доступных временных масштабах значительно больше и составляет примерно шесть–восемь порядков, что затрудняет сравнение наномеханических параметров материала, полученных данными методами. Относительно короткие времена моделирования также приводят к проблемам в исследовании свойств материала при низких температурах, при которых подвижность полимеров заторможена. Несмотря на это обстоятельство, моделирование способно дать прямую информацию о деформации материала под нагрузкой и помочь установить молекулярные механизмы, ответственные за его механический отклик при исследовании методом АСМ. Другое обстоятельство, препятствующее прямому сравнению данных, полученных в эксперименте и в моделировании, – упрощенная модель исследуемой системы в компьютерном моделировании и проблемы, связанные с параметризацией взаимодействий ее компонентов. Важно отметить, что корректный выбор параметров при атомистическом компьютерном моделировании является первостепенной и не всегда тривиальной задачей. Данные АСМ в рамках комплексного экспериментально-теоретического подхода могут служить основой для получения корректной модели и, как следствие, максимально достоверных результатов в моделировании.
Кроме того, в ряде случаев результаты АСМ сложны в интерпретации. Это может быть связано не только с неполным знанием структуры и состава исследуемого материала, но и со специфическими взаимодействиями поверхности материала с зондом, которые крайне сложно учесть в рамках эксперимента. Компьютерное моделирование может дать прямую информацию о характере данного взаимодействия и о структуре нанообъектов, детектируемых в АСМ, что служит ключом к интерпретации результатов, полученных в ходе эксперимента.
Таким образом, совместное использование обсуждаемых в обзоре методов и комплексный анализ результатов, полученных с их помощью, дают гораздо больше информации, чем каждый метод по отдельности. Такой подход позволяет объединить положительные стороны методов и компенсировать их недостатки. Однако с учетом описанных выше ограничений рассматриваемый экспериментально-теоретический подход должен применяться с полным пониманием особенностей входящих в его состав методов и исследуемых систем, а также с обязательным подтверждением как экспериментальных данных, так и результатов моделирования.
Дальнейшее развитие каждого метода по отдельности, увеличение точности и разрешения методов в будущем должны еще больше стимулировать их совместное использование. Более того, перспективы экспериментально-теоретического подхода еще полностью не реализованы. Так, объединение современных методов моделирования, предоставляющих информацию о свободной энергии системы, с АСМ позволит достичь более глубокого понимания молекулярных взаимодействий в полимерных системах, адгезионных свойств таких систем и их реакции на внешние механические воздействия.
Работа выполнена при финансовой поддержке Министерства науки и высшего образования Российской Федерации (договор № 14.W03.31.0014).
Список литературы
Lang S.Y., Shi Y., Hu X.C., Yan H.J., Wen R., Wan L.J. // Curr. Opin. Electrochem. 2019. V. 17. P. 134.
Inukai J. // Curr. Opin. Electrochem. 2020. V. 21. P. 345.
Marinello F., La Storia A., Mauriello G., Passeri D. // Trends Food Sci. Technol. 2019. V. 87. P. 84.
Smirnov M.A., Sokolova M.P., Geydt P., Smirnov N.N., Bobrova N.V., Toikka A.M., Lahderanta E. // Mater. Lett. 2017. V. 199. P. 192.
Sokolova M.P., Smirnov M.A., Samarov A.A., Bobrova N.V., Vorobiov V.K., Popova E.N., Filippova E., Geydt P., Lahderanta E., Toikka A.M. // Carbohydr. Polym. 2018. V. 197. P. 548.
Sokolova M.P., Smirnov M.A., Geydt P., Bugrov A.N., Ovaska S.S., Lahderanta E., Toikka A.M. // Polymers. 2016. V. 8. № 11. P. 403.
Sokolova M.P., Smirnov M.A., Bugrov A.N., Geydt P., Popova E.N., Lahderanta E., Svetlichnyi V.M., Toikka A.M. // Polymers. 2017. V. 9. № 7. P. 268.
Sokolova M.P., Bugrov A.N., Smirnov M.A., Smirnov A.V., Lahderanta E., Svetlichnyi V.M., Toikka A.M. // Polymers. 2018. V. 10. № 11. P. 1222.
Smirnov M.A., Nikolaeva A.L., Bobrova N.V., Vorobiov V.K., Smirnov A.V., Lahderanta E., Sokolova M.P. // Polym. Test. 2021. V. 97. P. 107156.
He C., Shi S., Wu X., Russell T.P., Wang D. // J. Am. Chem. Soc. 2018. V. 140. № 22. P. 6793.
Skibinski J., Frydrych J., Rebis J., Rozniatowski K. // J. Power Technol. 2017. V. 97. № 2. P. 103.
Li H.X., Russell T.P., Wang D. // Chinese J. Polym. Sci. 2021. V. 39. P. 651.
Wu X., Shi S., Yu Z., Russell T.P., Wang D. // Polymer. 2018. V. 146. P. 188.
Kiio T.M., Park S. // Int. J. Med. Sci. 2020. V. 17. № 7. P. 844.
Babu P.K.V., Radmacher M. // Front. Neurosci. 2019. V. 13. P. 600.
Zemła J., Danilkiewicz J., Orzechowska B., Pabijan J., Seweryn S., Lekka M. // Semin. Cell Dev. Biol. 2018. V. 73. P. 115.
Korneev D.V., Popova A.V., Generalov V.M., Zaitsev B.N. // Biophys. 2016. V. 61. № 3. P. 413.
Habibullah H. // Meas. J. Int. Meas. Confed. 2020. V. 159. P. 107776.
Суслов А.А., Чижик С.А. // Материалы, технологии, инструменты. 1997. V. 3. P. 78.
Krieg M., Fläschner G., Alsteens D., Gaub B.M., Roos W.H., Wuite G.J.L., Gaub H.E., Gerber C., Dufrêne Y.F., Müller D.J. // Nat. Rev. Phys. 2019. V. 1. № 1. P. 41.
Alsteens D., Gaub H.E., Newton R., Pfreundschuh M., Gerber C., Müller D.J. // Nat. Rev. Mater. 2017. V. 2. № 5.
Patel A.N., Kranz C. // Annu. Rev. Anal. Chem. 2018. V. 11. P. 329.
Liu Y., Vancso G.J. // Prog. Polym. Sci. 2020. V. 104. P. 101232.
Kumaki J. // Polym. J. 2016. V. 48. P. 3.
Xu H., Shirvanyants D., Beers K., Matyjaszewski K., Rubinstein M., Sheiko S.S. // Phys. Rev. Lett. 2004. V. 93. № 20. P. 1.
Hobbs J.K., Farrance O.E., Kailas L. // Polymer. 2009. V. 50. P. 4281.
Magonov S.N., Reneker D.H. // Annu. Rev. Mater. Sci. 1997. V. 27. № 1. P. 175.
Bao Y., Luo Z., Cui S. // Chem. Soc. Rev. 2020. V. 49. № 9. P. 2799.
Efremov Y.M., Okajima T., Raman A. // Soft Matter. 2019. V. 16. № 1. P. 64.
Miranda A., Gómez-Varela A.I., Stylianou A., Hirvonen L.M., Sánchez H., De Beule P.A.A. // Nanoscale. 2021. V. 13. № 4. P. 2082.
Lherbette M., Dos Santos Á., Hari-Gupta Y., Fili N., Toseland C.P., Schaap I.A.T. // Sci. Rep. 2017. V. 7. P. 8116.
Nguyen-Tri P., Ghassemi P., Carriere P., Nanda S., Assadi A.A., Nguyen D.D. // Polymers. 2020. V. 12. P. 1142.
Hollingsworth S.A., Dror R.O. // Neuron. 2018. V. 99. P. 1129.
Allen M.P., Tildesley D.J. // Comput. Simul. Liq. Oxford Scholarship Online. 2017.
Lyulin S.V., Larin S.V., Nazarychev V.M., Fal’kovich S.G., Kenny J.M. // Polymer Science C. 2016. V. 58. № 1. P. 2.
Volgin I.V., Larin S.V., Lyulin S.V. // Polymer Science C. 2018. V. 60. № 2. P. 122.
Marrink S.J., Corradi V., Souza P.C.T., Ingólfsson H.I., Tieleman D.P., Sansom M.S.P. // Chem. Rev. 2019. V. 119. № 9. P. 6184.
Perilla J.R., Goh B.C., Cassidy C.K., Liu B., Bernardi R.C., Rudack T., Yu H., Wu Z., Schulten K. // Curr. Opin. Struct. Biol. 2015. V. 31. P. 64.
Enkavi G., Javanainen M., Kulig W., Róg T., Vattulainen I. // Chem. Rev. 2019. V. 119. № 9. P. 5607.
Ilie I.M., Caflisch A. // Chem. Rev. 2019. V. 119. № 12. P. 6956.
Gartner T.E., Jayaraman A. // Macromolecules. 2019. V. 52. № 3. P. 755.
Cisneros G.A., Karttunen M., Ren P., Sagui C. // Chem. Rev. 2014. V. 114. № 1. P. 779.
Corradi V., Sejdiu B.I., Mesa-Galloso H., Abdizadeh H., Noskov S.Y., Marrink S.J., Tieleman D.P. // Chem. Rev. 2019. V. 119. № 9. P. 5775.
Nazarychev V.M., Larin S.V., Lukasheva N.V., Glova A.D., Lyulin S.V. // Polymer Science A 2013. V. 55. № 9. P. 570.
Falkovich S.G., Larin S.V., Lukasheva N.V., Nazarychev V.M., Tolmachev D.A., Glova A.D., Mezhenskaia D.A., Kenny J.M., Lyulin S.V. // Multifunct. Polym. Nanocomposites Based Cellul. Reinf. / Ed. by D. Pugla, E. Fortunati, J.M. Kenny. Amsterdam: Elsevier, 2016. P. 313.
Lukasheva N.V., Tolmachev D.A., Nazarychev V.M., Kenny J.M., Lyulin S.V. // Soft Matter. 2017. V. 13. № 2. P. 474.
Lukasheva N.V., Tolmachev D.A., Karttunen M. // Phys. Chem. Chem. Phys. 2019. V. 21. № 3. P. 1067.
Tolmachev D.A., Lukasheva N.V. // Polymer Science A. 2014. V. 56. № 4. P. 545.
Tolmachev D., Lukasheva N., Mamistvalov G., Karttunen M. // Polymers. 2020. V. 12. P. 1279.
Tolmachev D.A., Lukasheva N.V. // Langmuir. 2012. V. 28. № 37. P. 13473.
Falkovich S.G., Martinez-Seara H., Nesterenko A.M., Vattulainen I., Gurtovenko A.A. // J. Phys. Chem. Lett. 2016. V. 7. № 22. P. 4585.
Gurtovenko A.A., Karttunen M. // Langmuir. 2019. V. 35. № 42. P. 13753.
Gurtovenko A.A., Javanainen M., Lolicato F., Vattulainen I. // J. Phys. Chem. Lett. 2019. V. 10. № 5. P. 1005.
Kostritskii A.Y., Tolmachev D.A., Lukasheva N.V., Gurtovenko A.A. // Langmuir. 2017. V. 33. № 44. P. 12793.
Gurtovenko A.A., Mukhamadiarov E.I., Kostritskii A.Y., Karttunen M. // J. Phys. Chem. B 2018. V. 122. № 43. P. 9973.
Karttunen M., Rottler J., Vattulainen I., Sagui C. // Curr. Top. Membr. 2008. V. 60. P. 49.
Duboué-Dijon E., Javanainen M., Delcroix P., Jungwirth P., Martinez-Seara H. // J. Chem. Phys. 2020. V. 153. № 5. P. 50901.
Duboué-Dijon E., Delcroix P., Martinez-Seara H., Hladílková J., Coufal P., Křížek T., Jungwirth P. // J. Phys. Chem. B 2018. V. 122. № 21. P. 5640.
Wong-ekkabut J., Karttunen M. // Biochim. Biophys. Acta – Biomembr. 2016. V. 1858. № 10. P. 2529.
Lyubartsev A.P., Rabinovich A.L. // Soft Matter. 2011. V. 7. № 1. P. 25.
Rabinovich A.L., Lyubartsev A.P. // Polymer Science C. 2013. V. 55. № 1. P. 162.
Baljon A.R.C., Williams S., Balabaev N.K., Paans F., Hudzinskyy D., Lyulin A.V. // J. Polym. Sci., Polym. Phys. 2010. V. 48. № 11. P. 1160.
Mazo M.A., Manevitch L.I., Gusarova E.B., Shamaev M.Y., Berlin A.A., Balabaev N.K., Rutledge G.C. // J. Phys. Chem. B. 2008. V. 112. № 10. P. 2964.
Mazo M.A., Manevitch L.I., Gusarova E.B., Berlin A.A., Balabaev N.K., Rutledge G.C. // J. Phys. Chem. C. 2008. V. 112. № 44. P. 17056.
Mazo M.A., Shamaev M.Y., Balabaev N.K., Darinskii A.A., Neelov I.M. // Phys. Chem. Chem. Phys. 2004. P. 1285.
Glyakina A.V., Balabaev N.K., Galzitskaya O.V. // J. Chem. Phys. 2009. V. 131. № 4. P. 45102.
Zubova E.A., Balabaev N.K., Musienko A.I., Gusarova E.B., Mazo M.A., Manevitch L.I., Berlin A.A. // J. Chem. Phys. 2012. V. 136. № 22. P. 224906.
Balabaev N.K., Mazo M.A., Kramarenko E.Y. // Macromolecules. 2017. V. 50. № 1. P. 432.
Mazo M., Balabaev N., Alentiev A., Yampolskii Y. // Macromolecules. 2018. V. 51. № 4. P. 1398.
Lyulin A.V., Balabaev N.K., Baljon A.R.C., Mendoza G., Frank C.W., Yoon D.Y. // J. Chem. Phys. 2017. V. 146. № 20. P. 203314.
Balabaev N.K., Mazo M.A., Lyulin A.V., Oleinik E.F. // Polymer Science A. 2010. V. 52. № 6. P. 633.
Markelov D.A., Mazo M.A., Balabaev N.K., Gotlib Y.Y. // Polymer Science A. 2013. V. 55. № 1. P. 53.
Kondratyuk N.D., Pisarev V.V. // Fluid Phase Equilib. 2019. V. 498. P. 151.
Kondratyuk N.D., Norman G.E., Stegailov V.V. // Polymer Science A. 2016. V. 58. № 5. P. 825.
Guseva D.V., Komarov P.V., Lyulin A.V. // J. Chem. Phys. 2014. V. 140. № 11. P. 114903.
Komarov P., Chiu Y.T., Chen S.M., Reineker P. // Macromol. Theory Simul. 2010. V. 19. № 1. P. 64.
Glova A.D., Melnikova S.D., Mercurieva A.A., Larin S.V., Lyulin S.V. // Polymers. 2019. V. 11. P. 2056.
Birshtein T.M., Polotsky A.A., Glova A.D., Amoskov V.M., Mercurieva A.A., Nazarychev V.M., Lyulin S.V. // Polymer. 2018. V. 147. P. 213.
Glova A.D., Falkovich S.G., Larin S.V., Mezhenskaia D.A., Lukasheva N.V., Nazarychev V.M., Tolmachev D.A., Mercurieva A.A., Kenny J.M., Lyulin S.V. // Polym. Int. 2016. V. 65. № 8. P. 892.
Glova A.D., Larin S.V., Falkovich S.G., Nazarychev V.M., Tolmachev D.A., Lukasheva N.V., Lyulin S.V. // Soft Matter. 2017. V. 13. № 37. P. 6627.
Glova A.D., Larin S.V., Nazarychev V.M., Karttunen M., Lyulin S.V. // Macromolecules. 2020. V. 53. P. 29.
Glova A.D., Falkovich S.G., Dmitrienko D.I., Lyulin A.V., Larin S.V., Nazarychev V.M., Karttunen M., Lyulin S.V. // Macromolecules. 2018. V. 51. P. 552.
Sharma P., Roy S., Karimi-Varzaneh H.A. // J. Phys. Chem. B. 2016. V. 120. № 7. P. 1367.
Glova A.D., Melnikova S.D., Mercurieva A.A., Larin S.V., Nazarychev V.M., Polotsky A.A., Lyulin S.V. // Phys. Chem. Chem. Phys. 2021. V. 23. P. 457.
Lyulin S.V., Gurtovenko A.A., Larin S.V., Nazarychev V.M., Lyulin A.V. // Macromolecules. 2013. V. 46. № 15. P. 6357.
Tolmachev D.A., Boyko O.S., Lukasheva N.V., Martinez-Seara H., Karttunen M. // J. Chem. Theory Comput. 2020. V. 16. P. 677.
Shaytan A.K., Armeev G.A., Goncearenco A., Zhurkin V.B., Landsman D., Panchenko A.R. // J. Mol. Biol. 2016. V. 428. № 1. P. 221.
Meli M., Colombo G. // Int. J. Mol. Sci. 2013. V. 14. P. 12157.
Sugita Y., Kamiya M., Oshima H., Re S. // Methods Mol. Biol. 2019. V. 2022. P. 155.
Barducci A., Bonomi M., Parrinello M. // Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011. V. 1. P. 826.
Abrams C., Bussi G. // Entropy. 2014. V. 16. P. 163.
Do T.N., Choy W.Y., Karttunen M. // J. Chem. Theory Comput. 2016. V. 12. P. 395.
Piana S., Klepeis J.L., Shaw D.E. // Curr. Opin. Struct. Biol. 2014. V. 24. P. 98.
Bussi G., Laio A. // Nat. Rev. Phys. 2020. V. 2. P. 200.
Kahlen J., Peter C., Donadio D. // Cryst. Eng. Comm. 2015. V. 17. № 36. P. 6863.
Yang J., Calero C., Bonomi M., Martí J. // J. Chem. Theory Comput. 2015. V. 11. P. 4495.
Glova A.D., Volgin I.V., Nazarychev V.M., Larin S.V., Lyulin S.V., Gurtovenko A.A. // RSC Adv. 2019. V. 9. № 66. P. 38834.
Nazarychev V.M., Glova A.D., Volgin I.V., Larin S.V., Lyulin A.V., Lyulin S.V., Gurtovenko A.A. // Int. J. Heat Mass Transf. 2021. V. 165. P. 120639.
Lyubartsev A.P., Rabinovich A.L. // Biochim. Biophys. Acta Biomembr. 2016. V. 1858. № 10. P. 2483.
Kondratyuk N.D., Lankin A.V., Norman G.E., Stegailov V.V. // J. Phys. Conf. Ser. 2015. V. 653. P. 012107.
Kondratyuk N.D., Norman G.E., Stegailov V.V. // J. Chem. Phys. 2016. V. 145. № 20. P. 204504.
Kondratyuk N.D. // J. Phys. Conf. Ser. 2019. V. 1385. P. 012048.
Dong Y., Li Q., Martini A. // J. Vac. Sci. Technol. A Vacuum, Surfaces, Film. 2013. V. 31. P. 030801.
Isralewitz B., Gao M., Schulten K. // Curr. Opin. Struct. Biol. 2001. V. 11. № 2. P. 224.
Pleshakova T.O., Bukharina N.S., Archakov A.I., Ivanov Y.D. // Int. J. Mol. Sci. 2018. V. 19. № 4.
Parot P., Dufrêne Y.F., Hinterdorfer P., Le Grimellec C., Navajas D., Pellequer J.L., Scheuring S. // J. Mol. Recognit. 2007. V. 20. № 6. P. 418.
An Y., Manuguri S.S., Malmström. J. // Methods Mol. Biol. 2020. V. 2073. P. 247.
Häberle W. // J. Vac. Sci. Technol. B Microelectron. Nanom. Struct. 1991. V. 9. № 2. P. 1210.
Young T.J., Monclus M.A., Burnett T.L., Broughton W.R., Ogin S.L., Smith P.A. // Meas. Sci. Technol. 2011. V. 22. № 12.
Friesner R.A. // Proc. Natl. Acad. Sci. U. S. A. 2005. V. 102. № 19. P. 6648.
Ruipérez F. // Int. Rev. Phys. Chem. 2019. V. 38. № 3–4. P. 343.
Vanommeslaeghe K., MacKerell A.D. // Biochim. Biophys. Acta–Gen. Subj. 2015. V. 1850. № 5. P. 861.
Senn H.M., Thiel W. // Angew. Chemie Int. Ed. 2009. V. 48. № 7. P. 1198.
Lemke T., Peter C., Kukharenko O. // J. Chem. Theory Comput. 2018. V. 14. № 11. P. 5476.
Laio A., Parrinello M. // Proc. Natl. Acad. Sci. U. S. A. 2002. V. 99. № 20. P. 12562.
Torrie G.M., Valleau J.P. // J. Comput. Phys. 1977. V. 23. № 2. P. 187.
Izrailev S., Stepaniants S., Isralewitz B., Kosztin D., Lu H., Molnar F., Wriggers W., Schulten K. Computational Molecular Dynamics: Challenges, Methods, Ideas / Ed. by Deuflhard P., Hermans J., Leimkuhler B., Mark A.E., Reich S., Skeel R.D. Berlin: Springer-Verlag, 1998. P. 39.
Dou Z., Qian J., Li Y., Wang Z., Zhang Y., Lin R., Wang T. // Ultramicroscopy. 2020. V. 212.
Guzmán D.L., Roland J.T., Keer H., Kong Y.P., Ritz T., Yee A., Guan Z. // Polymer. 2008. V. 49. № 18. P. 3892.
Mücksch C., Urbassek H.M. // J. Chem. Theory Comput. 2016. V. 12. № 3. P. 1380.
Ozer G., Valecv E.F., Quirt S., Hernandez R. // J. Chem. Theory Comput. 2010. V. 6. № 10. P. 3026.
Noh S.Y., Notman R. // J. Chem. Phys. 2020. V. 153. P. 034115.
Schiller U.D., Krüger T., Henrich O. // Soft Matter. 2017. V. 14. № 1. P. 9.
Bahramian A. // Surf. Interface Anal. 2015. V. 47. № 1. P. 1.
Drechsler S., Balog S., Kilbinger A.F.M., Casalini T. // Soft Matter. 2019. V. 15. № 36. P. 7250.
Bizzarri A.R., Bonanni B., Costantini G., Cannistraro S. // ChemPhysChem 2003. V. 4. № 11. P. 1189.
Pieczywek P.M., Kozioł A., Płaziński W., Cybulska J., Zdunek A. // Food Hydrocoll. 2020. V. 104. P. 105726.
Garay A.S., Rodrigues D.E., Fuselli A., Martino D.M., Passeggi M.C.G. // J. Phys. Chem. B 2016. V. 120. № 13. P. 3414.
Jamil T., Gissinger J.R., Garley A., Saikia N., Upadhyay A.K., Heinz H. // Nanoscale. 2019. V. 11. № 23. P. 11183.
Jeong S., Pinals R.L., Dharmadhikari B., Song H., Kalluri A., Debnath D., Wu Q., Ham M.H., Patra P., Landry M.P. // Sci. Rep. 2020. V. 10. № 1. P. 7074
Włoch J., Terzyk A.P., Wiśniewski M., Kowalczyk P. // Langmuir. 2018. V. 34. № 15. P. 4526.
Connelly L., Jang H., Teran Arce F., Ramachandran S., Kagan B.L., Nussinov R., Lal R. // Biochemistry. 2012. V. 51. № 14. P. 3031.
Tian M., Li B., Shen L. // ACS Macro Lett. 2020. V. 9. № 12. P. 1759.
Rajput R., Balasubramani G.L., Srivastava A., Wahi D., Shrivastava N., Kundu B., Grover A. // Biochem. J. 2019. V. 476. № 12. P. 1817.
Bhat I.A., Roy B., Hazra P., Kabir-ud-Din // J. Colloid Interface Sci. 2019. V. 538. P. 489.
Cai B., Li Z., Chen C.L. // Acc. Chem. Res. 2021. V. 54. № 1. P. 81.
Oda Y., Kawaguchi D., Morimitsu Y., Yamamoto S., Tanaka K. // Sci. Rep. 2020. V. 10. P. 20914
Liekkinen J., de Santos Moreno B., Paananen R.O., Vattulainen I., Monticelli L., Bernardino de la Serna J., Javanainen M. // Front. Cell Dev. Biol. 2020. V. 8. P. 581016.
Yu J., Jackson N.E., Xu X., Brettmann B.K., Ruths M., De Pablo J.J., Tirrell M. // Sci. Adv. 2017. V. 3. P. 1497.
Brettmann B., Pincus P., Tirrell M. // Macromolecules. 2017. V. 50. № 3. P. 1225.
Onaran G., Yurtsever M., Yilgor I. // Comput. Mater. Sci. 2019. V. 167. P. 160.
Sevinis Ozbulut E.B., Seven S., Bilge K., Akkas T., Tas C.E., Yildiz B., Atilgan C., Menceloglu Y.Z., Unal S. // Polymer. 2020. V. 188. P. 122114.
Liu Z., Cao L., Zhou T., Dong Z. // Energy and Fuels 2020. V. 34. № 4. P. 4006.
Liang M., Wang Y., Sun S., Yang W. // J. Appl. Polym. Sci. 2020. V. 137. № 16. P. 48595.
Tang H., Zhao Y., Shan S., Yang X., Liu D., Cui F., Xing B. // Environ. Sci. Technol. 2018. V. 52. № 14. P. 7689.
Kolberg A., Wenzel C., Hackenstrass K., Schwarzl R., Rüttiger C., Hugel T., Gallei M., Netz R.R., Balzer B.N. // J. Am. Chem. Soc. 2019. V. 141. № 29. P. 11603.
Sotomayor M., Schulten K. // Science. 2007. V. 316. P. 1144.
Nakajima K., Ito M., Wang D., Liu H., Nguyen H.K., Liang X., Kumagai A., Fujinami S. // Microscopy. 2014. V. 63. № 3. P. 193.
Hosoya R., Morita H., Nakajima K. // Macromolecules. 2020. V. 53. № 15. P. 6163.
Sapra K.T., Qin Z., Dubrovsky-Gaupp A., Aebi U., Müller D.J., Buehler M.J., Medalia O. // Nat. Commun. 2020. V. 11. P. 6205.
Wang W., Kistler K.A., Sadeghipour K., Baran G. // Phys. Lett. Sect. A Gen. At. Solid State Phys. 2008. V. 372. № 47. P. 7007.
Ikeshima D., Miyamoto K., Yonezu A. // Polymer. 2019. V. 173. P. 80.
Onofrio N., Venturini G.N., Strachan A. // J. Appl. Phys. 2013. V. 114. P. 094309.
Kim H., Venturini G., Strachan A. // J. Appl. Phys. 2012. V. 112. P. 094325.
Zhang G., Jiang H., Fan N., Yang L., Guo J., Peng B. // Mod. Phys. Lett. B 2018. V. 32. № 18. P. 1850198.
Pawlak R., Vilhena J.G., D’astolfo P., Liu X., Prampolini G., Meier T., Glatzel T., Lemkul J.A., Häner R., Decurtins S., Baratoff A., Pérez R., Liu S.X., Meyer E. // Nano Lett. 2020. V. 20. P. 652.
Singh M.K., Kang C., Ilg P., Crockett R., Kröger M., Spencer N.D. // Macromolecules. 2018. V. 51. № 24. P. 10174.
Guo X., Li Q., Liu T., Kang R., Jin Z., Guo D. // Front. Mech. Eng. 2017. V. 12. № 1. P. 89.
Promyoo R., El-Mounayri H., Agarwal M. // Conf. Proc. Soc. Exp. Mech. Ser. 2018. V. 5. P. 51.
Promyoo R., El-Mounayri H., Varahramyan K. // Comput. Sci. Inf. Technol. 2014. P. 151.
Fang T.H., Weng C.I., Chang J.G. // Surf. Sci. 2002. V. 501. P. 138.
Cai W., Xu D., Qian L., Wei J., Xiao C., Qian L., Lu Z.Y., Cui S. // J. Am. Chem. Soc. 2019. V. 141. № 24. P. 9500.
Cai W., Lu S., Wei J., Cui S. // Macromolecules. 2019. V. 52. № 19. P. 7324.
Zabolotnaya E., Mela I., Williamson M.J., Bray S.M., Yau S.K., Papatziamou D., Edwardson J.M., Robinson N.P., Henderson R.M. // Proc. Natl. Acad. Sci. 2020. V. 117. № 26. P. 14936.
Soto P., Gaete P.S., Fuentes C., Lozano B., Naulin P.A., Figueroa X.F., Barrera N.P. // Front. Cell. Neurosci. 2020. V. 14.
Yang Z., Ge C., Liu J., Chong Y., Gu Z., Jimenez-Cruz C.A., Chai Z., Zhou R. // Nanoscale 2015. V. 7. № 44. P. 18725.
Miyamoto K., Ikeshima D., Furutani T., Xiao H., Yonezu A., Chen X. // Surf. Topogr. Metrol. Prop. 2019. V. 7. № 3. P. 035003.
Zhang X., Chen J., Li E., Hu C., Luo S.-Z., He C. // Front. Chem. 2020. V. 8. P. 600918
Youssefian S., Rahbar N. // J. Mech. Behav. Biomed. Mater. 2013. V. 18. P. 1.
Pan Y., Ge S., Rashid Z., Gao S., Erwin A., Tsukruk V., Vogiatzis K.D., Sokolov A.P., Yang H., Cao P.-F. // ACS Appl. Energy Mater. 2020. V. 3. № 4. P. 3387.
Meißner R.H., Wei G., Ciacchi L.C. // Soft Matter. 2015. V. 11. № 31. P. 6254.
Camilloni C., Provasi D., Tiana G., Broglia R.A. // Proteins Struct. Funct. Genet. 2008. V. 71. № 4. P. 1647.
Stock P., Monroe J.I., Utzig T., Smith D.J., Shell M.S., Valtiner M. // ACS Nano. 2017. V. 11. № 3. P. 2586.
Best R.B., Li B., Steward A., Daggett V., Clarke J. // Biophys. J. 2001. V. 81. № 4. P. 2344.
Lee W., Zeng X., Zhou H.-X., Bennett V., Yang W., Marszalek P.E. // J. Biol. Chem. 2010. V. 285. № 49. P. 38167.
Song Y., Feng W., Liu K., Yang P., Zhang W., Zhang X. // Langmuir. 2013. V. 29. № 12. P. 3853.
Mosquera-Giraldo L.I., Borca C.H., Parker A.S., Dong Y., Edgar K.J., Beaudoin S.P., Slipchenko L.V., Taylor L.S. // Biomacromolecules. 2018. V. 19. № 12. P. 4593.
Lo K.C., Hau K.I., Chan W.K. // Nanoscale 2018. V. 10. № 14. P. 6474.
Bassalah M.E.A., Cerdà J.J., Sintes T., Aschi A., Othman T. // Eur. Polym. J. 2017. V. 96. P. 55.
Ikai T., Wada Y., Takagi Y., Shinohara K. // Polym. Chem. 2016. V. 7. № 46. P. 7057.
Taylor D.E., Strawhecker K.E., Shanholtz E.R., Sorescu D.C., Sausa R.C. // J. Phys. Chem. A 2014. V. 118. № 27. P. 5083.
Mondal J., Halverson D., Li I.T.S., Stirnemann G., Walker G.C., Berne B.J. // Proc. Natl. Acad. Sci. 2015. V. 112. № 30. P. 9270.
Georgakilas V., Demeslis A., Ntararas E., Kouloumpis A., Dimos K., Gournis D., Kocman M., Otyepka M., Zbořil R. // Adv. Funct. Mater. 2015. V. 25. № 10. P. 1481.
Awasthi N., Kopec W., Wilkosz N., Jamróz D., Hub J.S., Zatorska M., Petka R., Nowakowska M., Kepczyn-ski M. // ACS Biomater. Sci. Eng. 2019. V. 5. № 2. P. 780.
Pérez-Fuentes L., Drummond C., Faraudo J., Bastos-González D. // Soft Matter. 2015. V. 11. № 25. P. 5077.
Casillas-Ituarte N.N., Cruz C.H.B., Lins R.D., DiBartola A.C., Howard J., Liang X., Höök M., Viana I.F.T., Sierra-Hernández M.R., Lower S.K. // J. Biol. Chem. 2017. V. 292. № 21. P. 8797.
Schlierf M., Rief M. // Biophys. J. 2006. V. 90. № 4. P. L33.
Harris N.C., Song Y., Kiang C.-H. // Phys. Rev. Lett. 2007. V. 99. № 6. P. 068101.
Guo S., Lad N., Ray C., Akhremitchev B.B. // Biophys. J. 2009. V. 96. № 8. P. 3412.
Gebhardt J.C.M., Bornschlögl T., Rief M. // Proc. Natl. Acad. Sci. 2010. V. 107. № 5. P. 2013.
Woodside M.T., Block S.M. // Annu. Rev. Biophys. 2014. V. 43. № 1. P. 19.
Drechsler A., Elmahdy M.M., Uhlmann P., Stamm M. // Langmuir. 2018. V. 34. № 16. P. 4739.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)