

Высокомолекулярные соединения (серия С), 2021, T. 63, № 2, стр. 190-211
ПОРИСТЫЕ МАТЕРИАЛЫ НА ОСНОВЕ ПОЛИЛАКТИДА: ПОЛУЧЕНИЕ, ОСОБЕННОСТИ ГИДРОЛИТИЧЕСКОЙ ДЕСТРУКЦИИ И ОБЛАСТИ ПРИМЕНЕНИЯ
Е. С. Трофимчук a, *, В. В. Поцелеев a, М. А. Хавпачев a, М. А. Москвина a, Н. И. Никонорова a
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, 1, стр. 3, Россия
* E-mail: elena_trofimchuk@mail.ru
Поступила в редакцию 16.02.2021
После доработки 19.04.2021
Принята к публикации 25.05.2021
Аннотация
Материалы на основе полилактида часто рассматриваются как альтернатива материалам из традиционных трудно разлагаемых полимеров. Особенно привлекательными представляются пористые мембраны, матриксы и каркасы из полилактида для применения в биомедицине. В обзоре рассмотрены физико-химические основы и структурно-морфологические возможности различных методов получения пористого полилактида: спекание, 3D-печать, электроформование, вспенивание, травление, используя процессы фазового разделения и ориентационного растяжения. Отдельное внимание уделено влиянию параметров пористой структуры на скорость гидролитической деструкции полимера и перспективам развития областей использования подобных пористых материалов.
ВВЕДЕНИЕ
Полимерные материалы широко применяются во многих отраслях промышленности и в быту, успешно заменяя стекло, металл, бумагу и т.п. Наиболее востребованные крупнотоннажные полимеры, преимущества которых заключаются в низкой себестоимости, термостойкости и хороших физико-механических характеристиках, как правило, являются продуктами синтеза из нефтяного сырья. Однако истощение запасов нефти и газа заставляют искать альтернативные источники их замены. Из-за длительного срока разложения большинства промышленных полимеров также остро встает экологическая проблема, связанная с утилизацией отходов. В настоящее время даже в развитых странах только порядка 30% использованных материалов из полимеров перерабатывается вторично, поэтому прогнозирование количества отходов пластмасс и мероприятия, направленные на их снижение, представляются крайне важными [1, 2]. Один из эффективных подходов для решения указанных задач – разработка и широкое применение биоразлагаемых полимеров. Они должны сохранять эксплуатационные характеристики только в течение периода использования, а затем претерпевать физико-химические и биологические превращения под действием факторов окружающей среды (света, температуры, влаги) в продукты метаболизма природных биосистем (бактерий, дрожжей, грибов, водорослей). При этом высокомолекулярные вещества разлагаются на низкомолекулярные (вода, углекислый газ и т.д.), гуминовые вещества и биомассу, тем самым совершается естественный круговорот веществ, способный поддерживать экологическое равновесие в природе.
Среди биоразлагаемых полимеров полилактид, который представляет собой термопластичный сложный алифатический полиэфир, занимает особое место [3]. Привлекательность полилактида заключается в том, что сырьем для его производства являются возобновляемые источники растительного происхождения, чем обусловлена относительно невысокая цена этого полимерного материала. Его модификация за счет создания высокой пористости с контролируемой морфологией и размером пор существенно расширяет области потенциального использования подобных материалов. В опубликованных за последние 10 лет обзорах [4–8] основное внимание уделено вопросам получения пористого полилактида для решения различных биомедицинских задач, таких как создание каркасов (скаффолдов), имплантатов и т.д. Не менее важными представляются другие области применения, в которых биоразлагаемые полимеры могут конкурировать и заменять пористые материалы, полученные на основе трудно разлагаемых полимеров, а именно, в качестве различных микрофильтрационных, первапорационных разделительных мембран, сорбентов, пенопластов. В настоящем обзоре представлены физико-химические основы методов создания пористого полилактида и пути их реализации. Экологические проблемы, в частности, связанные с утилизацией отходов полимерного материала, представляются актуальными и определяющими потенциальные перспективы его использования. В связи с этим в обзоре будет затронут также вопрос о влиянии параметров пористой структуры на способность и скорость гидролитической деструкции полилактида.
МЕТОДЫ ПОЛУЧЕНИЯ ПОРИСТЫХ МАТЕРИАЛОВ НА ОСНОВЕ ПОЛИЛАКТИДА
В литературе имеется большое количество работ по получению пористых материалов как на стадии синтеза макромолекул [9, 10], так и уже существующих полимеров [11–15]. В настоящем обзоре основное внимание уделено физико-химическим и механическим методам формирования пористых материалов из высокомолекулярного полилактида. В табл. 1 систематизированы наиболее распространенные методы создания пористой структуры полилактида и потенциальные области применения.
Таблица 1.
Методы получения, характеристики и области применения пористых полимерных материалов на основе полилактида
| Метод | Условия получения | Диаметр пор | Структурные особенности | Потенциальные области применения | Лите-ратура |
|---|---|---|---|---|---|
| Спекание | Прессование порошка или волокон при нагревании | 60–150 мкм | Замкнутые поры, пористость 30–40 об. %, размер пор определяется дисперсностью порошка | Прототипирование и изготовление каркасов для инженерии и регенерации костной и хрящевой ткани; имплантаты с высоким уровнем точности; системы доставки лекарств | [16–21] |
| Выдерживание частиц в субкритическом CO2 с последующим снижением давления | 40–70 мкм | Система взаимопроникающих пор, пористость 40–45 об. % | [23, 24] | ||
| 3D-печать | Послойное нанесение расплава филамента в соответствии с цифровой трехмерной моделью | 300–1000 мкм | Каркасная структура с открытыми порами, пористость 30–85 об. %, геометрия и размер пор определяется диаметром филамента и цифровой моделью | Быстрое прототипирование и изготовление трехмерных, в том числе малоинвазивных, каркасов для регенеративной медицины и тканевой инженерии | [28–37] |
| Электро- формование | Осаждение заряженной полимерной струи на противоположно заряженный коллектор | 250 нм–5 мкм | Система открытых, взаимосвязанных пор между неплотно упакованными монолитными или пористыми волокнами диаметром 50 нм–2 мкм, пористость 50–70 об. % | Каркасы для тканевой инженерии, имитирующие естественный внеклеточный матрикс; носители для доставки лекарственных препаратов; микрофильтрационные и разделительные мембраны; экосорбенты нефтепродуктов | [44–47] |
| Травление | Вымывание частиц солей натрия или шариков парафина из смеси с полилактидом | 30–400 мкм | Система взаимопроникающих пор, пористость 30–97 об. % | Каркасы для тканевой инженерии, в том числе с контролируемым выделением лекарственных препаратов; высокопористые подложки | [58–62] |
| Селективное вымывание фазы второго полимера из смеси с полилактидом | 30 нм–450 мкм | [65–68] | |||
| Селективная деструкция фазы второго полимера из смеси с полилактидом | 60–1000 мкм | [63, 64] | |||
| Вспенивание | Насыщение полимера газами под давлением в процессе экструзии, вспенивание при выходе из экструдера | 200–400 мкм | Сочетание замкнутых и открытых пор, пористость 75 об. % | Каркасы для инженерии костной ткани; носители для доставки лекарственных препаратов; высокопористый пенопласт в качестве упаковочных и конструкционных материалов, тепло- и звукоизоляторов | [72, 77] |
| Насыщение сверхкритическим СО2 с последующей декомпрессией | 50–400 мкм | Замкнутые или открытые поры, пористость 70–93 об. % | [71–74] | ||
| Нагрев термически нестабильных неорганических или органических соединений, введенных в объем полимера, с выделением большого количества газообразных продуктов | 90–500 мкм | Преимущественно ячеистые замкнутые поры, пористость 47–90 об. % | [78–80] | ||
| Фазовое разделение | Термически индуцированное разделение фаз по кристаллизационному механизму | 10 мкм и 100–200 мкм | Анизотропная пористая структура в виде пены с пористыми стенками, пористость 80–96 об. % | Пористые каркасы для тканевой инженерии; модельные системы для in vitro скрининга противоопухолевых препаратов; мембраны для гемодиализа и первапорации; пористые покрытия для контролируемого выделения целевых веществ, например, минеральных удобрений; экосорбенты маслопродуктов | [86–92] |
| Термически индуцированное разделение фаз по жидкостному механизму | 25–300 мкм | Разнообразие пористых структур: от замкнутых макропор до губчатой ячеистой и волокнистой взаимопрони-кающей структуры, пористость 85–93 об. % | [94–96] | ||
| Разделение фаз по жидкостному механизму при добавлении осадителя | 10 нм–200 мкм | Разнообразие пористых структур: от замкнутых макропор до губчатой ячеистой и волокнистой взаимопрони-кающей структуры, пористость до 90 об. % | [85, 97, 98] | ||
| Ориентационное растяжение | Предварительное введение эластомерных или жидких добавок с последующим одноосным растяжением на воздухе | 0.5–10 мкм | Замкнутые поры, локализованы преимущест-венно в объеме материала | Материалы для тканевой инженерии; основа для получения нанокомпозитов | [113–116] |
| Одноосное растяжение в присутствии ААС по механизму крейзинга | 10–30 нм | Взаимопроникающая открыто-пористая структура, пористость возрастает с увеличением степени деформации до 40–60 об. % | [120–125] |
Спекание полимерных порошков или волокон
Метод спекания достаточно прост и позволяет получать пористые материалы из различных неорганических (керамика, металлы, силикаты и т.д.) и полимерных (ПЭ, ПТФЭ, ПС и т.д.) материалов. Он основан на локальном сплавлении контактирующих частиц, в результате которого в местах соприкосновения исчезают межзеренные границы, однако между ними сохраняются поры (рис. 1).
Рис. 1.
Схема формирования пор в процессе спекания полимерных гранул и СЭМ-микрофотография получаемого пористого материала из ПЛГ [16]. Цветные рисунки можно посмотреть в электронной версии.

Существует два основных варианта проведения спекания [17, 18]. Наиболее часто этот процесс осуществляют, проводя прессование исходного порошка или волокон при температуре несколько выше температуры стеклования (на 5°C) для аморфных полимеров или вблизи точки плавления для кристаллических полимеров. При спекании сферических частиц сополимера поли(D,L-лактид-гликолида) (ПЛГ) (температура стеклования 57°C) размером 200–600 мкм при температуре 62°C было установлено [16], что диаметр пор в конечном пористом материале определяется размером исходных частиц и распределением их по размерам, а также временем спекания. Использование микросфер диаметром 200 мкм приводило к формированию материалов с размером пор 67 мкм, при увеличении размера исходных частиц до 400–600 мкм наблюдали возрастание среднего диаметра пор до 150 мкм. При этом пористость изменялась незначительно и составляла 32–39 об. %. Также было определено оптимальное время спекания 24 ч, поскольку при меньшем времени не удавалось получать прочные контакты между частицами, а при большем происходило постепенное слияние частиц и исчезновение пор.
Перспективным методом термического спекания, который достаточно широко используется для полилактида, его сополимеров и композитов, является селективное лазерное спекание [19–21], относящееся к аддитивным технологиям [22] и позволяющее быстро изготавливать 3D анатомические модели и имплантаты с высоким уровнем точности. В данном случае лазерный луч определенным запрограммированным путем проходит по поверхности свободно насыпанного или уплотненного порошка полимера с размером частиц от нескольких микрон до нескольких сотен микрон (обычно используют частицы диаметром 30–60 мкм). Спекание происходит в результате локального плавления на границе соприкосновения частиц порошка при действии лазерного луча, не затрагивая объем.
При низкотемпературном варианте прессование проводят после обработки полимера растворителем или смесью растворитель–осадитель, причем температура кипения для растворителя существенно ниже, чем для осадителя. Для этого метода обычно используют достаточно токсичные органические жидкости (дихлорметан, ацетон, этанол). Однако в работах [23, 24] показано, что низкотемпературное спекание полилактида и ПЛГ можно проводить в экологически безопасном и нетоксичном субкритическом CO2 (15–25 бар, 25°C). В таких условиях субкритический CO2 оказывает слабое пластифицирующее действие на полимеры, вызывая понижение их температур стеклования или плавления только в поверхностном слое микросфер (диаметр 100–250 мкм). В процессе снижения давления газа с умеренной скоростью происходит объединение дисперсных частиц с образованием пористого каркаса (пористость порядка 40 об. %), содержащего систему взаимопроникающих пор размером 40–70 мкм.
Основная область использования пористых материалов из полилактида, полученных методом спекания, – создание различных имплантатов необходимой геометрии, в том числе покрытых биоактивными веществами, ускоряющими регенерацию ткани, например факторами роста, белками, гидроксиапатитом и т.д. [18, 23, 25]. Структура полимерного матрикса влияет на процесс пролиферации, а именно, формирование клеток происходит по стенкам пор, постепенно заполняя их, что способствует организации образующихся клеток в виде концентрических слоев, подобно губчатой костной ткани, или в виде ориентированных волокон [17, 26].
3D-печать
Среди методов получения пористых каркасов, преимущественно используемых для инженерии костной ткани, в последние десять лет наиболее интенсивно развивается метод трехмерной (3D) печати [27]. 3D-печать относится к аддитивным технологиям, которая заключается в послойном построении изделия из жидкой полимерной струи на основе цифровой трехмерной модели, что позволяет быстро проектировать и изготавливать трёхмерные структуры с высокой точностью и детализированной биомиметикой (рис. 2).
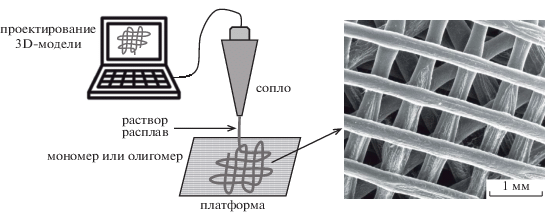
Печать может осуществляться расплавом из полимерного волокна, концентрированным раствором или суспензией (пастой), жидким мономером, олигомером или преполимером с проведением последующих реакций полимеризации или сшивания под действием УФ-облучения [29, 30]. Современные устройства для 3D-печати могут создавать каркасы в масштабе от нанометров с помощью адаптированного атомно-силового микроскопа, снабженного специальной иглой-соплом [31], до десятков миллиметров на коммерческом 3D-принтере [32]. Как правило, подобные матриксы содержат открытые и однородно распределенные по объему поры, размер которых задан с хорошей точностью (относительная погрешность около 5%). Для 3D-каркасов на основе полилактида размер пор обычно составляет не менее 300 мкм. Добавление к полимеру полиэтиленгликоля, обладающего свойствами пластификатора, позволяет снизить вязкость струи, и саму печать провести при более низкой температуре, что способствует лучшему контролю за процессом и уменьшению размера пор в одном из направлений до 75–150 мкм [33]. Анализ литературы показал, что объемная пористость материалов из полилактида, получаемых методом 3D-печати, как правило, тем выше, чем больше размер пор (рис. 3).
Рис. 3.
Зависимость размер пор–пористость для объемных каркасов полилактида, полученных методом 3D-печати.
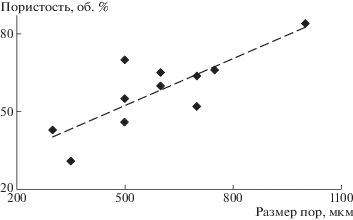
Важно, что свойства 3D-матриксов зависят не только от способа печати, но и от внутренней архитектуры и геометрии пор, которые бывают ортогональными (наиболее часто используются), треугольными, в виде сот и т.д. [28, 34–37]. Оптимальные параметры пористой структуры (форма, размер и суммарный объем пор) подбираются в соответствии с предполагаемым использованием пористого матрикса и поддержанием баланса между его механической прочностью и скоростью разложения [38]. Например, в работе [39] было установлено, что наиболее подходящий размер пор для каркаса с функцией регенерации костной ткани составляет 750 мкм при общей пористости 65 об. %, хотя каркас с размером пор 1000 мкм имел пористость на 20 об. % выше. Именно в таких пористых матриксах с оптимальным размером пор наблюдали максимальную скорость роста клеток остеобластов человека и протекание их дифференциации.
Метод 3D-печати также хорошо сочетается с другими способами получения пористых материалов на основе полилактида, например вспениванием в сверхкритическом CO2 [40], что позволяет создавать каркасы, содержащие как поры размером сотни микрон (300–700 мкм), так и диаметром порядка 10 мкм. Кроме того, вспенивание дополнительно приводит к росту объемной пористости на 10–15 об. %. Подобный комбинированный метод имеет хорошие перспективы для создания матриксов с иерархической структурой и их использования в тканевой инженерии.
Электроформование
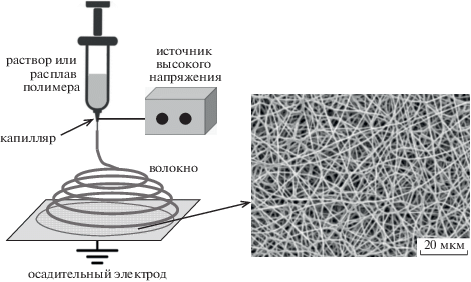
Электроформование или электроспиннинг представляет достаточно простой безфильерный метод производства тонких полимерных волокон диаметром от десятков нанометров до десятков микрометров. При таком способе к раствору или расплаву полимера, выдавливаемого из капилляра в виде струи, прикладывается высокое напряжение, которое индуцирует в ней одноименный электрический заряд. В результате происходит ускоренное движение полимерной струи к противоположно заряженной заземленной подложке (осадительный электрод), где она затвердевает в волокнистой форме при испарении растворителя или охлаждении и формируется пористый материал, похожий на вату или нетканый материал (рис. 4) [41–43].
Рис. 4.
Схема получения волокнистого материала методом электроформования и СЭМ-микрофотография получаемого матрикса из полилактида [44].
В этом процессе полимерная струя подвергается удлинению и утончению до 105 раз со скоростью порядка 103 с–1, кроме того, при определенном соотношении значений вязкости, поверхностного натяжения и плотности электрических зарядов она может дополнительно расщепляться на более тонкие струи. Морфологию подобных волокон и параметры получаемого пористого матрикса можно достаточно хорошо контролировать и регулировать характеристиками исходного раствора или расплава (вязкость, концентрация и ММ полимера, поверхностное натяжение, электропроводность), условиями (величина приложенного напряжения, расстояние между капилляром и осадительным электродом, тип подложки), температурой и влажностью [45]. Одна из проблем волокон, полученных методом электроформования, заключается в образовании каплеобразных утолщений, появление которых чаще всего происходит при использовании вязких растворов или достаточно высокомолекулярного полимера. Уменьшить этот эффект позволяет не только подбор оптимальных условий формования, но и особенная конструкция коллектора, позволяющая осуществлять при нагревании дополнительное механическое растяжение полимерных волокон [46].
Формование волокон полилактида наиболее часто проводится из растворов в гексафторизопропаноле или хлористом метилене при напряжении электростатического поля 10–30 кВ. В работе [47] были определены параметры пористой структуры материала, полученного из волокон полилактида диаметром 0.5–1.2 мкм. Его пористость составляла практически 70 об. %, а размер пор 3–5 мкм. Высокотемпературный отжиг подобных мембран при 90°С приводил к уменьшению среднего размера пор в 2 раза и одновременно к увеличению механической прочности, что позволило их использовать для микрофильтрации воды от частиц TiO2.
Одно из основных применений пористых волокнистых материалов из полилактида, полученных методом электроформования относится к области биомедицины, а именно, получение каркасов для тканевой инженерии, имитирующих естественный внеклеточный матрикс, а также носителей для доставки лекарств [48–52]. Однако поверхность волокон, полученных методом электроформования, не содержащих дисперсных добавок, обычно достаточно гладкая, а сами волокна прозрачны, что может стать препятствием хорошей адгезии клеток к подобным материалам, несмотря на их чрезвычайно высокую удельную поверхность. Действительно, в исследовании [53] было установлено, что наноразмерные поры на поверхности волокон полилактида играют важную роль в прикреплении эпителиальных клеток. В связи с этим свое активное развитие получили работы, посвященные созданию дополнительной пористой структуры на поверхности и в объеме электросформованных волокон. Необходимо отметить, что даже хорошо совместимые растворимые добавки и повышенная влажность могут быть причиной возникновения у волокон поверхностной шероховатости и пор. Для формирования пористых волокон наиболее часто используют методы, основанные на фазовом разделении, индуцированном изменением температуры или качеством растворителя в процессе его испарения или добавления осадителя [54]. Размер таких пор обычно составляет от нескольких десятков до нескольких сотен нанометров. Другой подход состоит в добавлении наноразмерных частиц иной природы, например γ-Fe2O3 [55]. Такой подход позволил получить мембраны с двухуровневой организацией пористой структуры, которые оказались эффективными для адсорбции масла (сорбционная емкость по отношению к моторному маслу составила около 270 г/г) и разделения смеси масло–вода. Существуют также работы, направленные на создание пор другого уровня непосредственно в нетканом материале. Для этого в качестве шаблонов, на которые происходит осаждение волокон полилактида, используют кристаллы льда, образующиеся на охлаждающемся осадительном электроде из влаги воздуха, с последующим их удалением методом сублимации [44]. Сформированные подобным образом каркасы содержат в себе трехмерную систему взаимосвязанных пор размером 10–500 мкм, что обеспечивает достаточно эффективное прорастание через них клеток фибробластов и кровеносных сосудов при исследовании in vivo. Отметим, что похожий подход получения пористых материалов с использованием криогенных технологий можно применять и в процессе традиционного “мокрого” метода формования волокон полилактида из раствора [56]. В данном случае также на первой стадии образуется система волокон полилактида диаметром 100– 400 нм и ледяных микросфер, которые действуют как макро-порообразователи (средний размер 100 мкм) и удаляются в процессе лиофильной сушки.
Травление
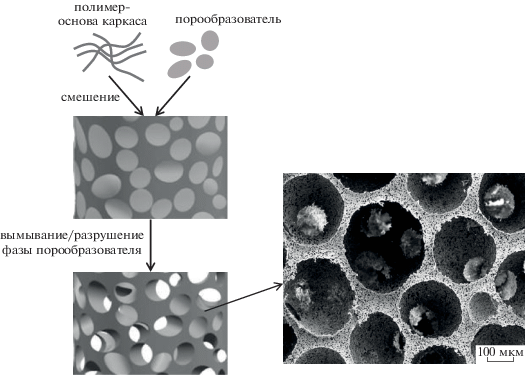
Метод травления (выщелачивание, вымывание), в основе которого лежит селективное удаление какой-то части полимерного или композиционного образца, в настоящее время широко используется для изготовления матриксов с пористостью выше 90 об. % [57]. Разработано несколько подходов для реализации данного метода. Для этого в полимер, который является основой каркаса, по растворной или расплавной технологии предварительно вводят вещества-порообразователи (низкомолекулярные соли, сахар, воск и т.д.) или другой полимер, которые затем селективно вымывают или разрушают, используя химические реакции (рис. 5).
Рис. 5.
Схема получения пористого материала методом травления и СЭМ-микрофотография получаемого пористого полилактида [58].
Наиболее распространенный и простой метод включает получение смеси полилактида с частицами нетоксичных солей натрия (например, хлорида, цитрата, тартрата) путем горячего прессования [59] или добавления их в раствор полимера (в хлороформе или хлористом метилене) [60, 61] с последующим формованием композиционного материала и длительным (от 2 до 5 суток) вымыванием из него соли в деионизированной воде. Параметры полученных пористых матриксов не зависят от типа использованной соли и определяются только ее массовой долей в композите и размером частиц. А именно, пористость возрастает с 50 до 93 об. % с увеличением содержания соли с 50 до 90 мас. % и практически не зависит от размера частиц. Диаметр образующихся пор близок к размеру исходных частиц соли, который обычно варьируется от десятков до сотен микрометров. Замечено, что при введении соли порядка 50–60 мас. % по растворной методике формируются асимметричные мембраны независимо от размера частиц. В случае использования горячего прессования из таких композитов вымывание соли было затруднено. Введение более 70 мас. % позволяло полностью вымывать соль из полимерной матрицы, при этом происходило образование системы взаимопроникающих открытых пор. Таким образом, этот метод позволяет независимо контролировать размер пор, однако в данном случае сложно регулировать структуру и степень взаимосвязанности формируемых пор.
Возможности идентификации морфологии пористой структуры возрастают, если вместо частиц соли в качестве удаляемого компонента использовать сферические частицы парафина заданного размера [58, 62]. Смесь полилактида с парафиновыми шариками получают по растворной технологии (раствор в пиридине или смеси диоксан/пиридин), из которой затем гексаном вымывают парафин. Данная методика позволяет получать матриксы пористостью до 95–99 об. %, содержащих систему контролируемых взаимосвязанных сферических пор диаметром от 100 до 500 мкм, размер которых определяется размером исходных парафиновых шариков. Дополнительная возможность модификации пористой структуры полимерного каркаса связана с использованием процессов фазового разделения при формовании матрицы [62]. В этом случае полимерная фаза формируется в виде нановолокон, между которыми имеются поры 0.2–1.5 мкм. Обнаружено, что адсорбция белков сыворотки и адгезия клеток остеобластов на подобную волокнистую структуру значительно выше, чем в неволокнистых каркасах, что обеспечивает благоприятную основу для роста тканей.
Другой подход к созданию пористых материалов методом травления заключается в получении полимер-полимерных композитов как смешением в общем растворителе [63], так и по расплавной технологии [64–68], причем в качестве второго компонента могут выступать полимеры, не совместимые с полилактидом (например, полистирол, поли-ε-капролактон, полиоксалаты, полигидроксибутират, полиэтиленоксид). В данном случае удается получать смеси со структурой непрерывных взаимопроникающих полимерных фаз. При нагревании выше температуры плавления или размягчения обоих полимеров происходит укрупнение фаз, что позволяет контролируемо управлять параметрами пористой структуры получаемого матрикса полилактида. Удаление второго полимера из композита осуществляют по методике селективного вымывания (растворения), например, полистирол циклогексаном [65, 66], поли-ε-капролактон уксусной кислотой [67], полиэтиленоксид водой [68] или используя селективную деструкцию [63, 64]. После удаления одной из двух фаз получается пористая структура второго полимера, состоящая из связанных между собой открытых пор объемом 30–80% и широко варьируемыми размерами от 30 нм до 1 мм. Полученные каркасы из полилактида оказались не цитотоксичными, с хорошей биосовместимостью и обладающими высокой доступностью пор для водных растворов биоактивных добавок. Такой подход представляется перспективным для создания материалов тканевой инженерии с контролируемым высвобождением лекарственного препарата.
Вспенивание
Вспенивание является одним из промышленных методов производства пористых полимерных материалов, часто называемых пенопластами. Обычно в качестве порообразователей используются газы (воздух, азот, аммиак, диоксид углерода), которые под давлением пропускают непосредственно через расплав полимера или которые образуются в результате физических процессов, например испарения, десорбции при нагреве и снижении давления, или при протекании химической реакции с выделением большого количества газообразных продуктов (рис. 6).
Рис. 6.
Схема получения пористого полимера методом вспенивания и СЭМ-микрофотографии получаемого материала из полилактида. Температура насыщения CO2 120 или 126оС (верхнее и нижнее фото соответственно), давление 5.5 МПа [69].
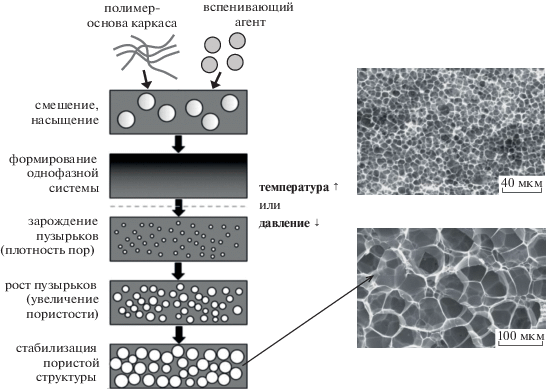
Чаще всего в качестве вспенивающего агента используют CO2 – нетоксичный, негорючий, достаточно инертный и дешевый газ, который часто применяют в экологически чистых, так называемых “зеленых” технологиях [69]. Количество поглощаемого СО2 полимером зависит от условий (давление, температура, время насыщения) и агрегатного состояния. При переходе от газообразного состояния к жидкому и сверхкритическому сорбция СО2 полилактидом увеличивается, достигая значений 6 мас. % при 15 ПМа, 40°С [70] и 20 мас. % при 27.8 МПа, 180°С [69]. Объем пор вспененного полилактида при использовании жидкого и газообразного СО2 не превышает 0.1–0.3 см3/г, а в сверхкритическом CO2 может достигать 2 см3/г. Особое внимание в качестве порообразователя привлекает сверхкритический CO2 [71], поскольку можно тонко контролировать скорость его диффузии и растворимость в полимерах и регулировать параметры формирующейся пористой структуры путем изменения давления, температуры, продолжительности экспозиции и скорости декомпрессии. Процесс вспенивания в сверхкритическом CO2 включает четыре стадии (рис. 6, схема): растворение газа в полимере (стадия насыщения), зарождение пузырьков (места образования пор) путем изменения термодинамического состояния системы за счет повышения температуры или понижения давления, рост пузырьков (размера пор), выход газа из объема полимера и стабилизация поры (стадия декомпрессии). Для получения стабильной пористой структуры необходимо, чтобы полимер в условиях вспенивания был твердым, но СО2 довольно хорошо растворялся. Для этого обычно насыщение осуществляют в условиях, близких к температуре стеклования или плавления соответственно для аморфных или кристаллических полимеров. Необходимо отметить, что CO2 оказывает значительное пластифицирующие влияние на полилактид [72], в результате чего температуры кристаллизации и стеклования полимера снижаются, что необходимо учитывать при выборе температуры вспенивания. В работе [72] изучали влияние температуры (от 5 до 55°С), давления (от 6 до 23 МПа) и ММ (от 13 × 103 до 96 × 103 ) на параметры пористой структуры аморфного полилактида. Увеличение давления и продолжительности насыщения СО2 способствовали интенсивному зародышеобразованию, что приводило к формированию мелких пор ячеистого типа. С ростом температуры и скорости диффузии, напротив, происходит увеличение размера и доли открытых и взаимосвязанных пор, к этому же приводит низкая скорость декомпрессии. В то же время нагревание и длительное время насыщения способствуют формированию однородной пористой структуры. Было отмечено, что вспененные материалы на основе более высокомолекулярного полилактида пористостью порядка 70% и размером пор 200–400 мкм оказались менее хрупкими [73].
Вспенивание частично кристаллического полилактида проводят при более высокой температуре 100–140°С, в результате чего получаются более стабильные и механически прочные материалы с общей пористостью выше 90 об. %, из которых порядка 75–80 об. % приходится на открытые поры размером 40–140 мкм [69, 74]. Дополнительные подходы, позволяющие варьировать параметры пористой структуры, а именно, снижать размер пор до 5 мкм и увеличивать их количество в единице объема до 1.5 раза, связаны с введением небольшого количества добавок в сверхкритическую среду (например, 1–4 мас. % ацетона) [75] или в полимер (например, 5 мас. % полиэтиленгликоля) [76].
Процесс вспенивания в автоклаве (дискретный режим производства) имеет ограничения из-за неэкономичности и в основном подходит для лабораторных экспериментов, а также для получения пористых имплантатов заданной геометрии. Более привлекательными для промышленности являются непрерывные процессы, например осуществление вспенивания при экструзии полимера. В данном случае газообразный пенообразователь подается под давлением в экструдер, где он смешивается с полимером [72, 77]. При выходе полилактида, содержащего газ, происходит падение давления, что приводит к фазовому разделению, образованию пор и их стабилизации в вязкой полимерной матрице. Было показано, что при использовании в качестве порообразователя СО2 его оптимальная концентрация в полилактиде должна составлять 7–8 мас. % [72]. В этих условиях формируются полимерные пены пористостью 75 об. % с концентрацией пор порядка 108 см–3 и размером 200–400 мкм. При меньшей концентрации CO2 (менее 5 мас. %) образуется мало пор – не более 105 см–3, при большей концентрации (выше 8 мас. %) – пористая структура оказывается нестабильной и со временем претерпевает значительную усадку.
При другом методе вспенивания, который осуществляют в процессе экструзии, применяют термически нестойкие химические порообразователи неорганической (карбонаты и бикарбонаты натрия и аммония в сочетании с лимонной кислотой) [78–80] и органической (ДАК) [78] природы. Вводить вспенивающие агенты можно на стадии экструзии, в гель полилактида и используя процесс набухания полимера в растворе исходных порообразователей. Последующее нагревание приводит к разложению введенных агентов с образованием большого количества газов, таких как CO2, NH3, N2, и формированию пор ячеистого типа размером 100–500 мкм. При этом в работе [80] отмечалось, что пористость полилактида и доля открытых пор зависит только от концентрации порообразователя.
Перспективы применения пористых материалов из полилактида, полученных методом вспенивания, находятся в области тканевой инженерии, в качестве каркасов для имплантатов и контейнеров для систем доставки лекарственных препаратов. Так, в работе [74] было показано, что в открыто-пористых системах с высоким объемом пор более 90%, в которых обеспечивается эффективный перенос питательных веществ, происходит интенсивная пролиферация клеток эмбриональных фибробластов. Также из вспененных CO2 под давлением сферических частиц полилактида методом спекания паром предлагается получать достаточно прочный микропористый материал типа биодеградируемого пенопласта, который можно рассматривать в качестве замены пористым трудно разлагаемым полиолефинам [69].
Фазовое разделение
В последние десятилетия методы фазового разделения для создания 3D пористых полимерных каркасов получили широкое развитие из-за своей универсальности, простоты и возможности контроля за морфологией, размером пор и степенью их взаимосвязанности. Методология фазового разделения заключается в изменении параметров однофазной системы раствора полимера (концентрация полимера, температура, термодинамическое качество растворителя и т.д.), что приводит к образованию фаз, обогащенных или обедненных полимером. После фазового разделения растворитель можно удалять различными способами: с помощью лиофильной сушки [81], сверхкритической сушки [82], а также используя замену растворителя с последующей сушкой в вакууме [83].
Изменение свободной энергии Гиббса при смешении полимера и растворителя с образованием гомогенного раствора можно вычислить по уравнению Флори–Хаггинса [84]
Рис. 7.
Схематические изображения фазовых диаграмм и морфологий формирующихся систем в результате фазового разделения для двухкомпонентной системы по механизму твердое тело–жидкость (а) и жидкость–жидкость (б), для трехкомпонентной системы (в), а также СЭМ-микрофотография получаемого пористого материала из полилактида (г) [85].
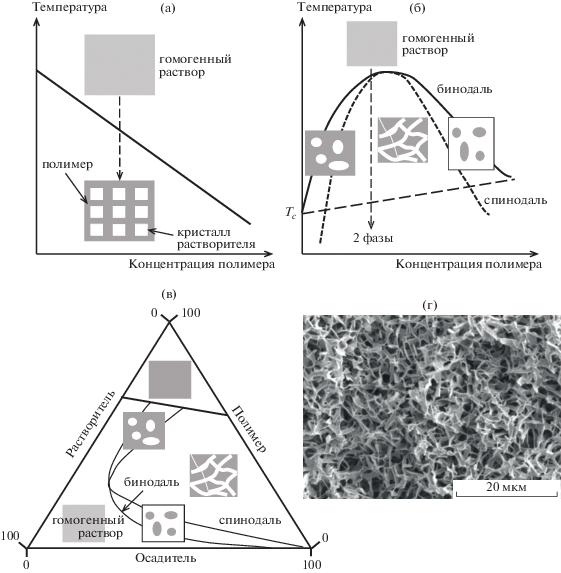
Чаще всего первый тип фазовой диаграммы (рис. 7а) характерен для системы, в которой при понижении температуры происходит кристаллизация растворителя, например, для растворов полилактида в 1,4-диоксане. При охлаждении раствора происходит кристаллизация диоксана (температура плавления 12°С), который затем удаляется методом лиофильной сушки или в вакууме [86–89]. В этом случае формируется анизотропная высокопористая структура в виде пен с объемом пор 80–96% и размером 100–150 мкм, стенки которых содержат более мелкие поры порядка 10–30 мкм [86, 90, 91]. Пористость полимерного материала уменьшается, а его плотность и количество остаточного растворителя увеличиваются с повышением начальной концентрации полилактида в растворе. Кроме того, кристаллизация полилактида способствует формированию более пористых и прочных пен. Полученные пористые матриксы считают перспективными для использования в клеточно-инженерных конструкциях [92] и в качестве носителей для биологически активных веществ с пролонгированным действием, например для белка костного матрикса, обладающего остеоиндуктивными свойствами [93].
При жидкостном механизме термически индуцированного фазового разделения получаемые пористые структуры более разнообразны. В данном случае граница, отделяющая однофазный гомогенный раствор от неустойчивого состояния, описывается бинодалью, однако внутри этого состояния имеется область метастабильного состояния, ограниченного спинодалью (рис. 7б). В области между бинодалью и спинодалью жидкостное фазовое разделение происходит по флуктуационному механизму через зарождение и рост неоднородностей, в результате которого образуются дисперсные частицы. Напротив, ниже спинодали разделение приводит к образованию взаимопроникающей сетки двух фаз, сопровождающееся гелеобразованием [94–96]. В работе [83] изучено влияние концентрации полилактида, состава растворителя (тетрагидрофуран/вода) и температуры охлаждения на морфологию формирующегося высокопористого материала (объем пор 85–93%). Было обнаружено, что увеличение концентрации полимера от 6 до 10 мас. % приводит к уменьшению среднего размера пор на порядок от 300 до 25 мкм. Аналогичный эффект наблюдается при уменьшении содержания воды в растворителе.
Более тонко регулировать морфологию пор и их размеры можно, используя двухкомпонентные растворители (смесь хорошего растворителя и осадителя). Фазовая диаграмма для таких систем представлена на рис. 7в. В этом случае фазовое разделение происходит вследствие добавления в гомогенный раствор осадителя и ухудшения термодинамического качества растворителя. Полученные пористые материалы характеризуются высокой пористостью более 90 об. %, удельной поверхностью до 54 м2/г и бимодальным распределением пор по размеру, а именно, 10–15 нм и от нескольких микрон до 200 мкм [85], и рассматриваются как перспективные материалы для использования в качестве имплантатов костной ткани. В работе [97] обнаружена корреляция между природой осадителя (совместимость с растворителем хлористым метиленом), вязкостью раствора и размером пор. Наиболее однородная пористая структура с наименьшим размером пор формируется при добавлении невязких жидкостей, совместимых с растворителем, но в которых полилактид не растворяется (например, изопропанол). Увеличение вязкости осадителя приводит к росту размера пор, а плохая совместимость с растворителем делает пористую структуру неоднородной.
Дополнительные возможности варьирования морфологии пористой структуры появляются в методе дробного осаждения, основанного на быстром помещении раствора полилактида в этанол на 48 ч, который является осадителем, с последующей сублимационной сушкой [98]. В данном случае структура материалов пористостью 91–98 об. % и размером пор 1–4 мкм варьировалась от зернистой, построенной из наночастиц, до сетчатой, образованной нанолентами или нановолокнами при уменьшении растворимости полилактида. Высокая концентрация исходных растворов способствовала формированию более тонкодисперсной структуры. Полученные пористые материалы являются высоко гидрофобными (их контактный угол смачивания 145°) и позиционируются как экосорбенты масла для разделения смесей масло–вода с сорбционной емкостью до 2900 мас. %.
Дополнительный контроль за морфологией и параметрами пористой структуры полилактида, возникающей в результате фазового разделения, возможен путем использования добавок гидрофильных полимеров (полиэтиленгликоль, ацетат целлюлозы) или наночастиц (SiO2, монтмориллонит, активированный уголь) [99–103]. Обнаружено, что пористость мембран полилактида и взаимосвязанность пор, образующихся в присутствии добавок, более высокая, что связывают со стабилизацией пор, а также за счет дополнительного вымывания этих добавок. Кроме того, повышается гидрофильность пористого материала, что делает его более водопроницаемым и привлекательным в качестве покрытий для гранул удобрения (например, мочевины) с контролируемой скоростью высвобождения [99] или в качестве сорбента токсичного красителя Родамина Б из водных растворов в широкой области рН 2–11.
Поскольку экологические проблемы в современном мире выходят на первый план, вопросы замены токсичных органических растворителей при получении пористого полилактида методом фазового разделения важны. В последнее время все большую актуальность приобретают подходы, в которых применяют экологически безопасные вещества. Для получения пористого полилактида в качестве подобного растворителя используют этиллактат, а в качестве осадителя – воду [81, 104]. В этом подходе морфология мембран зависела от времени испарения растворителя и изменялась от макропористой (размер пор десятки микрометров) до губчатой (размер пор единицы микрометров) и, наконец, плотной. Использование сверхкритической сушки для такой системы [105] позволяет значительно увеличить пористость полилактида до 90–95 об. % и удельную поверхность до 70–95 м2/г, а размер пор уменьшить до нанометрового уровня (10–100 нм).
Другой часто используемой экологически чистой средой является сверхкритический СО2, который применяют не только для осуществления сверхкритической сушки гелей полилактида, но и в качестве осадителя [102, 106]. Использование сверхкритического СО2 позволяет получить материалы с порами ячеистого типа размером до 100 мкм, однородно расположенными по всему сечению. При этом средний размер пор уменьшается с ростом давления и плотности CO2, с увеличением концентрации раствора полимера и понижением температуры. Предполагается, что подобные материалы имеют хороший потенциал в качестве альтернативных носителей лекарственных препаратов.
Таким образом, метод фазового разделения и его комбинации для получения пористого полилактида представляется достаточно эффективным способом, не требующим специального оборудования, при котором возможен контроль за параметрами пористой структуры и механических свойств материала.
Ориентационное растяжение
Достаточно часто в процессе ориентационного растяжения полимеров и композитов на их основе происходит формирование пор (рис. 8). В качестве основных механизмов порообразования выделяют следующие: кавитации как результат возникновения локального трехосного напряжения [107], крейзинг [108], отслаивание частиц наполнителя [109]. Известно, что материалы из полилактида (с низким содержанием D-изомерных звеньев) достаточно хрупкие, и величина их разрывного удлинения обычно не превышает 5–10% [110]. Связано это с низкой плотностью флуктуационной сетки зацепления “полужестких” цепей полилактида [111]. В работе [112] для увеличения плотности зацеплений и снижения хрупкости полимера предлагают вводить длинноцепочечный разветвленный полилактид в линейный полилактид. Дополнительно полученную смесь определенным образом ориентировали и структурировали по типу жестко-эластического материала, используя метод многоступенчатой экструзии с устройством умножения слоев [113]. В процессе одноосного растяжения подобного материала наблюдали его “побеление” вследствие формирования в нем пустот с размерами субмикронного и микронного уровня, которые увеличиваются вдоль направления растяжения.
Рис. 8.
Схемы формирования пористой структуры полимеров в процессе ориентационного растяжения по механизму образования кавитаций (а), крейзинга (б) и отслаивания частиц наполнителя (в).
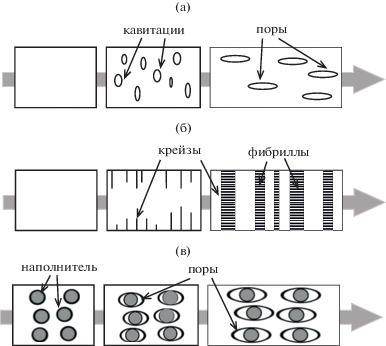
Другой подход снижения хрупкости полилактида и увеличения его деформируемости заключается в добавлении в объем полимера мягкой фазы (например, натурального каучука) [114] или жидкой фазы (например, олигомерного полидиметилсилоксана) [115], или ионных жидкостей [116]. В этих системах в процессе одноосного растяжения из-за облегчения возникновения кавитаций или в результате отслаивания частиц наполнителя происходит формирование пор также преимущественно микронного размера.
Достаточно универсальным структурно-механическим подходом к получению нанопористых полимерных материалов на основе широкого круга твердых аморфных стеклообразных и частично кристаллических полимеров является крейзинг в присутствии ААС [117, 118]. По определению ИЮПАК [119] крейзинг – это процесс образования в полимерном материале полостей (трещин), стенки которых соединены системой ориентированных разобщенных фибрилл, под действием механической нагрузки. В работах [120–124] были проведены систематические исследования стадий возникновения и развития фибриллярно-пористой структуры в аморфных пленках и волокнах полилактида по механизму крейзинга в присутствии различных жидких сред (алифатические спирты, водно-этанольные растворы, предельные углеводороды, кремнийорганические жидкости). Было показано, что такой процесс сопровождается формированием высокодисперсной ориентированной структуры по механизму классического крейзинга с диаметром пор и фибрилл порядка 20–30 нм и увеличением объемной пористости до 60 об. % с ростом степени деформации (рис. 9). Морфология таких пористых материалов в широком ряду по степени деформации (от предела текучести до 350–400%, когда полностью происходит переход исходного полимерного материала в фибриллы) представляет собой чередование недеформированных блочных частей и пористых крейзов. По данным малоуглового рентгеновского рассеяния при высоких степенях деформации в результате протекания процессов неполного коллапса высокодисперсной структуры крейзов образцы полилактида по всему объему приобретают достаточно однородную ориентированную фибриллярную кристаллическую структуру, содержащую щелевидные, вытянутые в направлении растяжения поры размером (10–15) нм × 125 нм [123].
Рис. 9.
Зависимость объемной пористости полилактида от степени деформации в этаноле в случае классического (1) и делокализованного (2) крейзинга.
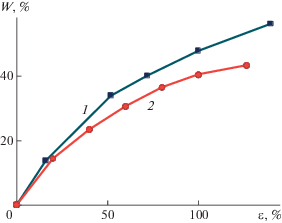
При деформации частично кристаллической пленки полилактида, кристаллизацию которой предварительно осуществляли в среде этанола при 50°C (степень кристалличности составляет 40–45%), формирование пористой структуры происходит по механизму делокализованного (межкристаллитного) крейзинга [125]. В этом случае пористая структура возникает одновременно во всем объеме образца. При степени деформации 100% объемная пористость пленки полилактида составляет порядка 40 об. % (рис. 9), она содержит популяции пор двух размеров порядка 10–20 нм и сотни нанометров. После удаления жидкой среды из деформированных образцов в изометрических условиях они становятся стабильными и сохраняют открыто-пористую структуру взаимопроникающих пор.
Полученные с использованием механизма крейзинга пористые матрицы полилактида и заполненные фосфатами кальция обнаружили высокую биоактивность по отношению к остеобластам MC3T3E1 [126]. Результаты исследований in vitro показали, что скорость роста популяции клеток в 5 раз выше на поверхности нанокомпозита полилактида, чем контрольного образца.
Таким образом, на сегодняшний день описано достаточно много разнообразных методов создания пористой структуры в полилактиде, параметры которой можно варьировать в широком диапазоне, а именно, величина объемной пористости может достигать 90–95 об. %, а размер пор от 10 нм до сотен микрон. Морфология пор также различна и меняется от закрытого ячеистого типа до системы открытых и взаимопроникающих пор. Поскольку полилактид интересен именно как экологически безопасный и биоразлагаемый полимер, задачи управления процессами деструкции пористых материалов на его основе, обладающими различными структурно-морфологическими типами и параметрами пористой структуры, актуальны для их контролируемого разложения в зависимости от условий и целей использования.
ВЛИЯНИЕ ПОРИСТОЙ СТРУКТУРЫ НА СКОРОСТЬ ГИДРОЛИТИЧЕСКОЙ ДЕСТРУКЦИИ ПОЛИЛАКТИДА
Пористые 3D-матриксы, волокна и пленки из полилактида и сополимеров на его основе представляют интерес в качестве материалов с контролируемыми сроками разложения, например для тканевой инженерии, как саморассасывающиеся хирургические шовные нити, оболочки для систем доставки лекарственных средств пролонгированного действия и т.п. Их разрушение обычно происходит в результате гидролитической деструкции сложноэфирных связей полимера во влажной среде. При этом продукты гидролиза представляют собой безопасные метаболиты, которые выводятся, не оказывая негативного влияния на организм и окружающую среду [64].
Знания особенностей гидролитического разложения полилактида и возможностей контроля за этими процессами важны для его промышленного производства и дальнейшего продвижения в области биомедицины [12]. Так, принято считать [127], что идеальная скорость разложения имплантата in vivo должна быть сопоставима или немного меньше скорости образования ткани. Например, после остеоинтеграции процесс формирования новой костной ткани и постепенной резорбции имплантата происходит в течение 0.5–1 года.
Деструкция может более интенсивно протекать либо на поверхности, либо в объеме материала, что определяется различными факторами: химическим и изомерным составом, молекулярно-массовыми характеристиками, наличием остаточного мономера, степенью ориентации и кристалличности полимера, морфологией и формой образца, а также условиями окружающей среды (рН, температура, влажность, присутствие ферментов, микроорганизмов и т.д.) [128]. При поверхностном разрушении реакция гидролиза преимущественно происходит на границе полимерного материала со средой во внешней диффузионно-кинетической области. Образующиеся продукты деструкции являются водорастворимыми и легко удаляются из зоны реакции, что приводит к постепенному уменьшению массы (объема) полимера. Нужно сказать, что подобный процесс поддается контролю, а скорость и степень разрушения полимера прямо пропорциональна изменению площади поверхности образца.
Полилактид является гидрофобным полимером, и для него при протекании гидролиза ММ обычно уменьшается быстрее, чем масса образца. Это связано с тем, что продукты разложения с М > 1 × 103 не растворяются в воде и остаются в объеме полимера. Следовательно, для материалов из полилактида более характерна объемная деструкция, когда реакция гидролиза сложноэфирных связей протекает одновременно во всем объеме (исключение составляет проведение гидролиза в сильнокислой при рН 2 или сильнощелочной среде при рН 12 и выше, когда процесс протекает преимущественно на поверхности). Причина заключается в автокаталитическом характере процесса, обусловленного невозможностью удаления продуктов деструкции – олигомеров молочной кислоты, концевые карбоксильные группы которых катализируют реакцию разрыва сложноэфирной связи. Установлено, что из-за медленной диффузии кислых продуктов деградации более толстые имплантаты разлагаются быстрее [129]. Подобный эффект был обнаружен при сравнении даже относительно тонких пленок из сополимера ПЛГ толщиной 10 и 100 мкм [130]. Важно отметить, что на начальных стадиях объемная деструкция носит вероятностный характер, часто зависит от скорости диффузии воды и доступности гидролитически активных центров, поэтому скорость разрушения полимерного материала в данном случае может изменяться неожиданным образом.
Для контроля над процессами деструкции полилактида используют различные подходы: гидрофилизацию поверхности материала, введение в объем гидрофильных наполнителей, в том числе наноразмерных, и ферментов, создание пористой структуры. В настоящем обзоре основное внимание будет уделено особенностям протекания гидролитического разложения и возможностям его контролируемого проведения в пористых материалах полилактида, полученных различными способами.
В работе [26] было показано, что значение Mw в непористом имплантате из сополимера ПЛГ за 8 недель выдерживания в деионизированной воде уменьшилось в 3 раза. В аналогичных по химическому составу материалах с порами 400–700 мкм, полученных методом спекания микросфер, за тот же период величина Mw снизилась менее чем в 2 раза независимо от размера пор. В качестве основной причины подобных различий указывается именно накопление и невозможность удаления из непористого имплантата кислых продуктов деструкции полимера.
Таким образом, скорость деструкции зависит не только от концентрации доступных для молекул воды гидролитически активных сложноэфирных групп полимера (а, значит, от скорости транспорта воды в объем материала), но и от возможности и скорости диффузии продуктов гидролиза в окружающую среду. В связи с этим основными параметрами пористой структуры, влияющими на разложение, являются не только объемная пористость, размер пор и их морфология, но в большей степени однородность распределения пор по размерам и по объему образца полилактида, а также их взаимосвязанность и доступность окружающей среды. В работе [127] исследовали процесс разложения in vitro трехмерных пористых 3D-матриксов из сополимера ПЛГ с пористостью 80–95% и размером пор 50–450 мм в натрий-фосфатном буфере (PBS) при 37°C в течение 26 недель. Было обнаружено, что образцы с большим размером пор и меньшей пористостью разрушались быстрее, чем образцы с меньшим размером пор и более высокой пористостью. Это объясняли тем, что материалы, содержащие относительно небольшое количество крупных пор, имеют более толстые стенки и меньшую площадь поверхности, что приводило к подавлению диффузии продуктов разложения и автокаталитическому характеру деструкции. Необходимо отметить, что какие-либо значимые изменения массы или механических характеристик для подобных пористых 3D-матриксов на основе полилактида [38] и его сополимеров ПЛГ [127] в результате выдерживания в растворе PBS при температуре 37°C, начинали происходить только через 8–12 недель. Отметим, что аналогичные сведения получены для матриксов из волокон полилактида диаметром 600–700 мкм, полученных методом электроспиннинга [50].
С повышением температуры реакция гидролиза сложноэфирных связей полилактида ускоряется, однако сохраняется характер влияния пористости на этот процесс. В работе [131] исследовали деструкцию пористых образцов с близкой пористостью (90–92 об. %), но различным распределением пор по размерам, полученных методом вымывания водорастворимой добавки (хлорид натрия). Реакцию гидролиза осуществляли в деионизированной воде при 60°C. В данном случае было отмечено, что непористые пластины и каркасы с большим размером пор (300–500 мкм) разрушаются быстрее – уже через 28 дней они теряли свою целостность. Уменьшение размера пор приводило к более медленной скорости потери массы образцами полилактида и изменению их структуры, и процесс продолжался примерно 49 дней. Наиболее устойчивыми к механическому разрушению оказались материалы с широким диапазоном размеров пор (90–500 мкм).
Более подробное исследование влияния геометрии и размера пор было проведено в работе [34]. Авторами описано поведение пористых каркасов из полилактида в среде, моделирующей внеклеточную жидкость организма, ионный состав которой близок к человеческой плазме крови, при температуре человеческого тела в течение 28 дней. Образцы были получены методом 3D-печати с использованием технологии быстрого прототипирования и содержали поры в виде сот, треугольной или квадратной формы с толщиной стенок 150, 250 и 350 мкм. Обнаружено, что через две недели в результате высаживания на стенках пор фосфатов кальция масса каркасов несколько увеличивалась, однако затем начинала падать в результате протекания процессов гидролитической деструкции. При этом наиболее интенсивно указанные процессы протекали в образцах с крупными порами треугольной формы (толщина стенок 150 мкм), а наименее интенсивно – в образцах с мелкими квадратными порами (толщина стенок 350 мкм). Симбатно этим процессам изменялась и прочность на сжатие, снижение которой для каркасов с крупными порами составляло более 20%, а с мелкими не превышало 6–8%. Исследование показало важность выбора оптимальных параметров пористой структуры каркасов, которые позволят не только сохранить их структурную стабильность и предотвратить преждевременную фрагментацию, но и смягчить вредное воздействие, вызванное снижением pH в процессе гидролитической деструкции.
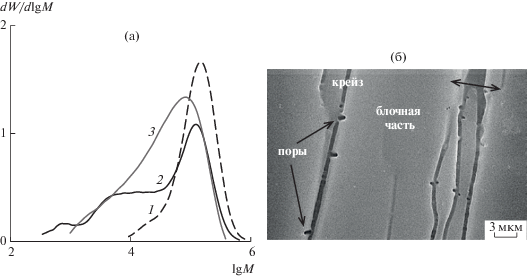
Описанные примеры влияния пористой структуры с уровнем размера пор десятки–сотни микрометров, сформированных самыми различными методами (спекание, 3D-печать, травление, вспенивание), показали, что пористость действительно способствует переводу реакции гидролиза полилактида в неавтокаталитический режим и значительному снижению скорости деструкции таких материалов. Однако в работе [132] было обнаружено, что создание открыто-пористой структуры с наноразмерными порами (10–25 нм) и высоко развитой удельной поверхностью фибрилл (75 м2/см3) по механизму крейзинга, напротив, ускоряет данный процесс. Связано это с ускорением разрыва химических связей при одновременном действии механического напряжения и гидролитически активной среды, а также благодаря высокой концентрации воды (20–30 мас. %), введенной в результате крейзинга в объем полимера, и доступных гидролитически активных сложноэфирных связей. Показано, что в отличие от исходной непористой изотропной пленки полилактида, для которой не наблюдали каких-либо значимых изменений молекулярно-массовых характеристик при выдерживании в растворе PBS при 37°C за весь период эксперимента (6 недель), для пористых структурно-модифицированных по механизму крейзинга материалов уже через одну–три недели происходило снижение среднечисловой и средневесовой ММ в несколько раз и увеличение дисперсности до 4–5. Локализацию процессов гидролитического разложения можно изменять путем варьирования структуры, формируемой в процессе крейзинга. Например, для пленок с чередованием крейзов и блочного полимера скорость деградации неодинакова по объему (наибольшая – на границе крейз–блочная непористая часть материала), что приводит к бимодальному характеру распределения ММ макромолекул в образцах (рис. 10а) и резкому их охрупчиванию через неделю выдерживания в PBS при 37°C. При этом рост пор, которые образуются в процессе деструкции, происходит вглубь блочного полимера (рис. 10б), что может указывать на ускоренное протекание деструкции в непористых частях образца, по-видимому, благодаря автокаталитическому характеру процесса. Для пленок полилактида, полностью перешедших в фибриллярно-пористую структуру при высоких степенях деформации (350%), с увеличением времени гидролитического разложения вид кривой ММР сохраняется (рис. 10а), но она постепенно смещается в область более низких ММ и становится шире. Для таких образцов процессы гидролитической деструкции протекают достаточно однородно по всему объему, и изменения структурно-механических параметров менее резкие, например, после шести недель гидролиза подобные пленки сохраняют свою прочность на уровне 90 МПа.
Рис. 10.
Кривые молекулярно-массового распределения исходного полилактида (1), полилактида, деформированного на 80 (2) и 350% (3), после выдерживания в течение пяти недель в растворе PBS при 37oC (а), а также СЭМ-микрофотография поверхности образца, деформированного на 80% (б), после выдерживания в течение пяти недель в растворе PBS при 37oC [132].
Для теоретического моделирования влияния различных параметров пористой структуры на процесс гидролитической деструкции полилактида в литературе описано несколько подходов. В работе [133] использовали алгоритм клеточного автомата для демонстрации влияния пористости каркаса (80, 90 и 95 об. %) на скорость разложения. Результаты показали, что образец с самой высокой начальной пористостью к концу теоретического эксперимента имел наибольшую остаточную массу, ММ и концентрацию сложноэфирных групп, а также наименьшее количество высвобождаемых и оставшихся в каркасе молекул олигомеров. Это свидетельствует о более легкой и быстрой диффузии олигомеров во время деструкции. В то же время моделирование не выявило образования каких-либо дополнительных полых структур ни в одном из рассмотренных пористых каркасов во время гидролиза, что указывает на отсутствие в них автокаталитического ускорения процесса.
Тот же алгоритм клеточного автомата был использован для моделирования влияния геометрического размера каркаса полилактида с начальной пористостью 90 об. % на его поведение в процессе гидролитической деструкции [134]. Четыре пористых образца разного размера были созданы с помощью метода повторяющихся единиц. Расчет показал, что потеря массы связана с геометрическим размером пористого каркаса и не зависит от ММ полимера. Это свидетельствует о том, что размерный эффект особенно важен при использовании образцов небольшого размера и с низкой пористостью.
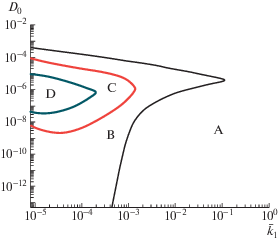
В работе [135] рассматривалось влияние еще одного важного параметра пористой структуры, а именно, взаимосвязанность и открытость пор (доступность окружающей среды). Материалы с закрытыми порами ячеистого типа и с открыто-пористой взаимопроникающей структурой из полилактида и его сополимеров ПЛГ разного состава были получены методом вспенивания сверхкритическим СО2 в контролируемых условиях. Для теоретического описания процессов деструкции в таких материалах использовали модель реакция–диффузия, которая учитывает автокаталитический механизм гидролиза и диффузию образующихся олигомеров. Для полиэфиров с произвольно распределенными изолированными порами эффективный коэффициент диффузии олигомеров зависит от величины объемной пористости, а скорость их диффузии в порах намного выше, чем в блочной непористой части полимерной матрицы. Важно отметить, что пористость в процессе деградации и вымывания олигомеров постепенно возрастает, начиная с ранних стадий деградации, что было продемонстрировано методом акустической микроскопии в работе [136]. Для таких систем с порами ячеистого типа важно соотношение двух параметров: k1, представляющего собой отношение скорости реакции некаталитического гидролиза к автокаталитической реакции, и Do, который вычисляется как отношение скорости диффузии к скорости автокаталитического гидролиза. На рис. 11 представлены диаграммы для биоразлагамых полиэфиров в широком диапазоне безразмерных коэффициентов диффузии и констант скорости некаталитического процесса разложения. Для полимеров, находящихся в зоне А, эффект от создания пористой структуры на скорость разложения незначителен. Но для полиэфиров, находящихся в зоне D, формирование пор является хорошим способом замедлить деградацию. Для подобных полимеров деструкция вспененного образца происходит намного медленнее, чем массивного непористого материала.
Рис. 11.
Диаграммы для биоразлагаемых полиэфиров в широком диапазоне безразмерных коэффициентов диффузии и констант скорости некаталитического процесса разложения [135]. Пояснения в тексте.
Деградация открыто-пористых материалов в основном определяется толщиной стенок пор, через которые происходит наиболее медленная стадия диффузии олигомеров. В этом случае предполагается, что путь диффузии олигомеров проходит от центральной части стенок пор к их поверхности, и размер стенок пор и их распределение по размерам становятся ключевыми структурными параметрами. А именно, чем тоньше толщина стенки поры, тем быстрее происходит диффузия олигомерных продуктов гидролиза, отсутствует их накопление, что обусловливает протекание гидролиза в режиме более медленного некаталитического процесса. Так, расчеты предсказывают, что снижение величины среднечисловой ММ для матрикса с толщиной стенки 2.5 мкм составляет порядка 10% за 50 дней, в случае увеличения толщины стенки до 32.5 мкм уменьшение величины Mn может достигать 60%.
Таким образом, придание полилактиду пористой структуры действительно является эффективным инструментом, который позволяет процесс гидролитической деструкции полимера переводить в контролируемый и предсказуемый режим протекания с целью создания функциональных материалов с прогнозируемыми сроками разложения.
ЗАКЛЮЧЕНИЕ
В настоящее время доля биоразлагаемых пластиков на мировом рынке чрезвычайно мала (порядка 1%), что связано с достаточно высокой их себестоимостью. Однако наблюдается тенденция к расширению этого рынка, и потенциал его огромен [137]. Основные области использования биопластиков включают как биомедицинские, так другие направления (например, в сельском хозяйстве, автомобилестроении, производстве игрушек, спортивного инвентаря и т.п.) [138, 139]. Наиболее частой сферой применения биоразлагаемых пластиков являются тара и упаковка для пищевых продуктов.
Из всего разнообразия биоразлагаемых материалов, которые сейчас производятся, полилактид занимает особое место, благодаря оптимальному сочетанию цены и его физико-механических характеристик. На рынке биопластиков на него приходится более 10% объема всей выпускаемой продукции, что составляет ежегодно более 200 тыс. тонн [137]. По прогнозам, к 2023 г. количество выпускаемой продукции на основе полилактида удвоится. Модификация данного полимера за счет создания высокой пористости с контролируемой морфологией и размером пор существенно расширяет области потенциального использования материалов на его основе. Совершенствование методов создания пористой структуры за счет комбинации различных подходов с учетом экологических проблем – приоритетное направление развития биоразлагаемых полимеров. Работы последних десятилетий в основном направлены на создание нового поколения изделий медицинского назначения: рассасывающихся шовных нитей, раневых покрытий, дифференцированных в зависимости от типа раны и фазы раневого процесса, 3D-матриц для тканевой инженерии, полимерных имплантатов, систем контролируемого выделения лекарственных препаратов [140, 141]. В последнее время пористые каркасы на основе полилактида с контролируемым размером пор и степенью их взаимосвязанности предлагается рассматривать в качестве основы для моделирования раковых опухолей [142]. Эти системы перспективны для in vitro скрининга противоопухолевых препаратов c имитацией in vivo условий.
Четкий контроль морфологии пористых материалов из полилактида открывает новые возможности использовать их как основу изделий биомедицинского назначения и как первопарационные, микрофильтрационные и разделительные мембраны, сорбенты, “умную” упаковку и дышащие текстильные изделия, а также пенопласты. Материалы из полилактида с пористостью выше 90% и большим многообразием морфологических видов, благодаря своей гидрофобности, высокой пористости и капиллярным эффектам в комбинации с биодеградируемостью потенциально позиционируются как экосорбенты масел и нефтепродуктов [68, 98]. Достаточно интересно применение пористых мембран из полилактида в сельском хозяйстве в качестве покрытий для гранул удобрений с их контролируемым высвобождением [99].
В перспективе материалы на основе биоразлагаемых полимеров могут составить реальную конкуренцию крупнотоннажным пластмассам, полученным из нефтехимического сырья, и успешно заменять их. В этом направлении более жесткие экологические требования к производству материалов и их свойствам способствуют успешному продвижению биопластиков.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-29-17016).
Список литературы
d’Ambrières W. // Field Actions Sci. Reports. 2019. V. 19. P. 12.
Lebreton L., Andrady A. // Palgrave Commun. 2019. V. 5. № 1. P. 1.
Casalini T., Rossi F., Castrovinci A., Perale G. // Front. Bioeng. Biotechnol. 2019. V. 7. P. 259.
Korzhikov V.A., Vlakh E.G., Tennikova T.B. // Polym. Sci. Ser. A. 2012. V. 54. № 8. P. 585.
Shah Mohammadi M., Bureau M.N., Nazhat S.N. // Biomedical Foams for Tissue Engineering Applications. Cambridge: Woodhead Publ. Limited, 2014.
Narayanan G., Vernekar V.N., Kuyinu E.L., Laurencin C.T. // Adv. Drug Delivery Revs. 2016. V. 107. P. 247.
Santoro M., Shah S.R., Walker J.L., Mikos A.G. // Adv. Drug Delivery Revs. 2016. V. 107. P. 206.
Donate R., Monzón M., Alemán-Domínguez M.E. // e-Polymers. 2020. V. 20. № 1. P. 571.
Wu D., Xu F., Sun B., Fu R., He H., Matyjaszewski K. // Chem. Rev. 2012. V. 112 № 7. P. 3959.
Yong X., Hu Q., Zhou E., Deng J., Wu Y. // ACS Biomater. Sci. Eng. 2019. V. 5. P. 5072.
Babaie E., Bhaduri S.B. // ACS Biomater. Sci. Eng. 2017. V. 4. № 1. P. 1.
Abbasi N., Hamlet S., Love R.M., Nguyen N.-T. // J. Sci.: Adv. Mater. Devices. 2020. V. 5. P. 1.
Ambekar R., Kandasubramanian B. // Ind. Eng. Chem. Res. 2019. V. 58. P. 6163.
Stucki M., Loepfe M., Stark W.J. // Adv. Eng. Mater. 2017. V. 20. № 1. Art. 1700611.
Santos-Rosales V., Iglesias-Mejuto A., García-González C.A. // Polymers. 2020. V. 12. № 3. P. 533.
Borden M., Attawia M., Khan Y., Laurencin C.T. // Biomaterials. 2002. V. 23. № 2. P. 551.
Shi X., Chen S., Zhou J., Yu H., Li L., Wu H. // Adv. Funct. Maters. 2012. V. 22. № 18. P. 3799.
Huang W., Li X., Shi X., Lai C. // Biomater. Sci. 2014. V. 2. № 9. P. 1145.
Zhou W.Y., Lee S.H., Wang M., Cheung W.L., Ip W.Y. // J. Mater. Sci. Mater. Med. 2018. V. 19. № 7. P. 2535.
Lee S. H., Zho W. Y., Wang M., Cheung W. L., Ip W.Y. // J. Biomimetics, Biomaterials Tissue Eng. 2008. V. 1. P. 81.
Duan B., Wang M. // MRS Bull. 2011. V. 36. № 12. P. 998.
Yeong W.-Y., Chua C.-K., Leong K.-F., Chandrasekaran M. // Trends Biotechnol. 2004. V. 22. № 12. P. 643.
Singh M., Sandhu B., Scurto A., Berkland C., Detamore M.S. // Acta Biomaterialia. 2010. V. 6. № 1. P. 137.
Bhamidipati M., Sridharan B., Scurto A.M., Detamore M.S. // Mater. Sci. Eng. C. 2013. V. 33. № 8. P. 4892.
Mazzoli A. // Med. Biol. Eng. Comput. 2013. V. 51. № 3. P. 245.
Borden M., Attawia M., Laurencin C.T. // J. Biomed. Mater. Res. 2002. V. 63. № 3. P. 421.
Gordeev E.G., Ananikov V.P. // Russ. Chem. Rev. 2020. V. 89. № 12. P. 1507.
Almeida C.R., Serra T., Oliveira M.I., Planell J.A., Barbosa M.A., Navarro M. // Acta Biomaterialia. 2014. V. 10. № 2. P. 613.
Wai-Yee Yeong, Chee-Kai Chua, Kah-Fai Leong // Trends in Biotechnology. 2004. V. 22. № 12. P. 643.
Wang X., Jiang M., Zhou Z., Gou J., Hui D. // Composites B. 2017. V. 110. P. 442.
Ventrici de Souza J., Liu Y., Wang S., Dörig P., Kuhl T.L., Frommer J., Liu G. // J. Phys. Chem. B. 2017. V. 122. № 2. P. 956.
Gregor A., Filová E., Novák M., Kronek J., Chlup H., Buzgo M., Blahnová V., Lukášová V., Bartoš M., Nečas A., Hošek J. // J. Biol. Eng. 2017. V. 11. № 1. P. 31.
Serra T., Planell J.A., Navarro M. // Acta Biomaterialia. 2013. V. 9. № 3. P. 5521.
Singh D., Babbar A., Jain V., Gupta D., Saxena S., Dwibedi V. // J. Braz. Soc. Mech. Sci. Eng. 2019. V. 41. P. 121.
Kowalczyk P., Trzaskowska P., Łojszczyk I., Podgórski R., Ciach T. // Colloids Surf. B. 2019. V. 179. P. 136.
Liulan L., Li M., Zhou Q. // Polym. Sci, Ser. A. 2019. V. 61. P. 855.
Serra T., Mateos-Timoneda M.A., Planell J.A., Navarro M. // Organogenesis. 2013. V. 9. № 4. P. 239.
Rodrigues N., Benning M., Ferreira A.M., Dixon L., Dalgarno K. // Proc. CIRP. 2016. V. 49. P. 33.
Fairag R., Rosenzweig D.H., Ramirez-Garcialuna J.L., Weber M.H., Haglund L. // ACS Appl. Mater. Interfaces. 2019. V. 11. № 17. P. 15306.
Zhou C., Yang K., Wang K., Pei X., Dong Z., Hong Y., Zhang X. // Materials Design. 2016. V. 109. P. 415.
Malakhov S.N., Chvalun S.N. // Russ. J. Gen. Chem. 2017. V. 87. P. 1364.
Ding J., Zhang J., Li J., Li D., Xiao C., Xiao H., Yange H., Zhuanga X., Chen X. // Progr. Polym. Sci. 2019. V. 90. P. 1.
Katsanevakis E., Wen X., Zhang N. // Adv. Polym. Sci. 2011. V. 246. P. 63.
Leong M.F., Rasheed M.Z., Lim T.C., Chian K.S. // J. Biomed. Mater. Res. A. 2009. V. 91. № 1. P. 231.
Bhattarai D.P., Aguilar L.E., Park C.H., Kim C.S. // Membranes. 2018. V. 8. P. 62.
Afifi A.M., Nakajima H., Yamane H., Kimura Y., Nakano S. // Macromol. Mater. Eng. 2009. V. 294. № 10. P. 658.
Li L., Hashaikeh R., Arafat H.A. // J. Membr. Sci. 2013. V. 436. P. 57.
Badami A.S., Kreke M.R., Thompson M.S., Riffle J.S., Goldstein A.S. // Biomaterials. 2006. V. 27. № 4. P. 596.
Dasari A., Quirós J., Herrero B., Boltes K., García-Calvo E., Rosal R. // J. Membr. Sci. 2012. V. 405–406. P. 134.
Hoveizi E., Nabiuni M., Parivar K., Rajabi-Zeleti S., Tavakol S. // Cell Biol. Int. 2013. V. 38. № 1. P. 41.
Santos L.G., Oliveira D.C., Santos M.S. L., Neves L.M.G., de Gaspi F.O.G., Mendonca F.A.S., Esquisatto M.A.M., Santos G.M.T., Mei L.H.I. // J. Nanosci. Nanotechnol. 2013. V. 13. № 7. P. 4694.
Braghirolli D.I., Steffens D., Pranke P. // Drug Discovery Today. 2014. V. 19. № 6. P. 743.
Ramesh B., Cherian K.M., Fakoya A.O.J. // Methods Molec. Biol. 2019. V. 2125. P. 119.
Huang Ch., Thomas N.L. // Polym. Revs. 2020. V. 60. № 4. P. 595.
Zhang D., Jin X., Huang T., Zhang N., Qi X., Yang J., Wang Y. // ACS Appl. Mater. Interfaces. 2019. V. 11. P. 5073.
Medeiros E.L.G., Braz A.L., Porto I.J., Menner A., Bismarck A., Boccaccini A.R., Lepry W.C., Nazhat Sh.N., Medeiros E.S., Blaker J.J. // ACS Biomater. Sci. Eng. 2016. V. 2. № 9. P. 1442.
Prasad A., Sankar M.R., Katiyar V. // Materials Today: Proc. 2017. V. 4. P. 2. P. 898.
Ma P.X., Choi J.-W. // Tissue Eng. 2001. V. 7. № 1. P. 23.
Hou Q., Grijpma D.W., Feijen J. // Biomaterials. 2003. V. 24. № 11. P. 1937.
Mikos A.G., Thorsen A.J., Czerwonka L.A., Bao Y., Langer R., Winslow D.N., Vacanti J.P. // Polymer. 1994. V. 35. № 5. P. 1068.
Mao D., Li Q., Li D., Chen Y., Chen X., Xu X. // Mater. Design. 2018. V. 142. P. 1.
Chen V.J., Ma P.X. // Biomaterials. 2004. V. 25. № 11. P. 2065.
Zorin I.M., Zorina N.A., Bilibin A.Y. // Polym. Sci. Ser. A. 2010. V. 52. № 2. P. 150.
Guo Z., Yang C., Zhou Z., Chen S., Li F. // RSC Adv. 2017. V. 7. № 54. P. 34063.
Yao D., Zhang W., Zhou J.G. // Biomacromolecules. 2009. V. 10. № 5. P. 1282.
Esquirol A.-L., Sarazin P., Virgilio N. // Macromolecules. 2014. V. 47. № 9. P. 3068.
Sarazin P., Roy X., Favis B.D. // Biomaterials. 2004. V. 25. № 28. P. 5965.
Sun X.-R., Cao Z.-Q., Bao R.-Y., Liu Z., Xie B.-H., Yang M.-B., Yang W. // ACS Sustainable Chem. Eng. 2017. V. 5. № 9. P. 8334.
Nofar M., Ameli A., Park C.B. // Mater. Design. 2015. V. 83. P. 413.
Залепугин Д.Ю., Тилькунова Н.А., Мишин В.С., Глухан Е.Н., Чернышова И.В., Королев В.Л. // Сверхкритические флюиды: Теория и практика. 2007. Т. 2. № 1. С. 61.
García-González C.A., Concheiro A., Alvarez-Lorenzo C. // Bioconjugate Chem. 2017. V. 26. № 7. P. 1159.
Reignier J., Gendron R., Champagne M.F. // Cellular Polymers. 2007. V. 26. № 2. P. 83.
White L.J., Hutter V., Tai H., Howdle S.M., Shakesheff K.M. // Acta Biomater. 2012. V. 8. № 1. P. 61.
Kuang T., Chen F., Chang L., Zhao Y., Fu D., Gong X., Peng X. // Chem. Eng. J. 2017. V. 307. P. 1017.
Kiran E. // J. Supercrit. Fluids. 2010. V. 54. № 3. P. 296.
Ju J., Peng X., Huang K., Li L., Liu X., Chitrakar C., Chang L., Gu Zh., Kuang T. // Polymer. 2019. V. 180. Art. 121707.
Nofar M., Park C.B. Polylactide Foams: Fundamentals, Manufacturing, and Applications. Oxford, Cambridge: William Andrew, 2018.
Shibryaeva L.S., Krasheninnikov V.G., Gorshenev V.N. // Polym. Sci. Ser. A. 2019. V. 61. № 2. P. 162.
Nam Y.S., Yoon J.J., Park T.G. // J. Biomed. Mater. Res. 2000. V. 53. № 1. P. 1.
Julien J.-M., Bénézet J.-C., Lafranche E., Quantin J.-C., Bergeret A., Lacrampe M.-F., Krawczak P. // Polymer. 2012. V. 53. № 25. P. 5885.
Punet X., Levato R., Bataille I., Letourneur D., Engel E., Mateos-Timoneda M.A. // Polymer. 2017. V. 113. P. 81.
Gay S., Lefebvre G., Bonnin M., Nottelet B., Boury F., Gibaud A., Calvignac B. // J. Supercrit. Fluids. 2018. V. 136. P. 123.
Önder Ö.C., Yilgör E., Yilgör I. // Polymer. 2016. V. 107. P. 240.
Rezabeigi E., Wood-Adams P.M., Drew R.A.L. // Polymer. 2014. V. 55. № 14. P. 3100.
Rezabeigi E., Wood-Adams P.M., Drew R.A.L. // Polymer. 2014. V. 55. № 26. P. 6743.
Schugens C., Maquet V., Grandfils C., Jerome R., Teyssie P. // Polymer. 1996. V. 37. № 6. P. 1027.
Maquet V., Blacher S., Pirard R., Pirard J.-P., Jérôme R. // Langmuir. 2000. V. 16. № 26. P. 10463.
Grigoriev T.E., Bukharova T.B., Vasilyev A.V., Leonov G.E., Zagoskin Y.D., Kuznetsova V.S., Gomzyak V.I., Salikhova D.I., Galitsyna E.V., Makhnach O.V., Tokaev K.V., Chvalun S.N., Goldshtein D.V., Kulakov A.A., Paltsev M.A. // BioNanoSci. 2018. V. 8. P. 977.
Загоскин Ю.Д., Григорьев Т.Е., Крашенинников С.В., Куевда Е.В., Губарева E.A., Бакиров А.В., Чвалун С.Н. // Докл. РАН. 2019. Т. 486. № 4. С. 433.
Yang F., Qu X., Cui W., Bei J., Yu F., Lu S., Wang S. // Biomaterials. 2006. V. 27. № 28. P. 4923.
Lombardo M.E., Carfì Pavia F., Vitrano I., Ghersi G., Brucato V., Rosei F., La Carrubba V. // Tissue and Cell. 2019. V. 58. P. 33.
Севастьянов В.И., Григорьев А.М., Басок Ю.Б., Кирсанова Л.А., Василец В.Н., Малкова А.П., Духина Г.А., Григорьев Т.Е., Загоскин Ю.Д., Токаев К.В., Токаев Т.К. // Вестн. трансплантологии и искусственных органов. 2018. Т. 20. № 2. С. 82.
Vasilyev A.V., Kuznetsova V.S., Bukharova T.B., Grigoriev T.E., Zagoskin Yu.D., Galitsina E.V., Fatkhudinova N.L., Babichenko I.I., Chvalun S.N., Goldstein D.V., Kulakov A.A. // IOP Conference Series: Earth and Environmental Science. 2020. V. 421. № 5. Art. 052035
Aubert J.H., Clough R.L. // Polymer. 1985. V. 26. № 13. P. 2047.
Hua F.J., Park T.G., Lee D.S. // Polymer. 2003. V. 44. P. 1911.
Yang F., Murugan R., Ramakrishna S., Wang X., Ma Y.-X., Wang S. // Biomaterials. 2004. V. 25. № 10. P. 1891.
Phaechamud T., Chitrattha S. // Mater. Sci. Eng. C. 2016. V. 61. P. 744.
Yin G., Zhao D., Zhang L., Ren Y., Ji S., Tang H., Zhou Z., Li Q. // Chem. Eng. J. 2016. V. 302. P. 1.
Wang X., Zhang X., Han X., Liu K., Xu C., Hu X., Jin Z. // J. Appl. Polym. Sci. 2020. V. 138. № 2. Art. 49649.
Liu Q., Zhang P., Na B., Lv R., Tian R. // J. Phys. Chem. C. 2014. V. 118. № 44. P. 25620.
Gao A., Liu F., Shi H., Xue L. // J. Membr. Sci. 2015. V. 478. P. 96.
Tsivintzelis I., Marras S.I., Zuburtikudis I., Panayiotou C. // Polymer. 2007. V. 48. № 21. P. 6311.
Sattar M., Hayeeye F., Chinpa W., Sirichote O. // J. Environment. Chem. Eng. 2017. V. 5. № 4. P. 3780.
Galiano F., Ghanim A.H., Rashid K.T., Marino T., Simone S., Alsalhy Q.F., Figoli A. // Clean Technol. Environmental Policy. 2018. V. 21. P. 109.
Salerno A., Domingo C. // Microporous Mesoporous Mater. 2014. V. 184. P. 162.
Tsivintzelis I., Pavlidou E., Panayiotou C. // J. Supercrit. Fluids. 2007. V. 40. № 2. P. 317.
Pawlak A., Galeski A., Rozanski A. // Progr. Polym. Sci. 2014. V. 39. № 5. P. 921.
Crazing in Polymers / Ed. by H.H. Kausch. Berlin; Heidelberg: Springer-Verlag, 1990.
Michler G.H., Baltá-Calleja F.J. // Nano- and Micromechanics of Polymers. Munich: Carl Hanser Verlag, 2012.
Farah S., Anderson D.G., Langer R. // Adv. Drug Delivery Revs. 2016. V. 107. P. 367.
Wu S. // Polym. Eng. Sci. 1990. V. 30. № 13. P. 753.
Guo Y., Peng S., Wang Q., Song X., Li C., Xia L., Wu H., Guo S. // Industr. Eng. Chem. Res. 2020. V. 59. № 26. P. 12096.
Cheng J., Li H., Cao Zh., Wu D., Liu Ch., Pu H. // Mater. Sci. Eng. C. 2019. V. 95. P. 292.
Bitinis N., Sanz A., Nogales A., Verdejo R., Lopez-Manchado M.A., Ezquerra T.A. // Soft Matter. 2012. V. 8. P. 8990.
Wang Y., Li K., Zhao X., Tekinalp H., Li T., Ozcan S. // Macromolecules. 2020. V. 53. № 11. P. 4568.
Chen B.-K., Wu T.-Y., Chang Y.-M., Chen A.F. // Chem. Eng. J. 2013. V. 215.
Volynskii A.L., Bakeev N.F. Solvent Crazing of Polymers. Amsterdam: Elsevier, 1995.
Yarysheva A.Y., Bagrov D.V., Bakirov A.V., Yarysheva L.M., Chvalun S.N., Volynskii A.L. // Eur. Polym. J. 2018. V. 100. P. 233.
Hatada K., Fox R.B., Kahovec J., Marechal E., Mita I., Shibaev V. // Pure Appl. Chem. 1996. V. 68. P. 2313.
Трофимчук Е.С., Мальцев Д.К., Седуш Н.Г., Ефимов А.В., Никонорова Н.И., Гроховская Т.Е., Чвалун С.Н., Волынский А.Л., Бакеев Н.Ф. // Докл. РАН. Физика. 2014. Т. 459. № 6. С. 689.
Trofimchuk E.S., Efimov A.V., Grokhovskaya T.E., Nikonorova N.I., Moskvina M.A., Sedush N.G., Dorovatovskii P.V., Ivanova O.A., Rukhlya E.G., Volynskii A.L., Chvalun S.N. // ACS Appl. Mater. Interfaces. 2017. V. 9. № 39. P. 34325.
Trofimchuk E.S., Nikonorova N.I., Moskvina M.A., Efimov A.V., Khavpachev M.A., Volynskii A.L. // Polymer. 2018. V. 142. P. 43.
Trofimchuk E.S., Efimov A.V., Moskvina M.A., Ivanova O.A., Nikonorova N.I., Zezin S.B., Bakirov A.V., Volynskii A.L. // Polymer Science A. 2018. V. 60. № 5. P. 845.
Khavpachev M., Trofimchuk E., Nikonorova N., Garina E., Moskvina M., Efimov A., Demina V., Bakirov A., Sedush N., Potseleev V., Cherdyntseva T., Chvalun S. // Macromol. Mater. Eng. 2020. V. 305. № 7. Art. 2000163.
Trofimchuk E.S., Moskvina M.A., Ivanova O.A., Potseleev V.V., Demina V.A., Nikonorova N.I., Bakirov A.V., Sedush N.G., Chvalun S.N. // Mendeleev Commun. 2020. V. 30. P. 171.
Fadeeva I., Trofimchuk E., Giretova M., Mal’tsev D., Nikonorova N., Fomin A., Rau J., Medvecky L., Barinov S. // Biomed. Phys. Eng. Express. 2015. V. 1. № 4. Art. 045011.
Wu L., Ding J. // J. Biomed. Mater. Res. A. 2005. V. 75. P. 767.
Gorrasi G., Pantani R. // Adv. Polym. Sci. 2018. V. 279. P. 119.
Grizzi I., Garreau H., Li S., Vert M. // Biomaterials. 1995. V. 16. P. 305.
Lu L., Garcia C.A., Mikos A.G. // J. Biomed. Mater. Res. 1999. V. 46. P. 236.
Odelius K., Höglund A., Kumar S., Hakkarainen M., Ghosh A.K., Bhatnagar N., Albertsson A.-Ch. // Biomacromolecules. 2011. V. 12. № 4. P. 1250.
Trofimchuk E.S., Moskvina M.A., Nikonorova N.I., Efimov A.V., Garina E.S., Grokhovskaya T.E., Ivanova O.A., Bakirov A.V., Sedush N.G., Chvalun S.N. // Eur. Polym. J. 2020. V. 139. Art. 110000.
Chao G., Xiaobo Sh., Chenglin Ch., Yinsheng D., Yuepu P., Pinghua L. // Mater. Sci. Eng. C. 2009. V. 29. № 6. P. 1950.
Guo Ch., Sheng X., Chu Ch., Dong Y. // Adv. Mater. Res. 2011. V. 311–313. P. 1741.
Song C., Zhang J., Cen L., Xi Z., Zhao L., Yuan W. // Industr. Eng. Chem. Res. 2020. V. 59. № 33. P. 14806.
Morokov E.S., Demina V.A., Sedush N.G., Kalinin K.T., Khramtsova E.A., Dmitryakov P.V., Bakirov A.V., Gri-goriev T.E., Levin V.M., Chvalun S.N. // Acta Biomater. 2020. V. 109. P. 61.
Bioplastics market data 2018. Report. European Bioplastics, nova-Institute (Hürth, Germany). https://www.european-bioplastics.org/wp-content/uploads/2016/02/Report_Bioplastics-Market-Data_2018.pdf
Jamshidian M., Tehrany E.A., Jacquot M., Desorby S. // Comprehensive Revs Food Sci. Food Safety. 2010. V. 9. № 5. P. 552.
Rogovina S.Z. // Polymer Science C. 2016. V. 58. № 1. P. 62.
Agwuncha S.C., Sadiku E.R., Ibrahim I.D., Aderibigbe B.A., Owonubi S.J., Agboola O., Babul Reddy A., Bandla M., Varaprasad K., Bayode B.L., Ray S.S. // Handbook of Composites from Renewable Materials. 2017. V. 8. P. 135.
Singhvi M., Zinjarde S., Gokhale D. // J. Appl. Microbiol. 2019. V. 127. № 6. P. 1612.
Lomardo M.E., Pavia F.C. // Tissue Cell. 2019. V. 58. P. 33.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)