

Высокомолекулярные соединения (серия С), 2021, T. 63, № 2, стр. 111-129
РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ С ОБРАТИМОЙ ДЕАКТИВАЦИЕЙ ЦЕПИ С УЧАСТИЕМ НИТРОКСИЛОВ И “ЗЕЛЕНАЯ” ХИМИЯ
М. Ю. Заремский a, *, Н. С. Мелик-Нубаров a
a Московский государственный университет имени М.В. Ломоносова, Химический факультет
119992 Москва, Ленинские горы, Россия
* E-mail: zaremski@mail.ru
Поступила в редакцию 27.03.2021
После доработки 14.05.2021
Принята к публикации 28.05.2021
Аннотация
Обзор является первой попыткой рассмотреть применение некоторых принципов “зеленой” химии в радикальной полимеризации с обратимой деактивацией цепи с участием нитроксилов. Обсуждаются результаты контролируемого синтеза легко деградируемых полимеров, а также полимеров, полученных в условиях “зеленой” химии методами фото-полимеризации, полимеризации в сверхкритическом диоксиде углерода и полимеризации мономеров из возобновляемого сырья.
ВВЕДЕНИЕ
Общая концепция “зеленой” химии, включающая двенадцать принципов, была впервые сформулирована P.T. Anastas и J.C. Warner в 1998 г. в “American Chemical Society” [1, 2]. В дальнейшем эта концепция развивалась и корректировалась, переосмысливалась последовательность ее принципов, однако их основные положения и количество оставались неизменными [3]. Суть этих принципов состояла в следующем.
1. Предотвращение: лучше предотвратить отходы производства, чем их перерабатывать. Количественной мерой данного принципа служит Е-фактор – отношение веса отходов к весу целевого продукта [4–6]. Его величина меняется в широком диапазоне от 0.1 в нефтепереработке до 100 в фармацевтике. Применительно к синтезу полимеров этот критерий прежде всего означает организацию процесса так, чтобы мономер максимально превращался в полимер [5].
2. “Экономия” атомов: методы синтеза должны быть такими, чтобы исходные вещества максимально входили в продукт. Этот принцип характеризуется показателем АЕ, равным отношению молекулярной массы продукта к сумме молекулярных масс всех исходных веществ [7]. Второй критерий можно рассматривать как конкретизацию первого. Он подразумевает выбор тех способов синтеза, в которых помимо целевого вещества не было бы никаких побочных продуктов. К примеру, с точки зрения “зеленой” химии для получения полимеров полимеризация предпочтительнее, чем поликонденсация, поскольку в последней обычно кроме полимера образуется низкомолекулярный продукт. Так, найлон-6, получаемый полимеризацией с раскрытием цикла капролактама, имеет показатель АЕ близкий к 100%, а его аналог найлон-6,6, производимый поликонденсацией, – 91.3% [5].
3. Нетоксичность исходных и конечных веществ: методы синтеза следует выбирать так, чтобы исходные и получаемые вещества были как можно менее вредными для человека и окружающей среды. В полимеризационных процессах этот критерий в большинстве случаев будет выполнен только наполовину. Хотя большинство производимых полимеров безопасны для природы и человека, мономеры, используемые в их синтезе, как правило, токсичны.
4. Дизайн безопасных веществ: новые химические продукты должны обладать необходимыми свойствами, но с меньшей токсичностью. В этом отношении методы контролируемой радикальной полимеризации имеют как плюсы, так и минусы. С одной стороны, именно методы контролируемой радикальной полимеризации – это мощнейший инструмент для получения полимеров и особенно сополимеров заданного строения, в том числе градиентных, мультиблочных с необходимыми свойствами [8–12]. С другой стороны, такие методы используют в основном токсичные контролирующие агенты. В наибольшей степени это относится к полимеризации с переносом атома, приводящей к загрязнению продуктов токсичными солями меди и других тяжелых металлов; в наименьшей – к полимеризации с участием обратимых ингибиторов – нитроксилов.
5. Нетоксичные растворители и вспомогательные вещества: в химическом производстве необходимо избегать применения растворителей и других вспомогательных веществ, или они должны быть нетоксичны. Применительно к контролируемой радикальной полимеризации это означает, во-первых, поиск нетоксичных контролирующих агентов, и во-вторых, проведение процесса в экологически чистых средах, к которым относят не только водные растворы, но и ионные жидкости, а также различные водно-дисперсионные среды [5].
6. Минимизация энергетических затрат: химический синтез необходимо проводить при температуре, близкой к комнатной, и при атмосферном давлении. В области контролируемой радикальной полимеризации решение этой задачи направлено в основном на поиск новых контролирующих агентов, способных к осуществлению фото-полимеризации.
Стоит отметить, что одна из первых попыток решить проблему минимизации энергетических затрат в полимеризационном синтезе была предпринята советскими химиками-теоретиками еще в 70-х годах прошлого века – была разработана модель непрерывного реактора с использованием саморазогрева реакционной смеси за счет тепла, выделяющегося в экзотермической реакции полимеризации [13].
На самом деле, энергетические затраты собственно на поддержание необходимой температуры полимеризации – это малая часть (<10%) затрат на весь процесс производства [14].
7. Использование возобновляемых ресурсов. Под возобновляемыми ресурсами понимаются сельскохозяйственное сырье и отходы других производств в отличие от нефти, газа и других природных ископаемых. Эта область в синтезе полимеров только недавно начала разрабатываться, хотя ресурс мономеров на основе биосырья достаточно разнообразен [15]. Количественной мерой данного критерия служит углеродный баланс ΔC – разность количества углерода, потребленного из воздуха и выделенного обратно в ходе синтеза и при расходе топлива. Должно выполняться правило ΔC ⪢ 0.
8. Сокращение промежуточных стадий: использование промежуточных стадий, например, защита реакционноспособных групп и удаление защиты, должно быть минимизировано, так как лишние стадии могут вызывать дополнительное загрязнение окружающей среды. В качестве успешного применения контролируемой радикальной полимеризации для решения данной задачи в обзоре [5] приведен пример синтеза поли-2-гидроксиэтилметакрилата. Ранее для этого применяли живую анионную полимеризацию с переносом группы [16], которая включает две дополнительных стадии – защиту ОН-группы мономера и удаление защиты после полимеризации. Использование полимеризации с переносом атома позволяет получить тот же полимер напрямую из мономера [17].
9. Катализ: лучше применять каталитическое количество веществ, чем стехиометрическое [18]. Под каталитическим подразумевается концентрация 10–9–10–3 моль/л [5]. Однако контролирующие агенты радикальной полимеризации не подходят под этот критерий. Например, галогениды металлов, которые используют в полимеризации с переносом атома, за исключением варианта ARGET ATRP [19], требуется применять в количестве на порядок выше верхней границы указанного интервала для эффективного контроля ММ полимера. Такому критерию удовлетворяют лишь металлоцены и пост-металлоцены, как, например, система Cp2ZrCl2–метилалюмоксан с призводительностью до 3 т ПЭ/ч г Zr [20].
10. Дизайн для биодеградации: после использования химические продукты должны легко разлагаться и не накапливаться в окружающей среде. В данном случае, все усилия полимерщиков-синтетиков были и остаются реализуемыми в обратном направлении – создании материалов устойчивых к любому внешнему воздействию. Среди биодгерадируемых синтетические полимеры занимают небольшую долю [21]. В “доковидную эпоху” мировое производство полимеров составляло 350 мегатонн в год, из них биодеградируемых немного меньше одной мегатонны [22].
11. Контроль в реальном времени для предотвращения загрязнения: необходимо продолжить разработку аналитических методологий, позволяющих осуществлять мониторинг и контроль в реальном времени в процессе производства во избежание образования опасных веществ.
12. Предотвращение аварий: вещества, используемые в химическом процессе, должны минимизировать вероятность аварии (выброса, взрыва, возгорания). Количественным выражением данного принципа служит параметр, равный количеству потерянных лет жизни на тонну продукта. В полимерном производстве этот показатель не сильно меняется для разных процессов и находится в интервале 0.002–0.005 [23].
Впервые применимость принципов “зеленой” химии к получению полимеров была всесторонне проанализирована в обзорной статье M.А. Dubе́ и S. Salehpour в 2014 г. [5]. Подводя итоги, авторы сделали вывод, что в выполнении принципов 2, 8, 9, 11 и 12 в полимерном синтезе уже достигнуты большие успехи; решение проблем, обозначенных в 1, 6 и 10, интенсивно разрабатывается; a наибольший потенциал на будущее заложен в принципах 3, 4, 5 и 7.
В настоящее время уже опубликовано несколько обзорных статей, в которых обсуждается применение принципов “зеленой” химии в процессах контролируемой радикальной полимеризации. Бόльшая часть из них относится к полимеризации с переносом атома – как самому “грязному”, с точки зрения “зеленой” химии, методу контролируемой полимеризации. Надо заметить, что первая обзорная статья в этой области появилась на несколько лет раньше [24], чем упомянутая выше. Естественно, основное внимание было уделено уменьшению содержания остатков катализаторов – солей меди – в полимерах, получаемых с помощью полимеризации с переносом атома, а также поиску альтернативных катализаторов переноса атома на основе железа или неметаллических соединений [25–30 ]. Известны обзоры, затрагивающие проблемы “зеленой” химии в процессах с обратимой и вырожденной передачей цепи [31–33]. В качестве “зеленого” направления в контролируемой радикальной полимеризации особо выделяется фотополимеризация как подход, который позволяет не только снизить энергетические затраты, но и в ряде случаев заменить токсичные контролирующие агенты на нетоксичные, а также избавиться от инициатора [24, 27, 31].
Насколько известно, подобного рода обзоры в области контролируемой полимеризации с участием нитроксилов в качестве контролирующих агентов до сих пор отсутствуют. Поэтому настоящую работу следует рассматривать как первую попытку проанализировать процессы радикальной полимеризации в условиях обратимого ингибирования с точки зрения подходов “зеленой” химии.
Привлекательность нитроксильных радикалов для контролируемого синтеза полимеров относительно принципов “зеленой” химии характеризуется их низкой токсичностью. Биологические эффекты нитроксилов определяются тем, что свободные радикалы отчасти имитируют активность фермента супероксиддисмутазы (СОД) [34]. Данный фермент восстанавливает супероксид анион-радикала (ОО–•), являющийся одним из наиболее опасных активных форм кислорода, образующегося в митохондриях в процессе клеточного дыхания. При этом формируется существенно менее сильный окислитель – пероксид водорода. В клетке нитроксильные радикалы легко восстанавливаются до соответствующих гидроксиламинов под действием супероксида, глутатиона или аскорбиновой кислоты [35–37]. Вследствие чего нитрокислы проявляют ярко выраженные антиоксидантные и радиопротекторные свойства [38, 39] и препятствуют развитию воспаления сосудов глаза [40]. В ряде работ отмечается противоопухолевая активность нитроксильных радикалов [41, 42].
Наличие неспаренного электрона позволяет использовать нитроксильные радикалы в медицине в исследованиях МРТ в качестве контрастирующих агентов, ускоряющих спиновую релаксацию, таким образом усиливая сигнал и увеличивая разрешение [43, 44].
Цитотоксичность нитроксилов, проявляемая на культурах клеток, регистрируется лишь в миллимольных концентрациях, причем пятичленные циклические нитроксильные радикалы более токсичны, чем шестичленные производные ТЕМПО [45]. Показано, что в миллимольных концентрациях нитроксилы демонстрируют мутагенные свойства, однако такие концентрации практически никогда не достигаются при исследовании биологических эффектов полимеров, синтезированных с помощью полимеризации с их участием [46]. Полимеры, полученные данным методом, не проявляют заметной цитотоксичности при концентрациях порядка 1 мг/мл [47]. Поэтому данный метод контролируемой радикальной полимеризации в наибольшей степени отвечает принципам “зеленой” химии.
ФОТОПОЛИМЕРИЗАЦИЯ С УЧАСТИЕМ НИТРОКСИЛОВ
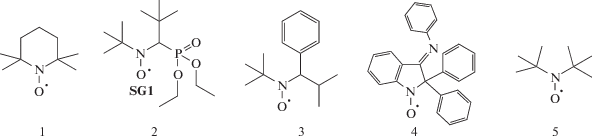
Полимеризация по механизму обратимого ингибирования с участием нитроксилов основана на обратимом захвате ими радикалов роста. Один из существенных недостатков данного метода состоит в необходимости поддержания высокой температуры [48–50]. Для большинства широко используемых нитроксилов: ТЕМПО (1), N-трет-бутил-N-[1-диэтилфосфоно-(2,2-диметилпропил)]нитроксил (SG-1, 2), трет-бутил-1-фенил-2-метилпропилнитроксил (ТФИН, 3), 2,2-дифенил-3-фенилимино-2,3-дигидроиндол-1-илоксил (4) и ди-трет-бутилнитроксил (5) необходима температура полимеризации 100–130°C. Даже для более эффективных агентов, таких как имидазолиновые нитроксилы и SG-1, механизм обратимого ингибирования реализуется выше 70–80°С. Поэтому поиск нитроксилов, “работающих” при низких значениях температуры остается актуальной задачей [51–54]:
Однако, как отмечалось выше, понижение температуры реакции считается не очень эффективным приемом для осуществления 6-го принципа “зеленой” химии – уменьшения энергетических затрат, поскольку поддержание температуры реакции – лишь небольшая часть общих технологических затрат производства. В этом отношении, представляет интерес фотополимеризация не столько как метод, позволяющий снизить энергетические затраты, но, что более существенно, – как метод, который может привести к практически полному прекращению протекания основной побочной реакции – реакции диспропорционирования между нитроксилом и радикалом роста, которая не только дает побочные продукты – гидроксиламины, но и уменьшает общий выход полимера, нарушает контроль ММ и уширяет ММР продукта. Особенно остро эта проблема стоит в полимеризации ММА и других метакриловых мономеров [55, 56].
Возможность осуществления фотополимеризации с участием нитроксилов была предсказана за десять лет до ее реализации [57]. Для этого использовали два подхода – введение хромофорной группы в нитроксил [58] и фотосенсибилизацию.
Первые попытки применения фотоактивных алкоксиаминовых инициаторов для контролируемой радикальной полимеризации оказались неудачными. В работе [58] были получены четыре инициатора на основе ТЕМПО: три соединения содержали бензоилформильную, бензофенонформильную и ксантон-3-илоксиацетную группу в нитроксиле и одно – бензоилбензойную группу в алкильном фрагменте. К сожалению, ни одно из них не инициировало фотополимеризацию стирола, и только алкоксиамин, имеющий в своем составе ксантон-замещенный нитроксил, инициировал полимеризацию ММА по “неживому” механизму однократного инициирования.
Введение более эффективной хинолиновой “антенны” в ТЕМПО, хотя и позволило осуществить фотополимеризацию стирола, но после 2 ч облучения реакционной смеси УФ-светом образовывалось не более 10% ПС с Mn = 2.7 × 103 и Đ = 1.6 [59]. С точки зрения контроля ММ, этот нитроксил не оправдал ожиданий и в полимеризации ММА, но он оказался эффективным в стерео-контроле макромолекул: в его присутствии доля синдио-триад в ПММА повышалась с 46 до 60% [60].
Исследование фотодиссоциации шести алкоксиаминовых инициаторов (6–11), содержащих фотоактивную бензофеноновую группу в различных положениях по отношению к связи N–О, методом ЭПР показало, что константа скорости фотолиза прямо пропорциональна интенсивности света и с точностью до порядка пропорциональна коэффициенту экстинкции алкоксиамина [61]:
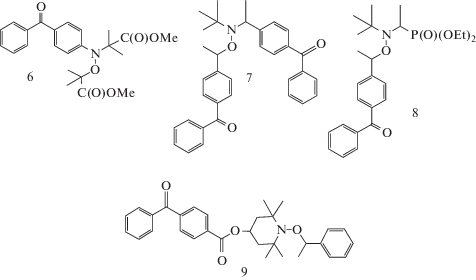
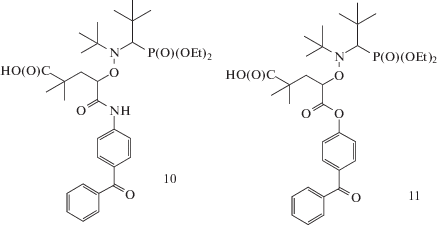
Главным фактором, влияющим на механизм фотолиза, является удаленность хромофорной группы от аминоксильной связи [62]. Если π-система хромофора непосредственно сопряжена с нитроксильной группой (соединение 6), то возможен распад по связям С–О и О–N. Для селективного раскрытия связи С–О наиболее благоприятно нахождение хромофора в α-положении по отношению к нитроксильной группе в инициаторах 7 и 8. При этом наличие второй группы бензофенона в нитроксильном фрагменте 7 не оказывает влияния на фотолиз инициатора. Наименьшей фотоактивностью, как и следовало ожидать, обладают инициаторы 9–11, с удаленной хромофорной группой.
Действительно, алкоксиамин 6, который дает при фотолизе не только нитроксильные, но и алкоксильные и азот-центрированные радикалы, легко инициирует фотополимеризацию бутилакрилата [58, 63]. Скорость процесса вчетверо выше, чем с участием инициатора 9. Конверсия достигает 80% за 8 мин при комнатной температуре. ММ полимера увеличивается с ростом конверсии, но оказывается вдвое выше теоретической, и дисперсность по ММ к концу полимеризации повышается до четырех. Однако такой же неконтролируемый процесс протекает и при фотополимеризации изоборнилакрилата, инициированной алкоксиамином 7, который фотолизуется исключительно с образованием нитроксилов [64]. Заметим, что термоинициированная полимеризация стирола при 120°С под действием алкоксиаминов 7 и 11 протекает в строго контролируемом режиме; в тех же условиях при полимеризации изоборнилакрилата контроль характеристик ММ значительно хуже [64]. В сочетании с триэтиламином соединение 7 работает как хорошо известная редокс-пара бензофенон–амин, инциирующая обычную радикальную полимеризацию [64].
На примере полимеризации бутилакрилата под действием алкоксиамина 12 было показано, что чем выше интенсивность УФ-облучения (λ = = 360 нм), тем выше скорость фотополмеризации, и тем ближе значения ММ к теоретическим; при этом дисперсность продукта изменялась от 1.5 до 2.0 [63]. Стирол в тех же условиях вообще не полимеризовался, а полимеризация ММА прекращалась при 35%-ной конверсии на стадии образования олигомеров. Замена бензофеноновой “антенны” на нафталиновую 13 или пиреновую 14 не привела к улучшению контроля ММ. В последнем случае облучение вели видимым светом:
В работе [65] была изучена фотополимеризация ММА под действием бинарной системы, состоящей как из фотоактивного инициатора, так и фотоактивного нитроксила, взятых в эквимольном количестве: 2,2-диметокси-2-фенилацетофенон и 4-замещеннный ТЕМПО. В качестве последнего использовалось три радикала:
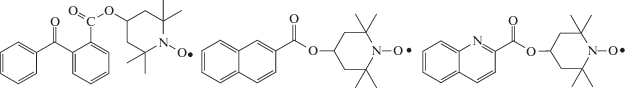
Для всех систем наблюдались близкие значения скорости полимеризации. ММ хотя и увеличивалась в ходе процесса, но была заметно выше теоретической, а дисперсность изменялась от 1.3–1.4 на начальных и средних конверсиях до значений выше 2.0 на глубоких.
Природа алкильного фрагмента алкоксиаминового фотоинициатора оказывает существенное влияние на его фотолиз. В ряду соединений, содержащих один и тот же изоиндолиновый нитроксильный фрагмент с антрахиноновой “антенной” [66]:
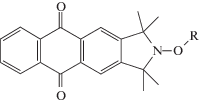 ,
,
где R – α-метилбензил (15), бензил (16), пентил (17), HOC(=O)(CH3)CН• (18), (CN)(CH3)CН• (19), EtOC(=O)(CH3)2C• (20); далеко не все способны распадаться под действием УФ-света. Только три из них – соединения 15, 20 и 16, т.е. низкомолекулярные модели нитроксильных аддуктов ПС и полиэтилметакрилата, диссоциируют на радикалы. Остальные, в том числе модели ПАН (19) и полиметилакрилата (18) были фотоустойчивы в течение 8 ч. Такое же различие было получено и для алкоксиаминов на основе изоиндолинового нитроксила с активной нафтильной “антенной”. Полоса поглощения данной группы сдвинута в коротковолновую область, т.е. в область квантов света с бόльшей энергией. Можно было ожидать, что стирол и алкилметакрилаты будут способны к фотополимеризации под действием фотоактивных производных этого нитроксила, а алкилакрилаты и акрилонитрил – нет. На практике оказалось, что ни стирол, ни бутил-акрилат вообще не полимеризуются при УФ-облучении в присутствии соединения 15 и его нафтил-замещенного аналога. Заметим, что в работе [67] синтезировано более тридцати различных изоиндолиновых алкоксиаминов, содержащих бензофеноновую, флуореноновую, антрахиноновую, нафтильную, антраценильную и другие группы, и изучена фотохимия; однако сведения об их применении в фотополимеризации, насколько известно, ограничиваются лишь несколькими, перечисленными выше.
Неэффективной оказалась попытка осуществить фотополимеризацию с использованием фотоактивных макронитроксилов, которые получаются при полимеризации in situ из нитронов с бензофеноновой (21), нафталиновой (22 и 23) и пиреновой “антенной” (24) [63, 66]:
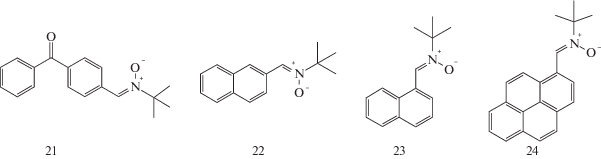
Полученные термической полимеризацией макроинициаторы ПС под действием систем ДАК-21 при 125°С и ДАК-22, ДАК-23 при 60°С, содержащие фотоактивную группу внутри цепи, работали как однократные фотоинициаторы полимеризации бутилакрилата с образованием блок-сополимера типа АВА:
Продукты содержали большое количество непрореагировавшего ПС. По механизму однократного фотоинциирования протекала и полимеризация бутилакрилата с участием ПС с бензофеноновой фотоактивной группой внутри цепи, полученного не полимеризационным способом, а по клик-реакции. Полистирол, синтезированный с системой ДАК-24, практически не инициировал фотополимеризацию. Заметим, что перечисленные образцы ПС аналогично ведут себя и при инициировании термической полимеризации бутилакрилата при 120°С.
Причиной нарушения контролируемого механизма фотополимеризации может быть деструкция ациклических нитроксилов под действием УФ-света. Так, облучение ди-трет-бутилнитроксила приводит к формированию нерадикального аддукта [68]:
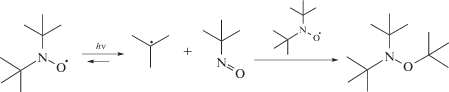
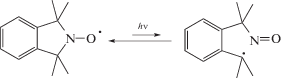
Напротив, циклические нитроксилы (ТЕМПО, проксил, изоиндолиновые) намного более стабильны, поскольку образующийся при их распаде нитрон легко захватывает находящийся вблизи С-центрированный радикал, что сдвигает равновесие диссоциации влево [69, 70]:
Длительное УФ-облучение ТЕМПО и его производных ведет не к его фотолизу, а к созданию гидроксиламина за счет отщепления нитроксилом протона из окружающей среды [71].
По мнению авторов, главная причина плохого контроля фотополимеризации с участием нитроксилов – низкая скорость реинициирования [66]. Не исключено, что нитроксил, нагруженный фотоантенной обладает и низкой ингибирующей активностью.
В свете изложенного выше, удивительными кажутся результаты китайских ученых [72]. Им удалось осуществить эмульсионную фотополимеризацию ММА под действием обычного нитроксила – 4-гидрокси-ТЕМПО и фотоинициатора Irgacure-184 при комнатной температуре. Несмотря на то что ни нитроксил, ни полимер не содержали хромофорных групп, авторы наблюдали линейный рост Mn полимера с конверсией и Đ = 1.27–1.36. К сожалению, механизм фотореинициирования не обсуждается.
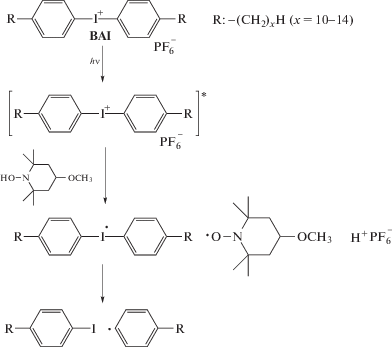
Второе направление – фотополимеризация с участием фотосенсибилизаторов – наиболее подробно было изучено в цикле трудов E. Yoshida [73] на примере полимеризации ММА, инициированной азоинициатором в присутствии 4-метокси-ТЕМПО и ряда катионных фотоинициаторов. Примером последнего служил бис-(алкилфенил)иодонийгексафторфосфат (алкил = С10–С14), который добавляли в количестве сопоставимом с нитроксилом. Первоначально предполагали, что эти соединения работают как фотокислотные генераторы в катионной полимеризации [74], но позже был доказан типичный радикальный фотосенсибилизационный механизм [75]. С введением соли диарилиодония скорость полимеризации и ММ полимера возрастали, при этом сильно уширялось ММР (Đ ~ 3.2). Сам сенсибилизатор в состав макромолекулы не входил: по данным спектроскопии ЯМР 1Н, полимер содержал на концах цепи осколок азоинициатора и нитроксил. При этом кинетика процесса подчинялась закону первого порядка по концентрации мономера, а Mn полимера линейно росла с конверсией.
Природа азоинициатора не оказывала значимого влияния на выход и молекулярно-массовые характеристики ПММА, которые определялись лишь мольным соотношением инициатор : нитроксил [76]. При эквивалентном соотношении полимеризация протекала количественно за 3 ч с образованием полимера с М = (30–40) × 103 и Đ > 3. При двукратном избытке метокси-ТЕМПО выход и ММ уменьшались в 2–3 раза, а дисперсия понижалась до 1.5–1.7.
Автором был предложен катион-радикальный механизм фотореинициирования полимеризации, включающий равновесие между полимер-нитроксильным аддуктом, радикалом роста ММА и катионом 4-метокси-ТЕМПО:
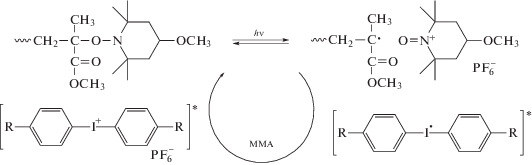
В пользу данного механизма свидетельствует тот факт, что при добавлении в систему трифениламина – вещества, разлагающего катионную форму нитроксила, фотополимеризация становилась неконтролируемой [75].
Аналогично вели себя и другие катионные фотоинициаторы – различные соли триарилсульфония, (η6-бензол)(η5-циклопентадиенил) Fe(II) гексафторфосфат [77, 78]. “Живой” механизм фотополимеризации ММА наблюдался и при замене 4-метокси-ТЕМПО на макронитроксил 4-(поли-ТГФ)–ТЕМПО [79].
Сообщалось о возможности использования данных систем для осуществления фотополимеризации ММА в ацетонитриле [77], а также дисперсионной полимеризации в среде метанол–вода [80]. Хотя в обоих случаях “живой” механизм полимеризации нарушался.
Аналогично протекала фотополимеризация глицидилметакрилата под действием трифлата (4-трет-бутилфенил)дифенилсульфония с участием нитроксила. В ходе реакции происходило раскрытие только двойных связей С=С мономера, но не эпоксицикла. Это доказывает радикальный механизм фотополимеризации [81].
Любопытно, когда в тех же условиях бинарную систему азо-инициатор–4-метокси-ТЕМПО заменяли на алкоксиаминовый инициатор 1-(циано-1-метилэтокси)-4-метокси-2,2,6,6-тетерметилпиеридин – аддукт осколка ДАК с тем же нитроксилом – контроль ММ полимера заметно уменьшился, а ММР уширилось [82]. При этом принципиально менялся механизм фотоинициирования полимеризации: соль иодония сама становилась инициатором, о чем свидетельствует наличие концевых фенильных групп в ПММА. Автором был предложен следующий механизм фотоинициирования [73]:
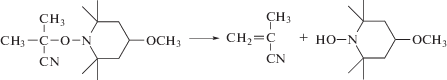
Таким образом, фотополимеризация хотя и позволяет осуществить полимеризацию с участием нитроксилов в более мягких условиях, но по степени контроля молекулярно-массовых характеристик она значительно уступает традиционной высокотемпературной. Тем не менее именно этот процесс уже активно применяется для получения люминисцентных покрытий, лазерной фотопечати и синтеза функциональных полимерных мультислоев на поверхности кремневых пластин [83–86]. Продолжаются и теоретические работы в этом направлении [87].
ДИСПЕРСИОННАЯ ПОЛИМЕРИЗАЦИЯ В СВЕРХКРИТИЧЕСКОМ ДИОКСИДЕ УГЛЕРОДА
Радикальная полимеризация в различных водно-дисперсионных средах (эмульсионная, микро- и мини-эмульсионная, суспензионная, осадительная) рассматривается в “зеленой” химии как экологически чистая альтернатива технологических процессов в массе или органических растворителях [24, 33, 88]. Контролируемая радикальная полимеризация с обратимой деактивацией цепи (с переносом атома и с нитроксильными радикалами) в гетерогенных условиях отличается особыми кинетическими эффектами, которые влияют на молекулярно-массовые характеристики продуктов [89, 90].
Различные виды дисперсионной полимеризации с участием нитроксилов интенсивно развивались практически сразу после открытия этого процесса. К настоящему времени накоплен огромный материал в этой области, обобщенный в нескольких обзорах [91–96]. В рамках данной работы мы ограничимся относительно новым и быстро развивающимся в течение последних 15 лет направлении – осадительной и дисперсионной полимеризации с участием нитроксилов в сверхкритическом СО2. Именно полимеризация с участием нитроксилов достигла существенно бо́льших успехов в этой области по сравнению с другими вариантами контролируемой радикальной полимеризации [92].
Для углекислого газа сверхкритическое состояние реализуется в относительно мягких условиях – Tcк = 31°C и Pcк = 7.4 MПa [97]. Этот нетоксичный, недорогой, негорючий и легко удаляющийся растворитель удовлетворяет принципам “зеленой” химии. Он характеризуется на порядок меньшей вязкостью и один–два порядка бóльшей диффузией реагентов в сравнении с обычными органическими средами. Почти все полимеры (за исключением фторированных и силиконовых) в сверхкритическом СО2 нерастворимы [92]. Поэтому полимеризация в сверхкритическом СО2 протекает в осадительном режиме.
Наиболее подробно полимеризация в сверхкритическом СО2 изучена на примере стирола в присутствии нитроксилов разной природы. Первые два патента на получение ПС с участием ТЕМПО в сверхкритическом СО2 были получены фирмой “Xerox” еще в 1996 г. [98, 99]. Последующие исследования проводились в аналогичных условиях (табл. 1). Полимеризация стирола под действием системы ДАК-SG-1 при 110°С идет до предельных конверсий; Mn нелинейно растет с конверсией, но Đ изменяется от 1.12 до 1.35 [100]. По молекулярно-массовым характеристикам ПС, полученный осадительной полимеризацией в сверхкритическом СО2, практически не отличается от полимера, синтезированного в тех же условиях в массе или в толуольном растворе при атмосферном давлении. Более того, при высоком содержании мономера (70 мас. %) его дисперсность даже ниже, чем у полученного в гомогенных условиях. Однако скорость осадительного процесса медленнее, чем в массе в 1.6 раза.
Таблица 1.
Условия полимеризации стирола в сверхкритическом СО2 и характеристики продукта
| Система | w*, % | Т, °С | P, МПа | Время, ч | Выход, % | Mn ×10–3 | Ð | Лите-ратура |
|---|---|---|---|---|---|---|---|---|
| ДАК-ТЕМПО–SO2 | – | 125 | 26.8 | 18 | – | 29.9 | 1.6 | [98] |
| Bz2O2–ТЕМПО– CF3(CF2)7SO3H | – | 125 | 26.7 | 24 | 86 | 1.67 | 1.6 | [99] |
| ДАК-SG-1 | 60 | 110 | 42 | 48 | 95 | 19.1 | 1.45 | [100] |
| 70 | 110 | 30 ± 5 | – | 82 | 23.8 | 1.36 | [101, 102] | |
| ДАК-ТФИН | 70 | 110 | 30 ± 5 | – | 29 | 16.0 | 1.26 | [101, 102] |
| соединение 25 | 40 | 110 | 30 | 61 | 39 | 20.0 | 1.29 | [103] |
| соединение 26 | – | – | – | 69 | 38 | 19.5 | 1.32 | – |
При замене нитроксила SG-1 на ТФИН скорость полимеризации в сверхкритическом СО2 понижалась примерно вдвое, ММ полимера возрастала и ММР уширялось [101]. Для достижения хорошего контроля ММ в сверхкритическом СО2 требовалось не менее двукратного избытка SG-1 и 3.3-кратного избытка ТФИН по отношению к ДАК. ММР полистирола, полученного с участием SG-1, не зависит от концентрации мономера, а с участием ТФИН – в 40%-ном растворе мономера ММР было уже, чем в 70%-ном. Такие отличия авторы связывали с разной растворимостью полярного SG-1 и неполярного ТФИН в сверхкритическом СО2 и стироле [102].
С целью увеличить растворимость нитроксила в сверхкритическом СО2 (и соответственно повысить контроль ММ) недавно были синтезированы новые алкоксиаминовые инициаторы для осадительной полимеризации стирола, содержащие перфторалкильные группы [103]:
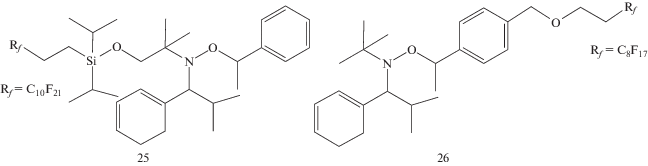
Однако скорость полимеризации оказалась ниже, чем с участием SG-1 или ТФИН. Молекулярно-массовые характеристики ПС не зависели от положения фторированной группы: в алкильном фрагменте инициатора или в нитроксиле (табл. 1) [104].
Интересно, что, когда в качестве инициаторов использовались полимерные алкоксиамины, содержащие аналогичные перфторалкилакрилатные заместители, полимеризация стирола вообще не шла.
В литературе описана осадительная полимеризация трет-бутилакрилата и диметилакриламида с участием нитроксилов в сверхкритическом СО2 [105]. Полимеризация первого мономера под действием системы ДАК-SG-1 (50 мас. %, 118°С, 30 МПа) протекает по механизму обратимого ингибирования: Mn линейно растет при увеличении конверсии и Đ составляет 1.3–1.7. Для второго мономера также наблюдается “живой” механизм полимеризации (20 мас. %, 120°С, 30 МПа). В обоих случаях необходим трехкратный избыток нитроксила по отношению к инициатору; доля “живых” цепей к концу процесса составляла 82%. Оба полимера способны реинициировать полимеризацию стирола с образованием блок-сополимеров [106].
Дисперсионную полимеризацию в сверхкритическом СО2 обычно осуществляют в присутствии амфифильного блок-сополимера; СО2-фильным блоком чаще всего служит полидиметилсилоксан. Наиболее подробно изучена дисперсионная полимеризация стирола под действием SG-1 [107–109].
В первом опыте дисперсионной полимеризации стирола с участием нитроксилов был использован “инициатор–стабилизатор”, в качестве которого применяли ПДМС VPS-0501, содержащий 6–10 азо-групп в цепи [107]:
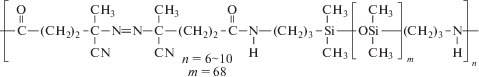
По мере распада азо-групп ПДМС инициирует полимеризацию, превращаясь при этом в блок сополимер, стабилизирующий дисперсию. Полимеризацию стирола проводили под действием системы VPS-0501/ SG-1(или ТЕМПО) при 100°С и давлении 42 МПа. В обеих системах конверсия была ниже 20%, а ММР – бимодальное. Последнее объясняли образованием двух типов блок-сополимера ПС–блок–ПДМС и ПС–блок–ПДМС–блок–ПС.
Чтобы осуществить контролируемую дисперсионную полимеризацию использовали несколько приемов: уменьшали концентрацию ПДМС с 20 до 1.5% и основное инициирование осуществляли с помощью ДАК; увеличивали отношение SG-1 к ДАК до 3.5; предварительно частично разлагали ПДМС, понижая число азо-групп в цепи до двух. В результате удалось получить блок-сополимер с выходом 90%, Mn = 5 × 104 и Ð = 1.6. При всех условиях наблюдали образование частиц неправильной формы с широким распределением по размерам.
Для полимера с унимодальным ММР в качестве стабилизатора брали полимер, получаемый дисперсионной полимеризацией с участием нитроксилов. Так, триблок-сополимер SG-1–ПС–блок–ПДМС–блок– ПС–SG-1, образующийся по методике, описанной выше, применяли в качестве инициатора–стабилизатора в полимеризации стирола в сверхкритическом СО2 [107]. Но и в этом случае наблюдали бимодальное ММР, что связывали с примесью монофункционального инициатора в блок-сополимере.
Полистирол с унимодальным ММР и Ð ~ 1.3 удалось получить, используя диблок-сополимер ПДМС–ПС–SG-1, который синтезировали полимеризацией с переносом атома с последующим радикальным замещением концевой группы Br на SG-1 [109]. Прослеживалось образование сферических частиц со средним диаметром 132 нм. Похожие результаты получили и в присутствии коммерчески доступного стабилизатора ПДМС–блок–ПММА (42 МПа, 110°С, 40%) [108].
Подход “инициатор–стабилизатор” оказался эффективным в дисперсионной сополимеризации ММА с 9% стирола в сверхкритическом СО2 (30 МПа, 70°С) [110]. Стабилизатором служил макроинициатор полигептадекафтордецилакрилат с концевой группой SG-1. Выход сополимера составил около 90%, Mn близка к теоретической и Đ = 1.26–1.31. Были получены сферические частицы с узким распределением по размерам 16 ± ± 2 мкм.
С помощью дисперсионной полимеризации с участием ТЕМПО представлен блок-сополимер ПС–блок–полибутилакрилат [111]. В качестве стабилизаторов синтезировали различные блок-сополимеры ПДМС–блок–ПС и поли(3,3,4,4,5,5,6,6,7,8,8,8-додекафтор-7-(трифторметил)октилметакрилат)–блок–ПС. Лучший контроль ММ наблюдали при использовании фторированного сополимера.
Для мономеров, плохо растворимых в сверхкритическом СО2, описана полимеризация в обратной суспензии [106, 112, 113]. Так, полимеризацию N-изопропилакриламида (НИПАМ) проводят при давлении 27–30 МПа, температуре 120°С и концентрации мономера 10 мас. % в присутствии ДАК и SG-1. В этих условиях НИПАМ плавится и образует капли суспензии в сверхкритическом СО2. В этих условиях достигается практически полное превращение мономера, Mn полимера линейно растет с конверсией и Đ изменяется от 1.3–1.4 в начале процесса до 2.0 на глубоких конверсиях. При последовательной полимеризации мономеров получили ряд блок-сополимеров полидиметилакриламид (ПДМА)–блок–ПНИПАМ, поли-трет-бутилметакрилат (ПТБА)–блок–ПНИПАМ, полиакриловая кислота–блок–ПНИПАМ и ПС–ПНИПАМ (табл. 2).
Таблица 2.
Молекулярно-массовые характеристики блок-сополимеров, полученных в сверхкритическом СО2 с участием нитроксила SG-1 [113]
| Блок-сополимер* | Mn, 103 | Ð |
|---|---|---|
| ПДМА58–ПНИПАМ82 | 13.4 | 1.5 |
| ПДМА58–ПНИПАМ117 | 21.4 | 1.6 |
| ПДМА58–ПНИПАМ217 | 29.2 | 2.1 |
| ПТБА62–ПНИПАМ81 | 20.6 | 1.4 |
| ПТБА62–ПНИПАМ192 | 30.7 | 1.3 |
| ПТБА62–ПНИПАМ254 | 34.6 | 1.3 |
| ПС62–ПНИПАМ266 | 34.2 | 1.5 |
В заключение данного раздела отметим практическое применение данного метода, направленное на синтез привитых сополимеров медицинского назначения [114]. Для чего прививали сополимер стирол–малеиновый ангидрид на высокомолекулярный хитозан в сверхкритическом СО2. Сначала путем химической модификации хитозана замещали 19% групп ОН на 4-гидрокси-ТЕМПО. Затем модифицированный хитозан инициировал привитую дисперсионную сополимеризацию стирола и малеинового ангидрида.
ПОЛИМЕРИЗАЦИЯ МОНОМЕРОВ БИОЛОГИЧЕСКОГО ПРОИСХОЖДЕНИЯ
Как уже отмечалось выше, полимеризация мономеров, вырабатываемых из возобновляемого природного сырья (как правило, растительного происхождения), рассматривается как одно из перспективных направлений в “зеленой” химии. В основном это мономеры для поликонденсации или ионной полимеризации с раскрытием цикла [115]; реже – для катионной и контролируемой радикальной полимеризации [116, 117].
Виниловые соединения биопроисхождения напрямую в контролируемой радикальной полимеризации используются крайне редко. Чаще всего специалисты находят компромиссное решение в виде мономеров, производимых химической модификацией биологического сырья, например производных итаконовой кислоты, (мет)акриловых эфиров терпенов, которые опробовали в полимеризации с переном атома и процессах с обратимой передачей цепи [115, 118–121].
Описан контролируемый синтез полимеров из терпеновых производных – цикладемолакрилата и метакрилата:
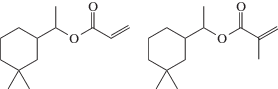 ,
,
а также тетрагидрогераниолакрилата и метакрилата:
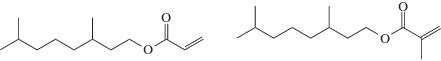
в толуоле и миниэмульсии под действием алкоксиамина “Dispolreg 007” 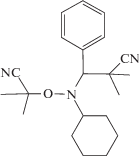 [122]. Полимеризация протекает до глубоких значений конверсии при 97°C. Контролируемый
режим реализуется до степени полимеризации Pn = = 200 при синтезе в растворе и Pn = 500 в миниэмульсии с дисперсностью Đ = 1.2–1.4.
[122]. Полимеризация протекает до глубоких значений конверсии при 97°C. Контролируемый
режим реализуется до степени полимеризации Pn = = 200 при синтезе в растворе и Pn = 500 в миниэмульсии с дисперсностью Đ = 1.2–1.4.
При последовательной полимеризации указанных метакрилатов получены блок-сополимеры типа “мягкий–жесткий” с Mn = (7–12) × 104, в которых эластичным блоком служило политетрагидрогераниоловое производное, а стеклообразным – полицикладемоловое производное [122].
Другой пример мономеров, синтезированных химической модификацией природного сырья,
– изоборнилметакрилат (ИБМА): 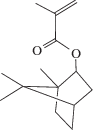 , получаемый из камфена и метакриловой кислоты, а также С-13–МА – смесь алкилметакрилатов
со средним числом атомов углерода в алкильной группе 13. Их полимеризация и сополимеризация
в толуольном растворе при 90–110°С, инициированная алкоксиамином “Dispolreg 007”,
протекает с неполной конверсией (<80%), что авторы связывали с высокой вязкостью полимеризата
[123]. В тех же условиях полимеризация в миниэмульсии происходит практически количественно.
Однако ММ в обоих случаях превышала теоретическую в несколько раз, а Đ составляла 1.39–1.66 при полимеризации в растворе и 1.50–1.73 в миниэмульсии. Аналогично
происходила и тройная сополимеризация указанных мономеров с метакрилоил-силсесквиоксаном
[124]. Последовательная полимеризация мономеров позволяет получить из них блок- и градиентные
сополимеры.
, получаемый из камфена и метакриловой кислоты, а также С-13–МА – смесь алкилметакрилатов
со средним числом атомов углерода в алкильной группе 13. Их полимеризация и сополимеризация
в толуольном растворе при 90–110°С, инициированная алкоксиамином “Dispolreg 007”,
протекает с неполной конверсией (<80%), что авторы связывали с высокой вязкостью полимеризата
[123]. В тех же условиях полимеризация в миниэмульсии происходит практически количественно.
Однако ММ в обоих случаях превышала теоретическую в несколько раз, а Đ составляла 1.39–1.66 при полимеризации в растворе и 1.50–1.73 в миниэмульсии. Аналогично
происходила и тройная сополимеризация указанных мономеров с метакрилоил-силсесквиоксаном
[124]. Последовательная полимеризация мономеров позволяет получить из них блок- и градиентные
сополимеры.
Миниэмульсионная блок-сополимеризация ИБМА и С-13–МА, инициированная макроинициатором – сополимером олигоэтиленгликольметакрилата с акрилонитрилом с концевой группой SG-1, также протекает по механизму обратимого ингибирования и сопровождается линейным ростом Mn продукта с конверсией, но Đ близка к двум [125].
Сравнительное исследование сополимеризации метилциннамата (метилового эфира коричной кислоты) под действием различных типов контролирующих агентов радикальной полимеризации – RuCpCl(PPh3)2 (полимеризация с переносом атома), 2-циан-2-пропилэтилтритиокарбоната (полимеризация с обратимой передачей цепи) и нитроксила ТФИН – показало возможность контроля во всех системах. Само производное как 1,2-дизамещенное виниловое соединение в гомополимеризацию не вступает, но легко сополимеризуется как со стиролом, так и с метилакрилатом. Во всех случаях рост цепи идет региоселективно путем атаки стирольного или акрилатного радикала по атому углерода, ближайшему к эфирному атому углерода метилциннамата, с образованием радикала роста стирола. Для указанных систем Mn линейно растет с конверсией и Đ = 1.05–1.20 при сополимеризации со стиролом и Đ = 1.2–1.4 – с метилакрилатом.
По механизму обратимого ингибирования с участием нитроксилов могут полимеризоваться и немодифицированные терпены – высокоактивные диены, такие как β-мирцен (7-метил-3-метилен-окта-1,6-диен) [126–128] – компонент эфирных масел лавра, иланг-иланга, дикого тимьяна, лимонника и можжевельника. Продукт его полимеризации ценен как низкотемпературный эластомер с температурой стеклования –75°С [129]:

Полимеризация β-мирцена под действием алкоксиаминового инициатора на основе SG-1 (“Bloc Builder”) протекает до 70%-ной конверсии за 200–400 мин [126]. Скорость реакции подчиняется закону первого порядка по мономеру, Mn продукта линейно растет с конверсией, а Đ близка к значению 1.2. Произведение константы скорости роста kp на константу равновесия К между “спящими” и растущими цепями характеризует “живость” процесса и по порядку величины kpK = = 4.3 × 10–5 c–1 отвечает значению, наблюдаемому при контролируемой полимеризации стирола. В тех же условиях β-мирцен легко сополимеризуется в контролируемом режиме со стиролом, образуя блок- и статистические сополимеры. В последнем случае его активность почти на порядок выше, чем у стирола (r1 = 1.88 ± 0.12, r2 = = 0.25 ± 0.04).
При сополимеризации β-мирцена с ИБМА под действием макроинициатора типа “Bloc Builder” на основе SG-1 при 100°С синтезированы градиентные сополимеры [127]. Активность β-мирцена выше, чем у ИБМА на два порядка (r1 = 1.90–2.16, r2 = 0.02–0.07), поэтому в ходе полимеризации формируются макромолекулы, “голова” которых обогащена терпеном, а “хвост” метакрилатом. Сам ИБМА в тех же условиях по механизму обратимого ингибирования не полимеризуется, поэтому его сополимеризация с мирценом происходит в затухающем режиме. Подробно контролируемый синтез статистических сополимеров с участием β-мирцена и указанного макроинициатора описан в диссертационной работе [128]. Интересно, что все синтезированные сополимеры имеют узкое ММР с ММ значительно ниже теоретической (табл. 3).
Таблица 3.
Молекулярно-массовые характеристики статистических сополимеров β-мирцена [113]
| Сомономер (М2) | Соотношение мирцен : М2 | Конверсия, % | Mn × 10–3 | Mn(теор) × 10–3 | Ð |
|---|---|---|---|---|---|
| Метилакрилат | 1 : 9 | 46 | 5.5 | 29 | 1.23 |
| трет-Бутилакрилат | 1 : 9 | 65 | 16.7 | 40 | 1.31 |
| трет-Бутилакрилат | 3 : 7 | 66 | 10.4 | 43 | 1.19 |
| Метилметакрилат | 1 : 1 | 85 | 6.4 | 39 | 1.25 |
| Метилметакрилат | 1 : 1 | 59 | 8.4 | 30 | 1.24 |
| Стирол | 9 : 1 | 47/39* | 12.6 | – | 1.25 |
| 7 : 3 | 41/37* | 8.4 | – | 1.24 | |
| 1 : 1 | 50/45* | 11.7 | – | 1.19 | |
| 3 : 7 | 54/45* | 13.9 | – | 1.24 | |
| 1 : 9 | 84/78* | 19.8 | – | 1.31 | |
| Изоборнилметакрилат | 9 : 1 | 86/36* | 18.3 | – | 1.57 |
| 7 : 3 | 84/32* | 10.9 | – | 1.35 | |
| 1 : 1 | 75/30* | 9.4 | – | 1.28 | |
| 3 : 7 | 78/35* | 7.5 | – | 1.29 | |
| 1 : 9 | 30/67* | 5.9 | – | 1.34 |
Похожим образом ведет себя другой, близкий по строению терпеновый 1,3-диен-фарнезен [130]:
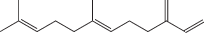
Его сополимеризация с глицидилметакрилатом под действием алкоксиамина типа “Bloc Builder” происходит аналогично сополимеризации β-мирцена с ИБМА. Однако при замене инициатора на “Dispolreg 007” контроль процесса теряется и образуется сополимер с бимодальным ММР.
СИНТЕЗ ЛЕГКО РАЗЛАГАЕМЫХ ПОЛИМЕРОВ
Синтетические биодеградируемые или легко гидролизуемые полимеры получают либо поликонденсацией, либо ионной полимеризацией гетероциклов [131–133]. Тем не менее известны несколько подходов, которые позволяют осуществить контролируемый синтез таких полимеров методами контролируемой радикальной полимеризации, подробно описанных в обзорах последних лет [134, 135]. Их можно свести к трем вариантам: внедрение легко деструктируемых звеньев в полимерную цепь, блок- или привитую сополимеризацию с образованием деградируемых блоков, и сшивание полимеров легко разрушаемыми сшивками.
Первая идея, которая была предложена для получения легко разлагаемых полимеров с помощью контролируемой полимеризации с участием нитроксилов, заключалась в вовлечении в этот процесс циклических кислородсодержащих виниловых мономеров – кетенацеталей. Надо отметить, что подобного рода синтезы уже были известны. Например, контролируемая полимеризация с раскрытием цикла 2-метилен-1,3-диоксепана под действием системы ди-трет-бутилпероксид–ТЕМПО при 125°С была описана еще в 1996 г. [136, 137]. Авторы предположили следующий механизм полимеризации:
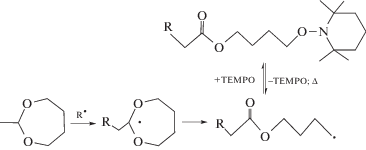
Практически одновременно была описана полимеризация с раскрытием двух циклов 2-метилен-1,4,6-триоксаспиро [4,4]нонана и 8,9-бензо-2-метилен-1,4,6-триоксаспиро[4,4]нонана в присутствии ТЕМПО, протекающая по механизму обратимого ингибирования [138, 139]:
Однако никаких исследований разложения полученных полимеров в то время не проводилось.
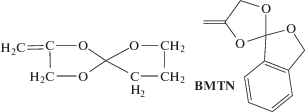
Целенаправленный синтез разлагаемых полимеров с участием кетенацеталей был осуществлен
в серии работ французских ученых [47, 140–143]. Для этого они синтезировали 5,6-бензо-2-метилен-1,3-диоксепан  и новый кетенацеталь 2-метилен-4-фенил-1,3-диоксалан
и новый кетенацеталь 2-метилен-4-фенил-1,3-диоксалан  .
.
Оказалось, что гомополимеризация этих мономеров, инициированная с помощью макроинициатора типа “Bloc Builder”, при 110–130°С протекает c плохим контролем ММ [140]. Напротив, при радикальной сополимеризации 2-метилен-4-фенил-1,3-диоксалана с ММА в тех же условиях при мольной доле ацеталя ≥20% Mn сополимера линейно увеличивается с конверсией, а ММР было тем у́же, чем выше доля ацеталя [141]. Сополимер деструктировал в 5%-ной щелочной среде при комнатной температуре в течение 1 ч до степени полимеризации ~3. Аналогичные результаты были получены при бинарной сополимеризации олигоэтиленгликольметакрилата с 2-метилен-4-фенил-1,3-диоксаланом, 5,6-бензо-2-метилен-1,3-диоксепаном или 2-метилен-1,3-диоксепаном [140, 142]. Как сополимеры, так и продукты их деструкции нетоксичны.
Подробно гидролитическая деструкция полученных сополимеров диоксалана с ММА (диоксалан-со-ММА) и олигоэтиленгликольметакрилатом (диоксалан-со-ОЭГМА) была изучена в сопоставлении с известными биодеградируемыми полимерами – полилактидом (ПЛ), поликапролактоном (ПКЛ) и сополимером лактида с гликолидом (С-со-ЛГ) [134]. Скорость гидролиза в фосфатном буфере (рН 7.4, 37°С) понижается в ряду: С-со-ЛГ > ПЛ > диоксалан-со-ОЭГМА > ПКЛ > > диоксалан-со-ММА.
При этом молекулярная масса С-со-ЛГ за один месяц уменьшается на 90%, а в случае диоксалана-со-ММА она понижается на 15–20% лишь за 1 год. Сополимеры диоксалан-со-ММА оказались такими же устойчивыми к биоразложению, как ПММА и олигоэтиленгликольметакрилат, а степень энзиматического разложения диоксалан-со-ОЭГМА под действием Candida Antartica составляла 13–15% за одну неделю.
Разработанный подход – контролируемый синтез разлагаемых сополимеров с участием кетенацеталей – применим и для тройных сополимеров [143]. Так, терполимеры ММА–АН–2-метилен-4-фенил-1,3-диоксалана и олигоэтиленгликольметакрилата–АН–2-метилен-4-фенил-1,3-диоксалана, полученные в тех же условиях и содержащие 40–70 мол. % диоксалана, легко гидролизуются в щелочной среде.
Другой подход к контролируемому синтезу разлагаемых полимеров, который заключается в привитой полимеризации виниловых мономеров на различные полисахариды (целлюлозу, хитозан, крахмал) с участием нитроксилов, кратко изложен в обзоре 2021 г. [144]. Продукты такого синтеза считать разлагаемыми нельзя, поскольку разлагаемой является только полисахаридная часть.
ПОЛИНИТРОКСИЛЫ – КАК КАТАЛИЗАТОРЫ
В заключение приведем несколько примеров катализа с участием полимерных нитроксилов на основе легко доступного ТЕМПО, которые напрямую к полимеризации с участием нитроксилов отношения не имеют, но важны с точки зрения девятого принципа “зеленой” химии.
Хорошо известно, что нитроксилы обладают каталитической активностью в реакции окисления спиртов до альдегидов, кетонов и кислот [145]. Главный недостаток этого процесса – необходимость очистки продуктов от стабильных радикалов. Для устранения этого недостатка применяют полимеры, содержащие нитроксильные радикалы в боковых группах (полимерные нитроксилы) [146]. Разумеется, их получают не радикальной, а ионной полимеризацией или химической модификацией.
Первая попытка применить такой полимерный нитроксил – ПС с боковыми радикалами ТЕМПО – вместо низкомолекулярного 4-метокси-ТЕМПО для окисления бензилового спирта была неудачной [147]. Выход бензальдегида не превышал 50% в сравнении со 100% в присутствии 4-метокси-ТЕМПО. Зато весьма успешным было использование продукта окисления коммерчески доступного олигомера “Chimassorb 944” [148]:
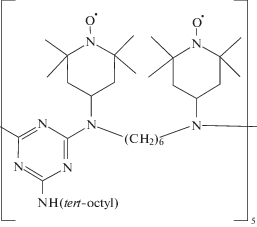
Выход альдегидов и кетонов при окислении двенадцати спиртов под действием гипохлорита натрия с участием этого олигомера был близок к количественному.
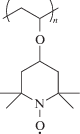
Эффективным гетерогенным катализатором окисления бензальдегида до бензойной кислоты, а целлюлозы до полиуроновой кислоты под действием гипохлорита натрия оказался полимер 4-оксивинил–ТЕМПО [149]:
В качестве катализаторов аэробного окисления бензилового спирта были синтезированы новые полиэфиркетоны и полиамиды, функционализированные ТЕМПО [150, 151]:
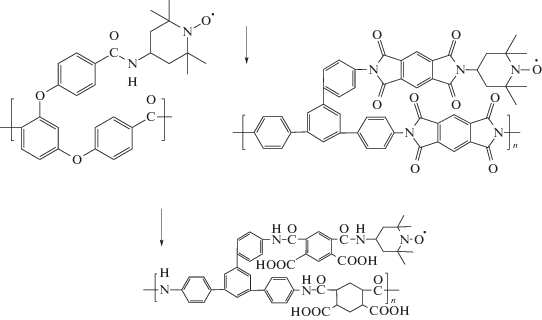
После шести циклов применения катализаторов их активность составляла 80–100%.
Как показало специальное исследование ПС и полиуретана с боковыми радикалами ТЕМПО в качестве катализаторов в реакции окисления различных спиртов под действием гипохлорида натрия и бис-(ацетоксииодбензола), полимерная природа на каталитическую активность нитроксила не влияет [152]. Никакого “полимерного эффекта”, т.е. увеличения активности катализатора за счет концентрирования каталитических центров на макромолекуле, во всех цитированных работах обнаружить не удалось.
ЗАКЛЮЧЕНИЕ
Контролируемый синтез полимеров методом обратимого ингибирования с участием нитроксилов интенсивно развивается уже почти 30 лет. Это направление стало не только “универсальным лабораторным инструментом” в руках химика-синтетика, позволяющим получать полимеры с необходимыми свойствами, но и вышло на “промышленные рельсы”. Расширяется применение этого метода в производстве адгезивов, покрытий, диспергаторов, компатибилизаторов [94]. Поэтому проблемы “зеленой” химии весьма актуальны и значимы для указанных процессов, хотя особое внимание им стали уделять только в последнее десятилетие.
В настоящем обзоре рассмотрены лишь некоторые, на взгляд авторов, самые актуальные и перспективные способы решения вопросов “зеленой” химии в процессах обратимого ингибирования нитроксилами. Проведенный анализ литературных данных показал, что в области дисперсионной полимеризации в сверхкритическом CO2 уже получены обнадеживающие результаты контролируемого синтеза полимеров. В фотополимеризации с участием нитроксилов, несмотря на большое количество исследований, желаемой степени контроля достичь пока не удалось. Работ в области контролируемого синтеза полимеров из возобновляемого сырья, а также легко деградируемых полимеров с участием нитроксилов крайне мало. Описаны лишь единичные примеры таких объектов. Однако первые результаты показывают перспективность этих направлений. Именно они, как нам кажется, и заслуживают основного внимания исследователей в ближайшие годы.
Работа выполнена в рамках проекта “Современные проблемы химии и физикохимии высокомолекулярных соединений” (Госбюджет № АААА-А21-121011990022-4).
Список литературы
Anastas P.T., Warner J.C. Green Chemistry: Theory and Practice. New York: Oxford Univ. Press, 1998. P. 30.
Anastas P.T., Zimmerman J.B. // Env. Sci. Technol. 2003. V. 37. № 5. P. 95.
Green Chemistry: Fundamentals and Applications / Eds by Suresh C. Ameta, Rakshit Ameta. New York: Apple Acad. Press CRC Press, a Taylor & Francis, 2013. Ch. 3.
Sheldon R.A. // Green Chem. 2007. V. 9. P. 1273.
Dube M.A., Salehpour S. // Macromol. React. Eng. 2014. V. 8. P. 7.
Sheldon R.A. // ACS Sustainable Chem. Eng. 2018. V. 6. № 1. P. 32.
Trost B.M. // Science. 1991. V. 254. P. 1471.
Matyjaszewski K. //Adv. Mater. 2018. V. 30. P. 1706441.
Destarac M. // Macromol. React. Eng. 2010. V. 4. P. 165.
Grishin D.F., Grishin I.D // Russ. J. Appl. Chem. 2011. V. 84. № 12. P. 2021.
Boyer C., Bulmus V., Davis T.P., Ladmiral V., Liu J., Perrier S. // Chem. Rev. 2009. V. 109. P. 5402.
Ivanov I.V., Meleshko T.K., Kashina A.V., Yakimansky A.V. // Russ. Chem. Rev. 2019. V. 88. P. 1248.
Stolin A.M., Merzhanov A.G., Malkin A.Ya. // Polym. Eng. Sci. 1979. V. 19. № 15. P. 1065.
Goikoetxea M., Heijungs R., Barandiaran M.J., Asua J.M. // Macromol. React. Eng. 2008. V. 2. № 1. P. 90.
Gandini A. // Macromolecules. 2008. V. 41. № 24. P. 9491.
Sogah D.Y., Hertler W.R., Webster O.W., Cohen G.M. // Macromolecules. 1987. V. 20. № 7. P. 1473.
Robinson K.L., Khan M.A., Banez M.V.D., Wang X.S., Armes S.P. // Macromolecules. 2001. V. 34. № 10. P. 3155.
Sheldon R.A. // Chem. Commun. 2008. V. 29. P. 3352.
Ribelli T.G., Lorandi F., Fantin M., Matyjaszewski K. // Macromol. Rapid Commun. 2019. V. 40. 1800616.
Kaminsky W. // Macromolecules. 2012. V. 45. P. 3289.
Song R., Murphy M., Li C., Ting K., Soo C., Zheng Z. // Drug Design, Development Therapy. 2018. V. 12. P. 3117.
Čolnik M., Hrnčič M.K., Škerget M., Knez Ž. // Chem. Ind. Chem. Eng. Q. 2020. V. 26. № 4. P. 401.
Roes A.L., Patel M.K. // Risk Anal. 2007. V. 27. P. 1311.
Tsarevsky N.V., Matyjaszewski K. // Chem. Rev. 2007. V. 107. P. 2270.
Lu C., Wang C., Yu J., Wang J., Chu F. // Green Chem. 2014. V. 16. P. 1854.
Surmacz K., Chmielarz P. // Materials. 2020. V. 13. P. 1717.
Achi N.E., Bakkour Y., Adhami W., Molina J., Penhoat M., Azaroual N., Chausset-Boissarie L., Rolando C. // Front. Chem. 2020. V. 8. P. 740.
Hu X., Zhu N., Guo K. // Adv. Polym. Technol. V. 2019. Article ID 7971683.
Dadashi–Silab S., Matyjaszewski K. // Molecules. 2020. V. 25. № 7. P. 1648.
Jablonský M., Škulcová A., Šima J. // Molecules. 2019. V. 24. P. 3978.
Phommalysack-Lovan J., Chu Y., Boyer C., Xu J. // Chem. Commun. 2018. V. 54. P. 6591.
Buss B.L., Miyake G.M. // Chem. Mater. 2018. V. 30. № 12. P. 3931.
Semsarilar M., Sébastien Perrier // Nature Chem. 2010. V. 2. P. 811.
Mitchell J.B., Samuni A., Krishna M.C., DeGraff W., Ahn M.S., Samuni U., Russo A. // Biochemistry. 1990. V. 29. P. 2802.
Chen K., Glockner J.F., Morse P.D., Swartz H.M. // Biochemistry. 1989. V. 28. № 6. P. 2496.
Glebska J., Skolimowski J., Kudzin Z., Gwozdzinski K., Grzelak A., Bartosz G. // Free Radic. Biol. Med. 2003. V. 35. P. 310.
Glebska J., Gwozdzinski K. // Curr. Top. Biophys. 1998. V. 22. P. 75.
Hahn S.M., Krishna C.M., Samuni A., DeGraff W., Cuscela D.O., Johnstone P., Mitchell J.B. // Canser Res., Suppl. 1994. V. 54. S. 2006.
Soule B.P., Hyodo F., Matsumoto K.-I., Simone N.L., Cook J.A., Krishna M.C., Mitchell J.B. // Antioxid. Redox. Signal. 2007. V. 9. № 10. P. 1731.
Zamir E., Zhang R.L., Samumi A., Kogan M., Pe’er J. // Free Radic. Biol. Med. 1999. V. 27. № 1/2. P. 7.
Lewandowski M., Gwozdzinski K. // Int. J. Mol. Sci. 2017. V. 18. P. 2490.
Кирилюк И.А., Святченко В.А., Морозов Д.А., Казачинская Е.И., Киселев Н.Н., Бакунова С.М., Войнов М.А., Локтев В.Б., Григорьев И.А. // Антибиотики и химиотерапия. 2012. Т. 57. № 1–2. С. 3.
Winalski C.S., Shortkroff S., Mulkern R.V. // Magn. Resonance Med. 2002. V. 48. № 6. P. 965.
Keana J.F.W., Pow S., Rosen G.M. // Magn. Resonance Med. 1987. V. 3. P. 83.
Durand G., Choteau F., Prosak R.A., Rockenbauer A., Villamena F.A., Puccia B. // New J. Chem. 2010. V. 34. P. 1909.
Guo X., Seo J.-E., Bryce S.M., Tan J.A., Wu Q., Dial S.L., Moore M.M., Mei N. // Toxicol. Sci. 2018. V. 163. № 1. P. 214.
Guegain E., Michel J.-P., Boissenot T., Nicolas J. // Macromolecules. 2018. V. 51. № 3. P. 724.
Nicolas J., Guillaneuf Y., Lefay C., Bertin D., Gigmes D., Charleux B. // Prog. Polym. Sci. 2013. V. 38. P. 63.
Graeme Moad, Ezio Rizzardo // Royal Soc. Chem., Polym. Chem. 2016. № 19. P. 1.
Zaremski M.Yu. // Polymer Science C. 2015. V. 57. P. 65.
Detrembleur C., Jerome C., Winter J.D., Gerbaux P., Clement J.-L., Guillaneuf Y., Gigmes D. // Polym. Chem. 2014. V. 5. P. 335.
Guillaneuf Y., Lamps J.-P., Catala J.-M., Gigmes D., Drockenmuller E. // J. Polym. Sci. A. 2012. V. 50. P. 3750.
Ruehl J., Hill N.L., Walter E.D., Millhauser G., Braslau R. // Macromolecules. 2008. V. 41. P. 1972.
Abreu C.M.R., Mendonca P.V., Serra A.C., Noble B.B., Guliashvili T., Nicolas J., Coote M.L., Coelho J.F.J. // Macromolecules. 2016. V. 49. № 2. P. 490.
Guegain E., Guillaneuf Y., Nicolas J. // Macromol. Rapid Commun. 2015. V. 36. № 13. P. 1227.
Zaremski M.Yu., Odintsova V.V. // Polymer Science. 2021. C. № 1 (в пeчaти).
Scaiano J.C., Connolly T.J., Mohta N., Pliva C.N. // Can. J. Chem. 1997. V. 75. P. 92.
Guillaneuf Y., Bertin D., Gigmes D., Versace D.-L., Lalevée J., Fouassier J.-P. // Macromolecules. 2010. V. 43. № 5. P. 2204.
Goto A., Scaiano J.C., Maretti L. // Photochem. Photobiol. Sci. 2007. V. 6. № 8. P. 833.
Su J., Liu X., Li M., Zhang T., Cui Y. // Int. J. Polym. Sci. 2016. Article ID 6482050.
Versace D.-L., Lalevée J., Fouassier J.-P., Guillaneuf Y., Bertin D., Gigmes D. // Macromol. Rapid Commun. 2010. V. 31. № 15. P. 1383.
Versace D.-L., Guillaneuf Y., Bertin D., Fouassier J.P., Lalevée J., Gigmes D. // Org. Biomol. Chem. 2011. V. 9. № 8. P. 2892.
Bosson J. // Ph.D.-Thesis. Aix-Marseille Université. 2015.
Morris J., Telitel S., Fairfull-Smith K.E., Bottle S.E., Lalevée J., Clément J.-L., Guillaneuf Y., Gigmes D. // Polym. Chem. 2015. V. 6. P. 754.
Su J., Liu X., Hu J., You Q., Cui Y., Chen Y. // Polym. Int. 2015. V. 64. № 7. P. 867.
Lin A. // Ph.D.-Thesis. Aix-Marseille Université. 2019.
Morris J.C. // PhD-Thesis, Aix-Marseille Université. 2016.
Anderson D.R., Keute J.S., Chapel H.L., Koch T.H. // J. Am. Chem. Soc. 1979. V. 101. № 7. P. 1904.
Bottle S.E., Chand U., Micallef A.S. // Chem. Lett. 1997. V. 26. № 9. P. 857.
Kalugina A.D., Mel’nikov M.Y. // High Energy Chem. 2007. V. 41. P. 385.
Keana J.F.W., Dinerstein R., Fredhelbmai T. // J. Org. Chem. 1971. V. 36. № 1. P. 118.
Liu X., Jing Y., Bai Y. // Front. Chem. China. 2008. V. 3. № 1. P. 41.
Yoshida E. // Polymers. 2012. V. 4. P. 1125–1156.
Yoshida E. // Colloid Polym. Sci. 2009. V. 287. P. 767.
Yoshida E. // Open J. Polym. Chem. 2014. V. 4. № 3. P. 47.
Yoshida E. // Colloid Polym. Sci. 2010. V. 288. P. 341.
Yoshida E. // Colloid Polym. Sci. 2010. V. 288. P. 239.
Yoshida E. // Colloid Polym. Sci. 2010. V. 288. P. 1639.
Yoshida E. // Colloid Polym. Sci. 2009. V. 287. P. 1417.
Yoshida E. // Colloid Polym. Sci. 2011. V. 289. P. 1625.
Yoshida E. // Polymers. 2012. V. 4. P. 1580.
Yoshida E. // Colloid Polym. Sci. 2010. V. 288. P. 7.
Telitel S., Telitel S., Bosson J., Lalevée J., Clément J.-L., Godfroy M., Fillaut J.-L., Akdas-Kilig H., Guillaneuf Y., Gigmes D. // Langmuir. 2015. V. 31. № 36. P. 10026.
Bonardi A.H., Dumur F., Gigmes D., Xu Y.Y., Lalevée J. // ACS Omega. 2020. V. 5. P. 3043.
Telitel S., Morris J., Guillaneuf Y., Clément J.-L., Morlet-Savary F., Spangenberg A., Malval J.-P., Lalevée J., Gigmes D., Soppera O. // ACS Appl. Mater. Int. 2020. V. 12. № 27. P. 30779.
Telitel S., Telitel S., Bosson J., Spangenberg A., Lalevée J., Morlet-Savary F., Clément J.-L., Guillaneuf Y., Gigmes D., Soppera O. // Adv. Mater. Int. 2014. V. 1. № 5. P. 1400067.
Huix-Rotllant M., Ferré N. // J. Phys. Chem. A. 2014. V. 118. № 25. P. 4464.
Charleux B., Ganachaud F. // Macromol. Eng. 2007. V. 1. P. 605.
Zetterlund P.B. // Polym. Chem. 2011. V. 2. P. 534.
Zetterlund P.B., Okubo M. // Macromolecules. 2006. V. 39. P. 8959.
Zetterlund P.B., Kagawa Y., Okubo M. // Chem. Rev. 2008. V. 108. P. 3747.
Zetterlund P.B., Aldabbagh F., Okubo M. // J. Polym. Sci. A. 2009. V. 47. P. 3711.
Save M., Guillaneuf Y., Gilbert R.G. // Austr. J. Chem. 2006. V. 59. P. 693.
Vivaldo-Lima E., Jaramillo-Soto G., Penlidis A. // Encyclopedia of Polymer Science and Technology. New York: John Wiley & Sons, Inc., 2016.
Zetterlund P.B., Thickett S.C., Perrier S., Bourgeat-Lami E., Lansalot M. // Chem. Rev. 2015. V. 115. P. 9745.
Cunningham M.F. // Progr. Polym. Sci. 2008. V. 33. P. 365.
Span R., Wagner W. // J. Phys. Chem. 1996. V. 25. P. 1509.
Odell P.G., Hamer G.K. Pat. 5,552,502 USA. 1996.
Odell P.G., Georges H., Hamer G.K. Pat. 0 735 051 A1, Eur. 1996.
McHale R., Aldabbagh F., Zetterlund P.B., Okubo M. // Macromol. Chem. Phys. 2007. V. 208. № 16. P. 1813.
Aldabbagh F., Zetterlund P.B., Okubo M. // Macromolecules. 2008. V. 41. № 7. P. 2732.
Aldabbagh F., Zetterlund P.B., Okubo M. // Eur. Polym. J. 2008. V. 44. № 12. P. 4037.
Petraitis J.M. // Ms.D. Thes. University of California Santa Cruz. 2017. https://escholarship.org/uc/item/3xm6d33n
Magee C., Earla A., Petraitis J., Higa C., Braslau R., Zetterlund P.B., Aldabbagh F. // Polym. Chem. 2014. V. 5. P. 5725.
O’Connor P., Zetterlund P.B., Aldabbagh F. // Macromolecules. 2010. V. 43. P. 914.
O’Connor P. // Ph.D.-Thes. National University of Ireland, Galway. 2012.
Ryan J., Aldabbagh F., Zetterlund P.B., Okubo M. // Polymer. 2005. V. 46. P. 9769.
McHale R., Aldabbagh F., Zetterlund P.B., Minami H., Okubo M. // Macromolecules. 2006. V. 39. P. 6853.
McHale R., Aldabbagh F., Zetterlund P.B., Okubo M. // Macromol. Rapid Commun. 2006. V. 27. P. 1465.
Grignard B., Phan T., Bertin D., Gigmes D., Jérôme C., Detrembleur C. // Polym. Chem. 2010. V. 1. P. 837.
Ramirez-Wong D.G., Posada-Velez C.A., Saldivar-Guerra E., Luna-Barcenas J.G., Ott C., Schubert U.S. // Macromol. Symp. 2009. V. 283–284. P. 120.
O’Connor P., Zetterlund P.B., Aldabbagh F. // J. Polym. Sci. A. 2011. V. 49. P. 1719.
O’Connor P., Yang R., Carroll W.M., Rochev Y., Aldabbagh F. // Eur. Poly. J. 2012. V. 48. P. 1279.
Garcґıa-Valdez O., Ramґırez-Wong D.G., Saldґıvar-Guerra E., Luna-Bґarcenas J.G. // Macromol. Chem. Phys. 2013. V. 214. P. 1396.
Satoh K. // Polymer J. 2015. V. 47. P. 527.
Yao K., Tang C. // Macromolecules. 2013. V. 46. P. 1689.
Wilbon P.A., Chu F., Tang C. // Macromol. Rapid Commun. 2013. V. 34. P. 8.
Szablan Z., Toy A.A., Davis T.P., Hao X., Stenzel M.H., Barner-Kowollik C. // J. Polym. Sci. A. 2004. V. 42. P. 2432.
Szablan Z., Toy A.A., Terrenoire A., Davis T.P., Stenzel M.H., Müller A.H.E., Barner-Kowollik C. // J. Polym. Sci. A. 2006. V. 44. P. 3692.
Anand V., Agarwal S., Greiner A., Choudhary V. // Polym. Int. 2005. V. 54. P. 823.
Meier M.A.R., Metzger J.O., Schubert U.S. // Chem. Soc. Rev. 2007. V. 36. P. 1788.
Noppalit S., Simula A., Billon L., Asua J.M. // Polym. Chem. 2020. V. 11. № 6. P. 1151.
Tajbakhsh S., Hajiali F., Marić M. // Ind. Eng. Chem. Res. 2020. V. 59. P. 8921.
Tajbakhsh S., Hajiali F., Marić M. // J. Appl. Polym. Sci. 2021. V. 138. № 13. e50095.
Tajbakhsh S., Marić M. // J. Polym. Sci. 2021.
Métafiot A., Kanawati Y., Gérard J.-F., Defoort B., Marić M. // Macromolecules. 2017. V. 50. P. 3101.
Métafiot A., Gagnon L., Pruvost S., Hubert P., Gérard J.-F., Defoort B., Marić M. // RSC Adv. 2019. V. 9. P. 3377.
Métafiot A. // Ph.D.-Thesis INSA, Lyon. 2019.
Sarkar P., Bhowmick A.K. // RSC Adv. 2014. V. 4. P. 61343.
Luk S.B., Milan M. // Macromol. React. Eng. 2019. P. 1800080.
Gross R.A., Kalra B. // Science. 2002. V. 297. P. 803.
Tian H., Tang Z., Zhuang X., Chen X., Jing X. // Prog. Polym. Sci. 2012. V. 37. P. 237.
Siracusa V., Rocculi P., Romani S., Rosa M. // Trends Food Sci. Technol. 2008. V. 19. P. 634.
Delplace V., Nicolas J. // Nature Chem. 2015. V. 7. P. 771.
Quan F., Zhang A., Cheng F., Cui L., Liu J., Xia Y. // Polymers. 2018. V. 10. P. 758.
Wei Y., Connors E.J., Jia X., Wang C. // Chem. Mater. 1996. V. 8. № 3. P. 604.
Wei Y., Connors E.J., Jia X., Wang C. // J. Polym. Sci. A. 1998. V. 36. № 5. P. 761.
Jia X., Li M., Han S., Wang C., Wei Y. // Mater. Lett. 1997. V. 31. P. 137.
Jia X., Wang J., Liu Y., Jin Z., Li M., Wei Y. // Polym. Int. 2000. V. 49. P. 1496.
Delplace V., Guégain E., Harrisson S., Gigmes D., Guillaneuf Y., Nicolas J. // Chem. Commun. 2015. V. 51. P. 12847.
Tran J., Guegain E., Ibrahim N., Harrisson S., Nicolas J. // Polym. Chem. 2016. V. 7. P. 4427.
Delplace V. M. // Ph.D.-Thes. L’université Paris-Sud. 2015.
Delplace V., Harrisson S., Tardy A., Gigmes D., Guillaneuf Y., Nicolas J. // Macromol. Rapid Commun. 2014. V. 35. P. 484.
Lamontagne H.R., Lessard B.H. // ASC Appl. Polym. Matter. 2020. V. 2. № 12. P. 5327.
Wertz S., Studer A. // Green Chem. 2013. V. 15. P. 3116.
Hansen K.-A., Blinco J.P. // Polym. Chem. 2018. V. 9. P. 1479.
Miyazawa T., Endo T. // J. Polym. Sci. A. 1985. V. 23. № 9. P. 2487.
Dijksman A., Arends I.W.C.E., Sheldon R.A. // Chem. Commun. 2000. P. 271.
Waskitoaji W., Suga T., Nishide H. // AIP Conf. Proc. 2009. V. 1169. P. 13. 150.
Shi Y., Nabae Y., Hayakawa T., Kakimoto M. // RSC Adv. 2015. V. 5. P. 1923
Nabae Y., Mikuni M., Takusari N., Hayakawa T., Kakimoto M. // High Perform. Polym. 2017. V. 29. P. 646.
Subhani M.A., Beigi M., Eilbracht P. // Adv. Synth. Catal. 2008. V. 350. № 18. P. 2903.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)