

Высокомолекулярные соединения (серия С), 2022, T. 64, № 1, стр. 3-29
РАДИКАЛЬНАЯ СОПОЛИМЕРИЗАЦИЯ С ОБРАТИМОЙ ДЕАКТИВАЦИЕЙ ЦЕПИ: СИНТЕЗ СОПОЛИМЕРОВ С КОНТРОЛИРУЕМОЙ ПОСЛЕДОВАТЕЛЬНОСТЬЮ ЗВЕНЬЕВ
Е. В. Черникова a, *, К. О. Минеева a
a Московский государственный университет имени М.В. Ломоносова. Химический факультет
119991 Москва, Ленинские горы, д. 1, стр. 3, Россия
* E-mail: chernikova_elena@mail.ru
Поступила в редакцию 28.02.2022
После доработки 13.03.2022
Принята к публикации 28.03.2022
- EDN: TNNFPB
- DOI: 10.31857/S230811472220002X
Аннотация
В обзоре рассмотрены вопросы контролируемого синтеза сополимеров с контролируемым распределением мономерных звеньев (статистические, градиентные, блок-статистические, блок-градиентные, мультиблочные сополимеры) в условиях радикальной полимеризации с обратимой деактивацией цепи и способы направленного изменения последовательности соединения звеньев в макромолекулах в ходе синтеза. Обсуждается влияние микроструктуры цепи на свойства таких сополимеров.
ВВЕДЕНИЕ
В последние десятилетия наблюдается бурное развитие направлений полимерной химии, позволяющих получать макромолекулы сложной архитектуры и строго заданного молекулярного строения. К ним относятся “живая” анионная полимеризация, псевдоживая катионная полимеризация, радикальная полимеризация с обратимой деактивацией цепи, их комбинации друг с другом и/или с реакциями клик-химии, клик-полимеризация и т.д. [1–20]. Синтез полимеров сложной архитектуры в большинстве случаев является достаточно трудоемким и имеет скорее академическое, а не прикладное значение. Вместе с тем контролируемый синтез более простых структур (линейных сополимеров с необходимым распределением звеньев в цепи) способен решить многие практические задачи и обеспечить получение полимеров с нужным комплексом свойств. Такие сополимеры могут представлять собой заготовки для конструирования макромолекул более сложной архитектуры, например разных вариантов привитых сополимеров или мультиблок-сополимеров. Свойства сополимеров одного состава, но с разной последовательностью звеньев в цепи обычно существенно различаются. В качестве примера можно привести разную способность к самоорганизации в растворе или блоке сополимеров статистического, градиентного, блочного и блок-статистического строения или отличающиеся физико-механические свойства статистических, чередующихся и блочных сополимеров [21–24].
Развитие методов “живой” анионной полимеризации и радикальной полимеризации с обратимой деактивацией цепи стартовало с контроля ММ и ММР, синтеза блок- и градиентных сополимеров, а также композиционно-однородных статистических сополимеров [25–30]. В начале ХХI века начались активные исследования в области получения сополимеров с точно заданным молекулярным строением, включая контролируемое распределение звеньев в цепи [31–33]. Очевидно, что оно в первую очередь определяется активностью мономеров в сополимеризации и составом мономерной смеси. Однако в ряде случаев возникает необходимость обмануть природу и получить из мономерной смеси не статистический, а, например, чередующийся сополимер или мультиблочный сополимер с короткими блоками. Решить эту задачу можно только в том случае, если активный центр макромолекулы “живет” до конца полимеризации или периодически “засыпает” и “оживает”. В ионной полимеризации полярность растворителя и объем противоиона (для неполярного растворителя) играют существенную роль в изменении кажущейся активности мономеров в сополимеризации и, как следствие, распределении мономерных звеньев [34]. Для радикальной полимеризации влияние растворителя и/или добавок, способных связываться с мономером или растущим центром, на микроструктуру цепи характерно в меньшей степени [35]. Однако, как оказалось, в радикальной полимеризации с обратимой деактивацией цепи появляются новые возможности управления составом сополимера, не доступные классическому радикальному процессу. В настоящем обзоре на примере радикальной полимеризации с обратимой деактивацией цепи мы рассмотрим, какие используются инструменты, как они позволяют менять последовательность мономерных звеньев в сополимерах и к изменению каких свойств сополимеров это приводит.
РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ С ОБРАТИМОЙ ДЕАКТИВАЦИЕЙ ЦЕПИ
Термин “радикальная полимеризация с обратимой деактивацией цепи”, рекомендованный ИЮПАК, вошел в научную лексику в 2010 г. [36]. Несмотря на то, что в зарубежной литературе он вытеснил прежние названия “живая”, “псевдоживая” или “квазиживая” радикальная полимеризация, в отечественной литературе данный термин пока еще до конца не прижился. Под радикальной полимеризацией с обратимой деактивацией цепи понимают цепную полимеризацию, в которой радикальные активные центры обратимо переходят в неактивное состояние (деактивация), в результате чего в системе устанавливается одно или несколько равновесий между активными центрами (макрорадикалами) и временно неактивными макромолекулами (“спящими” цепями). При этом роль квадратичного обрыва макрорадикалов в кинетике процесса и образовании макромолекул становится незначительной.
Деактивация макрорадикалов происходит в результате одной из трех реакций [37]:
а) обрыва на стабильном или малоактивном радикале
б) переноса атома от металлоорганического соединения
в) передачи цепи по механизму присоединения–фрагментации
Обратная реакции (активация) приводит к регенерации активного центра на той же макромолекуле и продолжению реакции роста цепи. Наличие стадий деактивации и активации отличает радикальную полимеризацию с обратимой деактивацией цепи как от “живой” (безобрывной) анионной, так и от классической радикальной полимеризации, но одновременно она имеет черты, характерные для каждой из них. Так, способность макромолекул превращаться в активные макрорадикалы (“оживать”) приводит к сужению ММР, росту ММ полимера при увеличении конверсии мономера и позволяет наращивать ММ готового полимера путем добавления к нему новой порции мономера, делая радикальную полимеризацию с обратимой деактивацией цепи похожей на живую анионную полимеризацию. Замена квадратичного обрыва макрорадикалов их обратимой деактивацией в свою очередь отличает ее от классической радикальной полимеризации.
Варианты и условия реализации радикальной полимеризацией с обратимой деактивацией цепи рассмотрены в многочисленных обзорах [2, 38–41]. Стоит остановиться на возможностях этого процесса в контролируемом синтезе сополимеров и его особенностях.
В простейшем случае бинарной сополимеризации в реакцию деактивации вступают макрорадикалы, содержащие концевые звенья мономеров М1 или М2, константы скорости которой будут зависеть от природы мономеров. Аналогично, константа скорости реакции активации также будет зависеть от природы мономеров. Следствием этого будет сосуществование нескольких равновесий деактивация–активация. Концентрация макрорадикалов с концевыми звеньями М1 и М2 может оказаться той же или отличной от классической радикальной сополимеризации в зависимости от соотношения констант равновесия и состава мономерной смеси. Для случаев обратимого обрыва и переноса атома, характеризующихся мономолекулярным механизмом активации макромолекул, такую схему, включающую четыре реакции роста цепи (для концевой модели) и два равновесия, можно представить следующим образом:
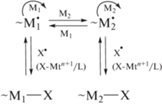
В сополимеризации с обратимой передачей цепи (ОПЦ) реакция активации бимолекулярная, поэтому с макрорадикалом с концевым звеном М1 могут взаимодействовать полимерные ОПЦ-агенты с тем же концевым звеном, соединенным с группой –SC(=S)Z, или с концевым звеном второго мономера, что увеличивает число возможных равновесий в системе уже для монофункционального ОПЦ-агента
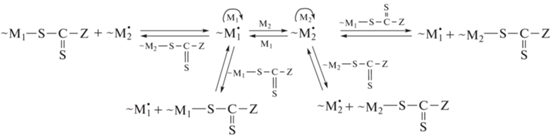
и еще больше для бифункционального ОПЦ-агента
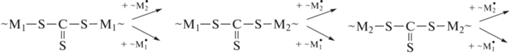
Наличие равновесий деактивация–активация и соотношение констант равновесия определяют кинетику сополимеризации и реализацию механизма обратимой деактивации цепи, т.е. степень контроля молекулярно-массовых характеристик. Однако они не должны влиять на состав сополимера, поскольку мономер в таких реакциях не участвует. Подробно кинетика сополимеризации и терполимеризации на примере механизма обратимого ингибирования с участием нитроксильных радикалов рассмотрены в работах [42–44]. Эту концепцию можно распространить и на сополимеризацию по механизму переноса атома и с некоторыми поправками на ОПЦ-сополимеризацию. Очевидно, что если в сополимеризацию вступают мономеры, для которых активация макрорадикалов происходит достаточно часто на протяжении полимеризации, то контроль молекулярно-массовых характеристик образующихся сополимеров будет высоким независимо от активности мономеров [45–48]. В обратном случае сополимеризация пройдет по пути классического радикального механизма или может даже затухать со временем [49]. Если константы равновесия (константы скорости деактивации и активации) для обоих мономеров существенно различаются, то механизм сополимеризации будет определяться активностью мономеров и составом мономерной смеси. В идеальном случае активный мономер, с более высокой вероятностью находящийся на конце растущей цепи, способен обеспечить контроль ММ в сополимеризации с неактивным мономером, если первый успешно полимеризуется по механизму обратимой деактивации цепи [50–54]. Таким образом, выбор агента деактивации–активации является критическим для успешного контролируемого синтеза сополимера.
Кроме того, при сополимеризации не в условиях азеотропа состав мономерной смеси постоянно меняется, в результате чего происходит изменение мгновенного состава сополимера. В классической радикальной сополимеризации это сопровождается образованием на высоких конверсиях макромолекул разного состава, т.е. композиционно неоднородного сополимера. В идеальном варианте полимеризации с обратимой деактивацией цепи благодаря многократным повторениям актов деактивации и активации цепи молекулы мономеров присоединяются к постоянно “оживающим” активным центрам, что приводит к изменению состава вдоль макромолекулы на протяжении сополимеризации, и все макромолекулы имеют близкий состав. Очевидно, что для синтеза композиционно однородных сополимеров ключевым моментом является большая частота “оживления” макромолекул. Если в сополимеризации участвуют мономеры, существенно различающиеся по активности, то в результате механизма обратимой деактивации цепи формируется градиентный сополимер [55]. Следовательно, правильный выбор агента деактивации–активации дополнительно контролирует композиционную однородность сополимеров.
К настоящему времени синтез градиентных сополимеров и статистических композиционно однородных сополимеров стал рутинной задачей для радикальной полимеризации с обратимой деактивацией цепи [1, 2]. В связи с этим более интересной и привлекательной задачей для исследователей в последние годы является разработка способов синтеза сополимеров, в которых кроме ММ и ММР контролируется микроструктура (последовательность мономеров) цепи.
ОБЩИЕ ПРИНЦИПЫ КОНТРОЛЯ МИКРОСТРУКТУРЫ ЦЕПИ СОПОЛИМЕРОВ
В любой сополимеризации последовательность включения мономеров в макромолекулу задается на стадии роста цепи и определяется константами сополимеризации (r1, r2) и соотношением мономеров в мономерной смеси.
Отсюда следует вывод о том, что в радикальной сополимеризации с обратимой деактивацией цепи, в которой “оживление” макромолекул происходит достаточно часто, контролировать микроструктуру цепи можно, изменяя направленным образом: состав мономерной смеси в ходе синтеза; относительную активность мономеров в сополимеризации. В результате такого воздействия можно получить градиентные сополимеры и блок-сополимеры разного строения:
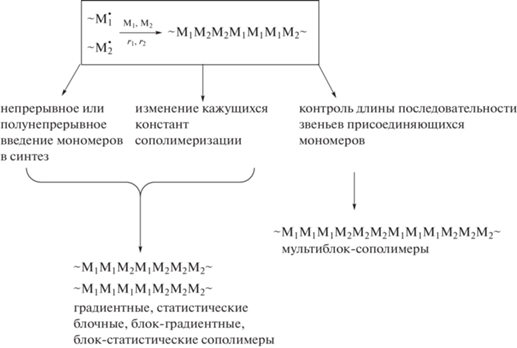
Возможна и более “тонкая” настройка микроструктуры, в ходе которой контролируется длина последовательности звеньев присоединяющихся мономеров. Данный вариант позволяет синтезировать мультиблок-сополимеры и создать заготовки для получения привитых сополимеров. Рассмотрим эти подходы подробнее.
Регулирование скорости введения мономеров в синтез
Первые сведения о синтезе градиентных сополимеров в условиях радикальной полимеризации с обратимой деактивацией цепи появились в конце 1990-х–начале 2000-х годов [56–59]. Спонтанно градиентные сополимеры образуются в случае использования разноактивных мономеров. Условием их формирования является существенное изменение мгновенного состава сополимера в течение сополимеризации [56, 57]. Однако для достаточно большого количества пар мономеров разница в их активностях невелика. В этом случае мгновенный состав сополимера можно регулировать, задавая скорость введения одного или обоих мономеров в сополимеризацию. Впервые такой прием был осуществлен в работе [60] на примере сополимеризации эквимолярной смеси стирола и ММА под действием комплексов Ru(II). По достижении конверсии стирола 52% в мономерную смесь ввели первую порцию ММА, при конверсии стирола 89% добавили вторую порцию ММА. В результате авторы фактически получили триблок-сополимер, в котором каждый блок представлял собой сополимер разного состава, т.е. состав макромолекулы изменялся вдоль цепи. Эту идею развили в работе [56] в сополимеризации в массе акрилонитрила (АН) со стиролом (rАН = 0.05, rстирол = 0.37 [61]) и н-бутилакрилата (БА) со стиролом (rБА = 0.12, rстирол = 0.88 [61]) под действием алкилгалогенидов и CuBr с органическими лигандами. Добавляя акрилонитрил или н-бутилакрилат с разной скоростью в полимеризацию стирола, авторам удалось получить сополимеры с разной степенью градиентности. Данный прием применяли также в полимеризации с участием нитроксильных радикалов и в ОПЦ-полимеризации. В табл. 1 приведены примеры синтеза градиентных, блок-градиентных, блок-статистических сополимеров разными вариантами полимеризации с обратимой деактивацией цепи с использованием подхода полунепрерывной полимеризации (дозированного введения одного из мономеров в синтез). Стоит отметить, что в течение сополимеризации можно добавлять как более активный [62], так и менее активный мономер [63], добиваясь нужного градиента в распределении звеньев путем выбора необходимой скорости введения мономера. При этом процесс ведут не только в гомогенных условиях, но и в гетерофазной, чаще эмульсионной, полимеризации. Использование такого приема позволяет в широких пределах изменять микроструктуру макромолекул и, как следствие, свойства образующихся сополимеров. Влияние строения градиентных сополимеров на их свойства обсуждается ниже.
Таблица 1.
Примеры использования полунепрерывной полимеризации для регулирования микроструктуры сополимеров
| Метод | Мономер 1 | Мономер 2 | Литература |
|---|---|---|---|
| Обратимое ингибирование | Стирол | 4-метилстирол | [64] |
| 4-ацетоксистирол | [65, 66] | ||
| 4-гидроксистирол | [65, 67–70] | ||
| БМА | [70] | ||
| 4-винилпиридин | [70] | ||
| ММА | [71, 72] | ||
| АК | [73, 74] | ||
| БА | ММА | [75] | |
| Стирол | [68–70, 76, 77] | ||
| ДМАА | [78, 79] | ||
| МА | Октадецилакрилат | [80] | |
| Полимеризация с переносом атома | трет-БА | ГФБМА | [81] |
| Стирол | [82] | ||
| БА | Изоборнилакрилат | [83] | |
| Трет-бутилметакрилат | [84] | ||
| ММА | Гидроксиэтилметакрилат | [85] | |
| Трет-бутилакрилат | [57] | ||
| АН | Стирол | [56] | |
| ОПЦ-полимеризация | ТФЭМА | МАЭФХ | [86] |
| Стирол | Акриловая кислота | [87] | |
| БА | [88–90] | ||
| Акриловая кислота | [63] | ||
| ММА | [91] | ||
| Гексафторбутилакрилат | [92] | ||
| БМА | Гексафторбутилметакрилат | [93] | |
| ПЭГМА | Тридекафтороктилакрилат | [94] | |
| АН | НИПАМ | [95] | |
| АК | [96] | ||
| ДМАА | АК | [97] | |
| МАК | [97] | ||
| БА | Винилацетат | [97] | |
| ХГПМА | [97] |
Примечание. АК – акриловая кислота, трет-БА – трет-бутилакрилат, БМА – бутилметакрилат, ГФБМА – 2,2,3,3,4,4,4-гептафторбутилметакрилат, ДМАА – N,N-диметилакриламид, НИПАМ – N-изопропилакриламид, МАК – метакриловая кислота, МА – метилакрилат, МАЭФХ – 2-метакрилоилоксиэтилфосфорилхолин, ПЭГМА – метиловый эфир полиэтиленгликольметакрилата, ТФЭМА – 2,2,2-трифторэтилметакрилат, ХГПМА – 3-хлор-2-гидроксипропилметакрилат.
Влияние растворителя и/или добавок на относительную активность мономеров
В радикальной сополимеризации реакционные среды, содержащие вещества, способные к специфическому взаимодействию с мономерами или радикалами роста путем образования водородных связей, донорно-акцепторных и координационных комплексов, также могут влиять на микроструктуру цепи и состав сополимера [98–101].
Сополимеризация метакриловой кислоты с ММА и стиролом или акриловой кислоты с метилакрилатом и стиролом в растворителях, способных к образованию водородных связей (ДМФА, пиридин, 1,4-диоксан, этанол, этиленгликоль), приводит к уменьшению содержания непредельных кислот в сополимерах за счет изменения относительной активности мономеров [102]. Оказалось, что АК и МАК в массе и в инертных растворителях (бензол, CCl4) существуют в виде димеров, но в растворителях (сильных акцепторах протонов) димеры разрушаются и заменяются Н-комплексами кислота–растворитель. В результате активность АК и МАК в сополимеризации падает. Аналогичным образом изменяется активность (мет)акриламидов в сополимеризации с ММА и стиролом в растворителях, способных и не способных образовывать Н-связи с мономером [103–105].
Влияние донорно-акцепторных взаимодействий в процессах с обратимой деактивацией цепи можно проиллюстрировать данными по сополимеризации 2-винилпиридина с винилацетатом [106] и винилацетата с акриловой и метакриловой кислотой в присутствии GeCl4 [107]. В массе независимо от состава мономерной смеси образуется большей частью поли(2-винилпиридин), а в присутствии бензойной кислоты – сополимер с регулярным чередованием звеньев обоих мономеров. В ОПЦ-полимеризации – это синтез чередующегося сополимера N-винилпирролидона с 1,1,1,3,3,3-гексафторизопропил-α-фторакрилатом в присутствии бензилдитиобензоата [108].
Используя такие приемы и радикальную полимеризацию с обратимой деактивацией цепи, можно получить сополимеры одинакового химического состава с узким ММР, но с разной микроструктурой цепи [109–112].
Впервые влияние полярности растворителя на закономерности радикальной сополимеризации ММА и стирола было описано в работе [113]. Рост полярности растворителя сопровождался уменьшением констант сополимеризации мономеров и увеличением их склонности к чередованию в сополимере. В дальнейшем стало понятно, что влияние растворителя проявляется тогда, когда хотя бы один из мономеров является полярной молекулой, например, акриловая и метакриловая кислоты, акриламид и т.д. [114–119]. Многочисленные примеры реализации эффекта растворителя приведены в обзоре [120]. Один из вариантов такого эффекта – эффект избирательной сольватации активного центра или bootstrap effect [121–123]. При сополимеризации разнополярных мономеров в малополярных и полярных растворителях их относительная активность и состав сополимеров значительно отличаются. Однако, если взять сополимеры одинакового среднего состава, синтезированные в разных растворителях, то они характеризуются одинаковой микроструктурой. Полученные результаты объясняют отличием локальных эффективных относительных концентраций мономеров возле активного центра от средних по объему. Данное явление наблюдается при сополимеризации в массе, когда один из сомономеров – хороший растворитель для образующегося сополимера, а второй – плохой. С ростом конверсии мономера состав мономерной смеси меняется, что приводит к изменению термодинамического качества такого растворителя [124]. С повышением ММ сополимера эффект избирательной сольватации усиливается [123, 125–128]. Он еще более выражен в сополимеризации в растворе: локальная концентрация растворителя и полярного мономера может отличаться от своих средних значений в объеме.
Естественно, что эффект избирательной сольватации проявляется и в радикальной полимеризации с обратимой деактивацией цепи при изменении природы растворителя. Так, при сополимеризации ММА и N,N-диметиламиноэтилметакрилата (ДМАЭМА) активность последнего выше, чем ММА в полимеризации с переносом атома в малополярном толуоле, и, наоборот, активность ДМАЭМА резко падает при переходе к полярным ДМФА или метанолу [129].
Необычное проявление эффекта избирательной сольватации обнаружено в работе [130] при ОПЦ-сополимеризации ДМАА и 2-(N-бутилперфтороктанфторсульфонамидо)этилакрилата. Состав сополимеров, образующихся в трифтортолуоле под действием несимметричных низкомолекулярного и полимерного (на основе полидиметилсилоксана) тритиокарбонатов, существенно отличается. Причем с увеличением доли фторакрилата в мономерной смеси, это различие усиливается. Предполагается, что гидрофобный полидиметилсилоксан сольватирован фторированным мономером, вследствие чего его локальная концентрация возрастает и состав сополимера обогащается этим мономером. Влияние полимерного аддукта с нитроксильным радикалом на константы сополимеризации стирола и акриловой кислоты в 1,4-диоксане описаны в работе [131]. В отличие от низкомолекулярного аддукта применение полиакриловой кислоты (ПАК) с нитроксильным радикалом в сополимеризации приводит к четырехкратному увеличению активности АК и трехкратному понижению активности стирола в сополимеризации. Таким образом, гидрофильный полимер способствует обогащению сополимера АК. В работах [109, 132, 133] показано, что в полярном растворителе (ДМФА) гидрофобный полимерный ОПЦ-агент на основе полистирола и гидрофильный полимерный ОПЦ-агент на основе ПАК изменяют относительную активность АК и стирола: ПАК повышает активность стирола, а ПС – активность АК. При этом ММ полимерного ОПЦ-агента также влияет на относительную реакционную способность мономеров. Такое же сильное воздействие на активность мономеров в присутствии полимерных ОПЦ-агентов на основе ПАК и ПС наблюдается в малополярном 1,4-диоксане. Однако данный эффект уменьшается с ростом конверсии мономера и тем быстрее, чем выше доля АК в мономерной смеси [23].
Таким образом, варьируя химическую природу полимерного ОПЦ-агента, его ММ и полярность растворителя, можно получить широкий спектр микроструктур сополимеров (статистическая, градиентная, блок-статистическая и др.) из мономерной смеси одного состава.
“Точечное” введение мономеров в синтез
Рассмотренные выше способы регулирования микроструктуры цепи можно отнести к синтезу полимеров с контролируемой последовательностью звеньев (sequence-controlled polymers), в которых последовательность мономеров в цепи регулируется в какой-то степени, но не полностью [134, 135]. Однако известны полимеры (белки, олигонуклеотиды и т.д.), в которых последовательность мономерных звеньев четко задана и одинакова для всех макромолекул (sequence-defined polymers) [134]. С развитием радикальной полимеризации с обратимой деактивацией цепи исследования способов создания макромолекул с четко заданной последовательностью мономеров стали все больше привлекать внимание исследователей.
Наиболее простым вариантом является синтез строго чередующихся сополимеров на основе пары мономеров с электронно-донорным и электронно-акцепторным заместителями, в которой по крайней мере один мономер не склонен к гомополимеризации. Типичными примерами служат сополимеры стирола с малеиновым ангидридом или N-замещенными малеимидами [136–139]. В данном случае механизм обратимой деактивации позволяет синтезировать чередующиеся сополимеры с узким ММР, а при избытке стирола в мономерной смеси – блок-чередующиеся сополимеры [140–143].
Идея использования малеинового ангидрида и N-замещенного малеимида для конструирования заданной последовательности цепи оказалась привлекательной. В работе [144] ее применили для создания трехлучевого блок-сополимера АВС:
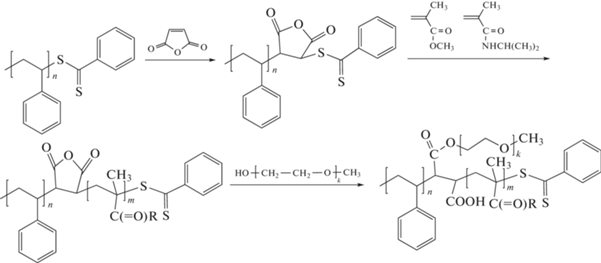
Этот подход развили в работе [145] для синтеза сополимера стирола с четырьмя замещенными малеимидами (N-пропил, N-бензил-, N-метил- и N-3,5-бис-(трифторметил)фенилмалеимид)), которые последовательно вводили в реакцию в выбранные моменты времени. Таким образом сополимеризацией с переносом атома удалось получить сополимеры заданной микроструктуры, в которых короткие блоки чередующихся сополимеров стирола и замещенного малеимида были разделены блоками полистирола:
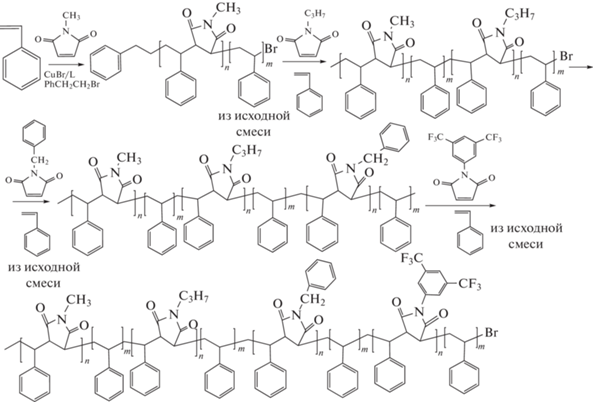
Позже эти же авторы существенно расширили ряд функциональных N-замещенных малеимидов [146]:
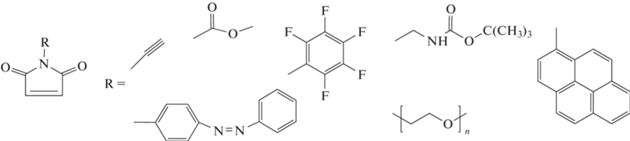
Данные производные оказались удобными “инструментами” для дальнейшей модификации боковых заместителей и для прививки к цепи полистирола других полимеров в точно заданных местах макромолекулы.
Общая идея рассматриваемого подхода заключается в добавлении на определенном этапе синтеза, например, при выбранной конверсии мономера, второго мономера, не склонного к гомополимеризации. В результате второй мономер или оказывается на конце растущей цепи, или внутри блока чередующегося сополимера определенной длины, после чего полученный полимер вступает в полимеризацию с новой порцией первого мономера или другого мономера, или смеси мономеров, способных к полимеризации и т.д. Один из примеров практической реализации данного подхода описан в работе [147]. Методом ОПЦ-полимеризации авторы синтезировали олигомерный ПС с узким ММР (Mn = 1.4 × 103, Đ = 1.07) и высоким содержанием “живых” цепей. Затем добавили к нему малеиновый ангидрид, и средний прирост степени полимеризации составил 1.5. На следующем этапе нарастили на макромолекуле примерно 10 звеньев стирола из расчета, чтобы малеиновый ангидрид оказался примерно в центре цепи. После этого данные операции повторили еще несколько раз, и в итоге получили полистирол с Mn = 5.7 × 103 и Đ = 1.35, в котором приблизительно каждые 10 звеньев стирола были разделены звеном малеинового ангидрида или коротким блоком чередующегося сополимера. Реакцией этерификации со стеариловым спиртом в результате получили привитой сополимер с известным расположением коротких алкильных привитых цепей.
Пример синтеза макромолекул сложной архитектуры с использованием рассмотренного выше подхода методом ОПЦ-полимеризации приведен в работе [148], в которой описано получение и свойства сополимера следующего строения: (A–прив–D)m(B–чер–C)nDk (A – ДМАЭМА, B – производное стирола, C – производное малеимида, D – НИПАМ или ММА, или стирол, или МА). Свойства таких сополимеров отличаются от линейных аналогов и определяются их химическим составом, плотностью прививки и соотношением длины блоков.
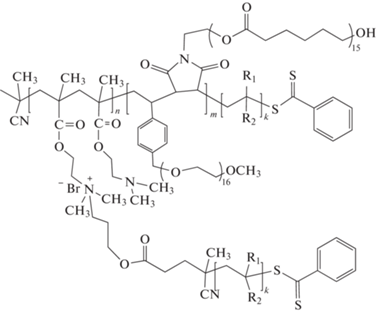
Здесь R1 = H, R2 = C(=O)NHCH(CH3)2; R1 = CH3, R2 = COOCH3; R1 = H, R2 = COOCH3; R1 = H, R2 = C6H5.
На практике невозможно точно поместить звено нужного мономера в конкретное место полимерной цепи. Этому препятствует статистическая природа цепной полимеризации. В результате неопределенность в расположении данного мономера в макромолекуле увеличивается пропорционально квадратному корню из ММ полимера [149]. В упомянутой работе авторы провели математический анализ расположения звеньев в макромолекулах сополимеров АnBmCn и АnBnАnCnАnDnАnCnАnBn разного состава, образующихся в ходе полимеризации с обратимой деактивацией цепи (например, ОПЦ), включая сополимеры с мономерами, не способными к гомополимеризации. Расчеты показывают, что с увеличением длины центрального блока повышается вероятность найти звено этого блока в центре цепи. Так, для сополимера А10BmC10 только 17.6% макромолекул содержат звено B в центре цепи при m = 1. При m = 2 эта величина возрастает примерно вдвое и равна 33.6%, и наконец для m = 11 она равна 95%. С увеличением числа блоков количество дефектных цепей (не удовлетворяющих требованию наличия заданного числа блоков известной длины, соединенных в определенном порядке) растет. При этом дефектность выше для мультиблок-сополимеров, включающих короткие блоки, и она понижается с ростом их длины. Данная оценка позволяет сформулировать требования к условиям синтеза сополимеров заданной микроструктуры.
Предложенный подход пригоден большей частью для стирола (и его производных) и малеинового ангидрида/малеимида, однако он не подходит для большинства виниловых мономеров, для которых константы сополимеризации с малеиновым ангидридом или малеимидом отличны от нуля. Однако он удобен для введения этих мономеров в качестве концевого звена цепи. В таком случае можно выбрать подходящий заместитель R в структуре малеимида, чтобы придать ему нужную функциональность. Так, в работе [150] с использованием ОПЦ-полимеризации были синтезированы ПДМАЭМА и ПНИПАМ с концевым звеном трифторацетата N-2-аминоэтилмалеимида, а также блок-сополимеры, блоки которых были разделены звеном замещенного малеимида. Звено малеимида с концевой аминогруппой можно использовать для биоконъюгации, присоединения флуоресцентного красителя и т.д.
Сополимеры стирола и малеиновой кислоты представляют интерес для различных биомедицинских применений. В работе [151] предложен любопытный способ получения периодического сополимера АВВ (малеиновая кислота–стирол–стирол) сочетанием ОПЦ-полимеризации и итеративного подхода с последующим гидролизом звеньев малеинового ангидрида.
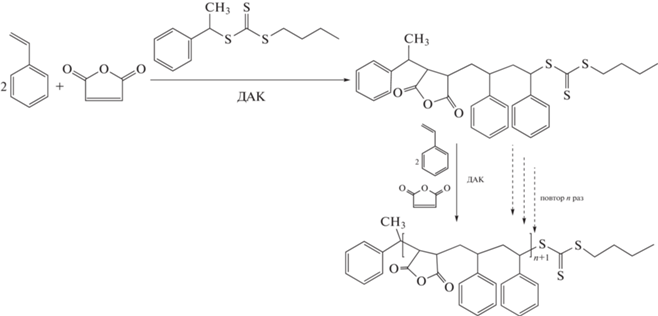
Суть его заключалась в том, что в полимеризацию вводили 2 мольных эквивалента стирола, 1 эквивалент малеинового ангидрида в расчете на 1 эквивалент ОПЦ-агента. После практически полного исчерпания мономеров добавляли их следующую порцию в том же соотношении. При этом в реакцию вводили необходимое количество инициатора. Число повторений стадий добавления мономеров определялось величиной степени полимеризации (10–50), которую необходимо было получить. Мольная доля малеинового ангидрида в конечном сополимере соответствовала требуемому соотношению 2 : 1. Однако авторы наблюдали некоторую неоднородность в распределении звеньев и небольшой разброс в ММ, которые усиливались с ростом ММ сополимера.
Параллельно исследовалась возможность конструирования полимеров путем включения в макромолекулу единичного звена мономера (single unit monomer insertion, SUMI). Известные примеры в этой области связаны с использованием ОПЦ-полимеризации. Первые сведения о такого рода синтезе были представлены в работах Zard S.Z. с соавторами [152–154] и связаны с взаимодействием ксантатов (R–O–C(=S)S–R′) с неактивными мономерами (винилацетат, аллиловые соединения) под действием УФ-облучения, фото- и термоинициаторов. Этот подход был в дальнейшем распространен на дитиобензоаты и тритиокарбонаты и на более активные мономеры (стирол, акриловые мономеры) [155–160]. Несмотря на то, что реакция протекала с высоким выходом, в ряде случаев наблюдалось образование различных побочных продуктов, включая олигомеры и продукты рекомбинации радикалов инициатора; образование первых обусловлено относительно невысокой константой передачи на ОПЦ-агент [160]. Для некоторых комбинаций мономерных пар удавалось добиться последовательного введения двух мономерных звеньев (например, стирол и малеиновый ангидрид) [134]. Впоследствии ОПЦ-полимеризация с фотоинициированным переносом электрона/энергии была применена для синтеза коротких олигомеров стирола, акрилатов и других мономеров с контролируемой последовательностью звеньев и узким ММР [161–164]. Например, в работе [164] продемонстрирована возможность последовательного введения трех разных мономерных звеньев в молекулу ОПЦ-агента и получение моно- и телехеликов, содержащих одну или две концевые карбоксильные группы соответственно. При наличии подходящих функциональных заместителей в уходящей группе ОПЦ-агента такие олигомеры могут рассматриваться как заготовки для создания более сложных молекулярных структур:
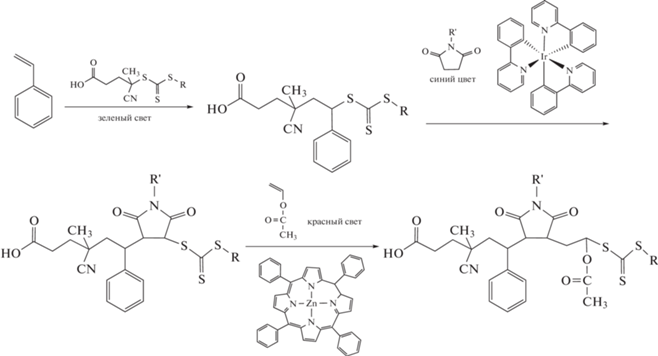
(R = C12H25, CH2CH2COOH; R' = C2H5,  )
)
Основные достижения в синтезе полимеров и олигомеров с контролируемой последовательностью мономерных звеньев путем “точечного” введения мономеров в макромолекулу обобщены в обзорах [134, 135, 165].
В заключение этого раздела стоит привести еще два подхода к конструированию макромолекул с точно заданной последовательностью звеньев.
Первый основан на последовательных многокомпонентных реакциях, в которых каждый мономер содержит функциональную группу для определенной клик-реакции [166]. Сочетанием набора таких функциональных мономеров можно получать линейные, разветвленные и древообразные сополимеры с заданной последовательностью звеньев.
Во втором подходе используют гибридную сополимеризацию, сочетающую два разных механизма при использовании одного и того же вещества и двух мономеров, способных полимеризоваться только по одному из механизмов. Так, сочетание механизма ОПЦ и анионного раскрытия цикла, где в качестве ОПЦ-агентов используют симметричные и несимметричные тритиокарбонаты, дитиобензоаты и дитиокарбаматы, а мономеров – стирол или акриламиды и циклические сульфиды, описано в работах [166–169]. В случае циклического сульфида и дитиокарбонильного соединения (оно же – ОПЦ-агент) полимеризация протекает с раскрытием цикла по анионному механизму в присутствии (C6H5)4P+Hal–:
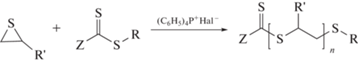
При полимеризации винилового мономера под действием ОПЦ-агента и радикального инициатора образуется полимер с концевыми группами ZC(=S)S– и R–. Затем к полученному полимеру добавляют циклический сульфид и (C6H5)4P+Hal– и проводят полимеризацию сульфида, в результате чего образуется диблок-сополимер. Если в реакционной смеси одновременно содержатся радикальный инициатор и (C6H5)4P+Hal–, то анионный механизм можно активировать нагреванием реакционной смеси, а радикальный – УФ-облучением [169]. Это позволяет получать ди-, три- и мультиблок-сополимеры путем последовательного изменения способа активации полимеризации.
Аналогичная идея лежит в основе сочетания псевдоживой катионной и ОПЦ-полимеризации [170–173]. Для активации катионного механизма в реакционную систему, включающую мономеры, полимеризующиеся по ОПЦ (метилакрилат) или по катионному механизму (изобутилвиниловый эфир), и ОПЦ-агент, вводят кислоту Льюиса, а для радикального механизма – радикальный инициатор. Такую полимеризацию можно проводить последовательно, заменяя один активатор на другой с образованием блок-сополимера и даже мультиблок-сополимера или вводя их одновременно и получая сополимер.
Наконец, в работе [174] был предложен уникальный ОПЦ-агент, позволяющий осуществлять как ОПЦ-полимеризацию, так и полимеризацию с раскрытием цикла. В своей кислой и основной форме он был способен обеспечить полимеризацию по ОПЦ-механизму неактивных и активных мономеров и с раскрытием цикла – мономеров, полимеризующихся под действием нуклеофильных и электрофильных соединений соответственно.
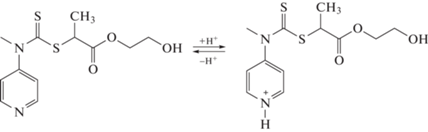
Это позволяет синтезировать мультиблок-сополимеры с заданной длиной блоков и узким ММР путем последовательного введения нужных мономеров и изменения рН среды.
Таким образом, к настоящему времени в руках экспериментаторов появился широкий набор инструментов, основанный на применении радикальной полимеризации с обратимой деактивацией цепи, для создания макромолекул необходимого состава с нужной последовательностью звеньев в цепи.
ГРАДИЕНТНЫЕ СОПОЛИМЕРЫ И ИХ СВОЙСТВА
Синтез градиентных сополимеров
Градиентные сополимеры представляют собой уникальный класс сополимеров, в которых состав плавно изменяется вдоль цепи и один конец макромолекулы обогащен преимущественно одним мономером, а другой – вторым мономером [21, 175–180]. Такое строение отличает градиентные сополимеры

от статистических

и блочных

и придает им необычный комплекс свойств.
Градиентные сополимеры в условиях радикальной полимеризации с обратимой деактивацией цепи образуются в двух случаях: 1) самопроизвольно при сополимеризации мономеров, сильно различающихся по активности (например, активный и неактивный мономер – стирол и N-винилпирролидон [54], акриловая кислота и винилацетат [112]); 2) вынуждено при сополимеризации мономеров близкой активности, когда мономеры вводят в реакцию с разной скоростью, постоянно меняя таким образом состав мономерной смеси (например, стирол и акриловая кислота [73], акрилонитрил и НИПАМ [95], акрилонитрил и акриловая кислота [96]). Оба варианта синтеза пригодны для всех способов реализации радикальной полимеризации с обратимой деактивацией цепи: полимеризации с участием стабильных или малоактивных радикалов [64–67, 70, 71, 74, 76–80, 181–187], полимеризации с переносом атома [47, 56, 81–83, 85, 188–197] и ОПЦ-полимеризации [58, 88, 81, 93, 94, 97, 198–203 ].
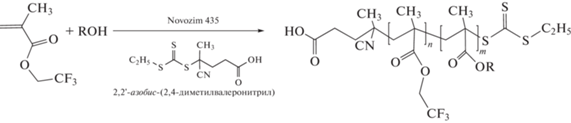
В полунепрерывной сополимеризации мономеров близкой активности чаще один мономер находится в исходной реакционной смеси, а второй вводят в нее с постоянной скоростью. Однако в ряде случаев для создания искусственного градиента используются и другие приемы. Один из них, предложенный в работах группы J.M. Torkelson, заключается в том, чтобы постепенно увеличивать скорость ввода второго мономера в течение полимеризации [69, 70, 77, 185, 204–207]. Другой прием основан на применении безэмульгаторной эмульсионной ОПЦ-полимеризации, в которую последовательно вводят порции стирола и БА разного состава [88]. Роль стабилизатора и полимерного ОПЦ-агента выполняет диблок-сополимер полистирола и ПАК, содержащий 5 и 20 звеньев стирола и АК соответственно. В случае линейного градиента мольная доля вводимого стирола последовательно уменьшается, а для градиента V-типа она сначала уменьшается и затем увеличивается до начального значения. Третий прием заключался в том, что второй мономер образуется in situ по реакции переэтерификации, катализируемой ферментом Novozim® 435 [208]. Трифторэтилметакрилат взаимодействует со спиртом (гексанол, бензиловый спирт, монозащищенный ПЭГ350, (±)-2-октанол) и частично превращается в соответствующий эфир. В присутствии инициатора и ОПЦ-агента запускается ОПЦ-полимеризация и в результате образуется градиентный сополимер
Описанные выше стратегии синтеза градиентных сополимеров все же не позволяют строго контролировать профиль изменения содержания мономеров вдоль полимерной цепи. Для решения этой задачи ряд исследователей осуществил кинетическое моделирование сополимеризации с учетом механизма обратимой деактивации цепи и способа реализации синтеза – непрерывный или полунепрерывный [84, 90, 192, 209–226]. Существенным моментом является знание констант сополимеризации, средних констант элементарных стадий, зависящих от текущего состава мономерной смеси и вязкости реакционной среды. С учетом того, что для многих пар мономеров полный набор требуемых констант неизвестен, авторам приходится использовать поправочные коэффициенты для достижения соответствия между экспериментальными и прогнозируемыми величинами. В работах [84, 90, 215–223] авторы моделировали последовательность звеньев вдоль цепи в зависимости от типа процесса (непрерывный или полунепрерывный) и механизма полимеризации (обратимое ингибирование [220–223] или ОПЦ-полимеризация [224–226]). Сверка соответствия рассчитанного профиля градиентности и экспериментального проводится обычно путем определения изменения среднего состава сополимера методами спектроскопии ЯМР 1Н, ИК-спектроскопии или путем нахождения распределения триадного состава звеньев методом спектроскопии ЯМР 13С.
Свойства градиентных сополимеров
Свойства градиентных сополимеров обсуждаются в многочисленных обзорах и статьях, например в работах [21, 55, 178].
Градиентные сополимеры, как и блок-сополимеры, обладают способностью к самоорганизации в растворе, массе или тонких пленках, однако их поведение различается. Например, блок-градиентные сополимеры ПАК–блок–П(АК–град–стирол) в отличие от блок-сополимера ПАК–блок–ПС растворяются в водно-щелочных средах уже при комнатной температуре и образуют наноразмерные агрегаты при умеренных значениях рН и высокой ионной силе раствора. Они обладают рН-чувствительными свойствами и способны обратимо изменять агрегационное число при изменении рН или ионной силы [49, 73, 123, 183, 227, 228]. В то же время блок-сополимеры ПАК–блок–ПС формируют замороженные агрегаты, устойчивые к изменениям внешней среды; исключение составляют блок-сополимеры с короткими блоками [229]. Аналогичной способностью образовывать динамические агрегаты обладают блок-градиентные сополимеры АК и БА в отличие от триблок-сополимеров ПАК–блок–ПБА–блок–ПАК [230–235]. Градиентные и блок-градиентные сополимеры могут быть стимулочувствительными и образовывать агрегаты при изменении рН или температуры [215, 236–238].
Самоорганизация градиентных сополимеров может происходить и в процессе синтеза, если реакционная среда является термодинамически плохим растворителем для образующегося сополимера. Этот процесс в чем-то близок к полимеризации, вызывающей самоорганизацию (polymerization-induced self-assembly) [239]. Суть ее заключается в следующем. При полимеризации мономера под действием “живого” полимера, полученного одним из вариантов полимеризации с обратимой деактивацией цепи, в растворителе, термодинамически хорошем для “живого” полимера и плохом для растущего блока, происходит постепенная потеря растворимости последнего. Следствием этого является самоорганизация блок-сополимера в растворителе, селективном по отношению к одному из блоков, в частицы нано- или микронного размера определенной морфологии, в которых происходит дальнейший рост второго блока. Аналогичный процесс наблюдается и в том случае, если провести сополимеризацию с обратимой деактивацией цепи мономеров, значительно различающихся своей активностью, в селективном растворителе по отношению к одному из гомополимеров, образуемых из данных мономеров [54, 112, 240–244].
Поведение градиентных и блочных амфифильных сополимеров (например, стирола и АК, БА и АК, октафторпентилакрилата и АК) на межфазной границе также разное [23, 133, 184, 236, 245]. Так, градиентные сополимеры стирола и АК характеризуются наибольшим межфазным давлением на границе раздела хлороформ–вода по сравнению с диблок-сополимерами того же состава и ММ [184]. Аналогичные явления характерны и для градиентных сополимеров фторакрилата и АК [245]. Структурирование наблюдается и в тонких пленках, формирующихся на стеклянной подложке испарением растворителя, для градиентных сополимеров стирола и АК в отличие от их статистических аналогов [23].
Свойства и способность к фазовому разделению в блоке статистических, градиентных и блочных сополимеров определяются параметрами парных взаимодействий (термодинамической совместимостью/несовместимостью гомополимеров) [236, 246–250]. Например, в работе [247] проанализировано поведение статистических композиционно однородных и неоднородных сополимеров, градиентных, блочных и блок-градиентных сополимеров стирола и метилакрилата, а также стирола и трет-бутилакрилата. Микроструктура цепи сополимеров стирола и МА и композиционная (не)однородность влияют на температуру стеклования сополимеров. Градиентные сополимеры образуют оптически прозрачные пленки, как и статистические однородные сополимеры. Кроме того, в ходе микрофазового разделения образуются аналогичные по составу фазы, как и в статистическом неоднородном сополимере. В результате механические свойства этих сополимеров оказываются близкими. Для пары стирол – трет-БА различия в свойствах статистических и градиентных сополимеров менее заметны вследствие близких значений параметра взаимной растворимости мономерных звеньев в полимере. В этом случае микрофазное разделение наблюдается только для блочного и блок-статистического сополимеров, для которых проявляются две температуры стеклования на кривых ДСК. В градиентных сополимерах смешение мономеров вдоль цепи (в отличие от блочных) приводит к большему увеличению совместимости между фазами, чем в блочных [251].
Профиль градиента, а также ММ сополимера влияют на фазовое поведение сополимеров, что проявляется, в частности, в изменении температур и ширины фазовых переходов, релаксации энтальпии в области стеклования [65–70, 74, 77, 252]. Например, температура стеклования статистических и градиентных сополимеров стирола и 4-метилстирола одинакового состава не различается, а ширина и положение пиков восстановления энтальпии отличаются существенно [65]. У первых они уже и интенсивнее, а у вторых шире, и максимум сдвинут в область низких температур. Для сополимеров разнополярных мономеров различие в поведении проявляется ярче. Так, градиентные и блочные бинарные сополимеры стирола с 4-ацетокси- или 4-гидроксистиролом характеризуются двумя температурами стеклования [67]. Для диблок-сополимеров температурные переходы из стеклообразного в высокоэластическое состояние узкие, а для градиентных – широкие. Причем ширина перехода растет с увеличением степени градиентности сополимера. В отличие от градиентных сополимеров статистические сополимеры стирола и 4-гидроксистирола близкого состава (~60 мол. % стирола) характеризуются одной температурой стеклования, и температурный интервал стеклования заметно уже [66]. Интересно, что уменьшение содержания стирола в градиентном сополимере приводит к уширению интервала стеклования и наблюдается только одна температура стеклования. В отличие от статистического сополимера градиентный, добавленный в количестве 5 мас. % к смеси полистирола (80 мас. %) и поликапролактона (20 мас. %), оказывается пригодным для компатибилизации смеси гомополимеров.
Проявление одной или двух температур стеклования у градиентных сополимеров зависит не только от степени градиентности и состава сополимера, но и от термодинамической (не)совместимости компонентов. В работе [69] методом малоуглового рентгеновского рассеяния изучили микрофазное разделение в сополимерах стирола с 4-гидроксистиролом и стирола с БА разной микроструктуры. Микрофазное разделение в высокомолекулярных градиентных сополимерах стирола с 4-гидроксистиролом аналогично блок-сополимерам. С уменьшением ММ градиентных сополимеров оно становится менее выраженным. Для сополимеров стирола с БА те же тенденции, однако микрофазное разделение проявляется слабо. Сравнение сополимеров стирола с 4-гидроксистиролом (компоненты сильно несовместимы) и стирола с БА (умеренно несовместимы) разной микроструктуры методом ДМА показало, что в обоих случаях для выбранных составов блок-сополимеры характеризуются двумя температурами стеклования, а статистические и градиентные – одной [68]. При этом ширина температурного интервала стеклования у градиентных сополимеров в несколько раз больше, чем у статистических. Повышение несовместимости компонентов сопровождается уширением области стеклования. Более детальное исследование влияния степени градиентности (линейная, сигмоидальная, блок-градиентная, блок-статистическая) на процессы стеклования проведено в работе [70] на примере сополимеров стирола с БА, трет-БА, 4-винилпиридином, н-бутилметакрилатом (БМА) и 4-ацетоксистиролом. Полученные результаты хорошо коррелируют с оценками теории самосогласованного поля. Чем выше степень несовместимости компонентов, тем шире температурный интервал стеклования, и наибольшего значения он достигает у сигмоидального градиента. Для слабо несовместимых компонентов (стирол–БМА) последовательность звеньев в градиенте практически не влияет на стеклование сополимера. Сочетание методов спектроскопии диэлектрической релаксации и ДСК позволило на примере сополимеров стирола и БА объяснить разный температурный интервал стеклования у статистических и градиентных сополимеров отличием в спектрах релаксации сегментов [77]. Возможность варьировать ширину температурного интервала стеклования, изменяя химическую природу сомономеров, состав, степень градиентности и ММ, делает градиентные сополимеры привлекательными для использования в качестве амортизационных материалов.
Применение градиентных сополимеров может быть достаточно разнообразным и не ограничивается демпферными материалами [68]. Наряду с блок-сополимерами они могут быть использованы в качестве компатибилизаторов смесей несмешивающихся полимеров [66, 92, 206, 253–256], стабилизаторов гетерофазной полимеризации [181, 182, 227, 257], материалов с памятью формы [258, 200 ], наноконтейнеров и биоконъюгатов для применения в медицине [259–261], ПАВ для экстракции производных металлов в сверхкритическом СО2 [29], для модификации межфазной границы [262], получения разделительных мембран [94, 263], проводящих материалов и фотовольтаики [91, 264–266]. К сожалению, только некоторые из них были реализованы на практике, что, по-видимому, обусловлено необходимостью удешевления и упрощения синтеза, чтобы сделать его технологически эффективным и экономичным.
МУЛЬТИБЛОЧНЫЕ СОПОЛИМЕРЫ И ИХ СВОЙСТВА
Мультиблок-сополимеры – это блок-сополимеры, содержащие в своем составе более трех блоков. С точки зрения контролируемого синтеза их можно получать либо последовательной полимеризацией нового мономера после исчерпания предыдущего (с выделением или без выделения промежуточных полимеров), либо путем сборки готовых блоков в единую конструкцию с помощью клик-реакций. Впервые об использовании радикальной полимеризации с обратимой деактивацией цепи для получения мультиблок-сополимеров сообщили в 1999 г. [267], однако подробное описание такого синтеза появилось позже [268]. Авторы синтезировали пентаблоки стирола и ММА ПММА–блок–ПС–блок–ПММА–блок–ПС–блок–ПММА и ПС–блок–ПММА–блок–ПС–блок–ПММА–блок–ПС полимеризацией с переносом атома в массе под действием бифункционального инициатора, что позволило осуществить реакцию в три стадии. Однако дисперсность блок-сополимеров была относительно высокой (1.8–2.0). Позже для синтеза пентаблок-сополимера ДМАЭМА и ММА применили ОПЦ-полимеризацию с участием монофункционального дитиобензоата [269]. В отличие от полимеризации с переносом атома в ОПЦ-процессе на каждой последующей стадии требуется дополнительно вводить инициатор для образования активных центров. Следствием этого является уширение ММР от стадии к стадии, и дисперсность конечного блока составила 1.8. Независимо, важность оптимизации концентрации инициатора продемонстрирована в работе [270], в которой показано, что при неправильном выборе соотношения [ОПЦ-агент] : [инициатор] происходит не только образование “мертвых” цепей, но и потеря функциональности концевых групп от исходного ОПЦ-агента. Решением проблемы может быть увеличение доли “живых” цепей за счет уменьшения концентрации инициатора или сокращения числа стадий. В работе [271] рассмотрены оба варианта на примере дитиобензоата и симметричного тритиокарбоната, с использованием которых пентаблок-сополимеры образуются в пять и три стадии соответственно. Действительно, уменьшение концентрации инициатора позволило понизить дисперсность на пятой стадии до 1.2, а при проведении пяти стадий с участием тритиокарбоната синтезирован гептаблок-сополимер с дисперсностью ниже 1.2. Согласно другому подходу, описанному в работе [272], уменьшения числа стадий можно добиться применением полифункциональных ОПЦ-агентов, например
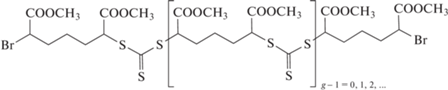
Однако в этом случае следует ожидать высокую композиционную неоднородность и дисперсность конечного продукта, поскольку макромолекула может расти неравномерно. Действительно, полученные блок-сополимеры характеризовались широким, в ряде случаев полимодальным ММР, что подтверждает высказанное соображение.
Следовательно, необходимо менять стратегию синтеза и добиваться композиционной однородности мультиблок-сополимеров. Первые шаги в этом направлении были предприняты в полимеризации с переносом атома и ОПЦ-полимеризации в начале 2010-х годов. Так, сообщается об итеративном методе полимеризации с переносом электрона с участием Cu(0) (вариант полимеризации с переносом атома), позволяющем с высокой степенью контролировать микроструктуру мультиблок-сополимера A–B–C–D–E–… с высоким выходом без необходимости очистки промежуточных продуктов [273]. Реализация подхода была продемонстрирована как на последовательной гомополимеризации МА, в которой по окончании полимеризации мономера добавлялась его следующая порция, так и на синтезе мультиблок-сополимера ПМА–блок–ПБА–блок–ПЭА–блок–ПЭГА–блок–ПЭА–блок–ПБА (ПЭА – полиэтилакрилат, ПЭГА – поли-2-этилгексилакрилат):
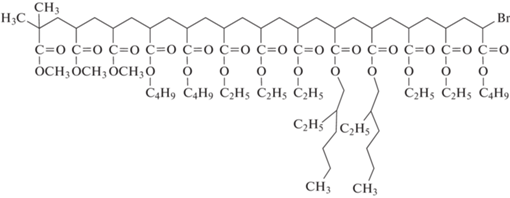
Правда, блок-сополимером данный продукт можно назвать весьма условно, так как длина каждого блока составляет от 1 до 5 в зависимости от стадии, что обусловлено применением очень высокой концентрации инициатора этил-2-бромизобутирата. Тем не менее, авторам удалось получить продукт высокой композиционной однородности. Кроме того, показаны разные варианты модификации концевой группы (Br) для последующего использования в клик-реакциях. Этот же прием был использован в работе [274] для синтеза звездообразного мультиблок-сополимера, каждый из пяти лучей которого состоял из четырех коротких блоков полиакрилатов с разными алкильными заместителями. Близкая идея реализована в работе [275] при синтезе пентаблок-сополимеров на основе глицидилакрилата и 3-триметилсиланилпроп-2-инилового эфира акриловой кислоты, в которых длина блоков была выше за счет уменьшения соотношения инициатор : мономер в четыре раза. Используя глицидильную и триметилсилильную группы, авторы затем провели их модификацию путем тиол-галоген, тиол-эпокси и азид-алкин клик-реакций:
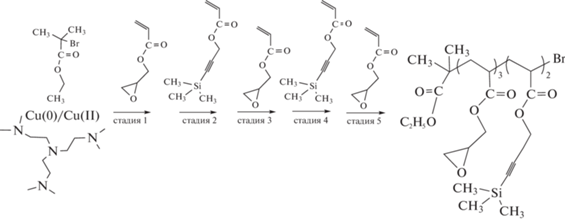
Описанный выше подход был усовершенствован в работе [276]. Путем аккуратного выбора соотношения концентраций инициатора, CuBr2 и лиганда авторам удалось найти условия для реализации первой и последующей стадий с максимальным выходом “живых” цепей и конверсией мономера 95–100%, наращивая на каждом этапе блок длиной в ~100 звеньев. Тем не менее на хроматограммах промежуточных и конечных продуктов блок-сополимеризации видны небольшие плечи в низкомолекулярной области, отвечающие “мертвому” полимеру, содержащемуся в блоках-предшественниках. Следующий шаг в развитии этого направления – синтез линейных триблок-сополимеров в воде с узким ММР (Đ = = 1.1), высоким выходом на каждой стадии и содержанием “живых” цепей более 90% [277], а затем и мультиблок-сополимеров на основе ДМАА, НИПАМ, ДЭАА и монометилового эфира полиэтиленгликольакрилата (ПЭГА) [278], а также 4-акрилоилморфолина (АМ) [279] и звездообразных мультиблок-сополимеров [280]. Наконец, повысить эффективность получения мультиблок-сополимеров удалось, применив в полимеризации с переносом электрона УФ-облучение (λмакс = 360 нм) [281]. С увеличением длины блоков, естественно, количество “живых” цепей уменьшается. В связи с этим число блоков, которые можно нарастить без заметного уширения ММР, падает с ростом их степени полимеризации Pn. Если макромолекула состоит из 11 блоков, Pn каждого из которых равна 3, построенных из метил-, этил-, гидроксиэтил- и солкетальакрилата, то дисперсность конечного блок-сополимера Đ равна 1.19. В случае 8 блоков из тех же мономеров с Pn каждого из которых 10, дисперсность Đ = 1.10. При Pn = 25 каждого из 6 блоков Đ = 1.15 и при Pn = 100 каждого из 5 блоков Đ = 1.21. Преимущество этого подхода – возможность мгновенного прекращения инициирования путем выключения облучения и возможность проведения реакции при комнатной и пониженной температуре, что было реализовано в работах [282–285].
Наряду с полимеризацией с переносом атома (электрона) ОПЦ-полимеризация также активно стала использоваться для конструирования мультиблок-сополимеров. Так, впервые методом ОПЦ удалось синтезировать гекса-, додека- и икосаблок-сополимеры [286]:
ПБА25–блок–ПАМ5–блок–П(АМ7–стат–ГЭА3) –блок–ПАМ5–блок– П(АМ7–стат–АК5) –блок–ПНИПАМ19,
ПДМАА10–блок–ПАМ10–блок–ПДЭАА10–блок–ПНИПАМ10–блок–ПДМАА10–блок–ПАМ10–блок–ПДЭАА10–блок–ПНИПАМ10–блок– ПДМАА10–блок–ПАМ10–блок–ПДЭАА10–блок–ПНИПАМ10,
ПАМ10–блок-ПАМ3–блок–ПДЭАА3–блок–ПАМ3–блок–ПДМАА3–блок–ПАМ3–блок– ПДЭАА3–блок–ПАМ3–блок–ПДМАА3–блок–ПАМ3–блок–ПДЭАА3–блок–ПАМ3–блок– ПДМАА3–блок-ПАМ3–блок–ПДЭАА3–блок–ПАМ3–блок–ПДМАА3–блок–ПАМ3–блок– ПДЭАА3–блок–ПАМ3–блок–ПДМАА3,
где ГЭА – гидроксиэтилакрилат. В дальнейшем условия синтеза мультиблок-сополимеров удалось оптимизировать так, чтобы конверсия на каждой стадии составляла не менее 99%, а количество “живых” цепей не менее 97% [287, 288]. Ключевым моментом в этом синтезе является выбор мономеров с высокой константой скорости роста для достижения высокого выхода, соотношения концентраций инициатора и ОПЦ-агента, а также температуры для практически полного разложения инициатора на каждой стадии. Это позволяет уменьшить прирост “мертвых” цепей и минимизировать длину “переходной зоны” между блоками, образующейся за счет сополимеризации остаточного с предыдущей стадии мономера с добавленным на новой стадии мономером. Данное соображение было доказано моделированием образования декаблок-сополимера и сравнением с экспериментально полученными данными [289].
Сократить число стадий можно путем повышения функциональности ОПЦ-агента. Так, описан трехстадийный синтез пентаблок-сополимера ПГЭМА–блок–ПДМАЭМА–блок–ПЭГМА–блок–ПДМАЭМА–блок–ПЭГМА и ПДМАЭМА–блок–ПЭГМА–блок–ПДМАЭМА–блок–ПЭГМА–блок–ПДМАЭМА с использованием тритиокарбоната [290]:
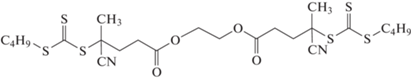
Следующим шагом в оптимизации синтеза стало фантастическое увеличение скорости реакции – каждая стадия завершалась в течение ~3 мин при протекании реакции на воздухе [291]. Этого удалось достичь применением низкотемпературного инициатора 2,2'-азобис-[2-(2-имидазолин-2-ил)пропан]дигидрохлорида (VA-044) и высокой температуры синтеза. Данный процесс можно проводить и на уровне микроколичеств реагирующих веществ, сохраняя его преимущества: высокую скорость и толерантность к кислороду воздуха [292].
Масштабирование синтеза мультиблок-сополимеров методом ОПЦ с помощью последовательности проточных реакторов осуществлено в работе [293]. Количество реакторов определяется типом продукта (гомополимер, диблок-, триблок-, тетраблок-сополимер и т.д.). Время реакции при 100оС в каждом реакторе составляет 40 мин. Другой вариант масштабирования реализован в работе [294], согласно которому конструкция предполагает возможность как циркуляции реакционной смеси по замкнутому контуру системы, так и подачу в нее нового мономера и выделение продуктов. Авторы использовали низкотемпературный инициатор VA-044 и проводили каждую стадию при 70оС в течение 2 ч. Этим способом синтезированы блок-сополимеры ПАМ20–блок–ПДМАА20–блок–ПДЭАА20, ПАМ10–блок–ПДМАА10– блок–ПДЭАА10–блок–ПАМ10–блок–ПДМАА10–блок–ПДЭАА10 и ПАМ10–блок–ПГЭАА10–блок–ПНИПАМ10–блок–ПАМ10–блок–ПГЭАА10–блок– ПНИПАМ10. В обоих случаях необходимо учитывать вязкость реакционной среды, чтобы поток с промежуточными продуктами мог смешиваться с вводимыми мономерами и перемещаться по контуру системы.
Другой интересный вариант синтеза мультиблок-сополимеров [295] основан на идее, предложенной в работе [208], которая заключается в сочетании реакции переэтерификации трифторэтилакрилата с различными спиртами, катализируемой ферментом, и ОПЦ-полимеризации. В данном случае ОПЦ-полимеризация инициировалась переносом электрона под действием УФ-облучения при комнатной температуре, и в реакционную систему последовательно вводили трифторэтилакрилат и необходимые спирты. В результате удалось получить пентаблок- и триблок-сополимеры
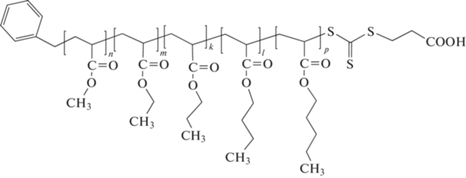
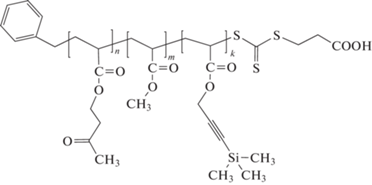
Катализируемая ферментами ОПЦ-полимеризация, приводящая к синтезу мультиблок-сополимеров, описана в работе [296]. В результате реакции пиранозооксидазы с D-глюкозой вырабатывается перекись водорода, взаимодействующая с пероксидазой хрена, что приводит к появлению радикалов ацетилацетона, инициирующих последовательную блок-сополимеризацию ДМАА, АМ, ПЭГА и ПЭГМА. Особенностью этого процесса является его толерантность к кислороду воздуха, что исключает стадии дегазации или продувания смеси инертным газом.
ОПЦ-полимеризация с переносом электрона под действием УФ-облучения желтым LED светом (λ = 560 нм), в присутствии 5,10,15,20-тетрафенил-21Н,23Н-порфирина цинка в качестве фотокатализатора, подобно описанной выше [160–164], была использована для синтеза мультиблок-сополимеров [297]. Этот подход применим для широкого круга мономеров (акриламиды, акрилаты, метакрилаты, стирол), и верхняя граница контроля степени полимеризации Pn составляет 1000 (Đ < 1.3). При более высоких значениях Pn молекулярно-массовое распределение полимеров значительно уширяется. Данный метод синтеза использовали для получения мультиблок-сополимеров, обладающих антимикробной активностью [298].
Наряду с линейными мультиблок-сополимерами предложены варианты синтеза мультиблок-сополимеров более сложной архитектуры. Например, в работе [299] описан интересный способ получения привитых сополимеров путем модификации боковых заместителей и синтеза полимерного ОПЦ-агента содержащего группы ZC(=S)S как на конце цепи, так и в боковых заместителях. В результате при добавлении в реакционную среду нового мономера и свежей порции инициатора наращивается блок гомополимера не только в основной, но и боковых цепях:
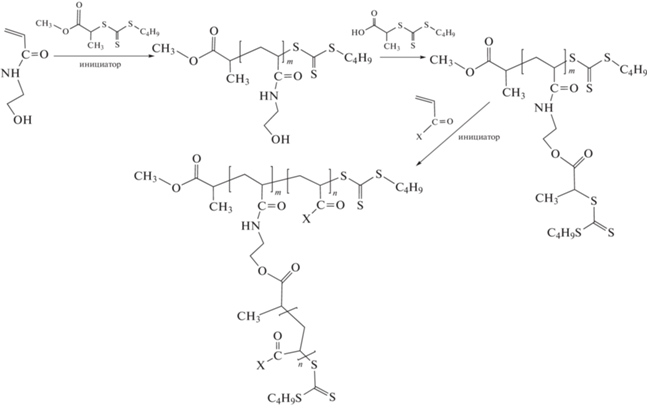
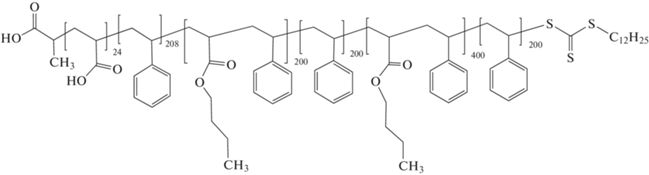
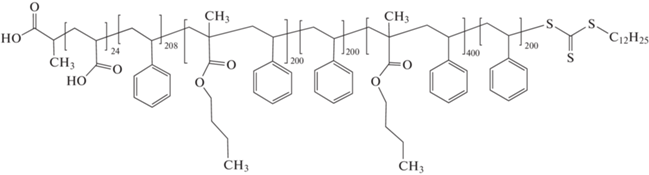
В серии исследований [300–302] разработан вариант эмульсионной ОПЦ-полимеризации, позволяющий осуществлять синтез мультиблок-сополимеров с высокой эффективностью и контролем длины каждого блока. По сути это хорошо известный вариант затравочной полимеризации. На примере трех важнейших классов мономеров – акрилатов, метакрилатов и стирола показана возможность синтеза мультиблок-сополимера с Mn до 140 × 103 и продолжительностью каждой стадии не более 3 ч. Благодаря термодинамической несовместимости блоков их сегрегация происходила внутри образующихся полимерных частиц и приводила к образованию структурированных нанообъектов:
ПАК–блок–ПС–блок–П(БА–со–стирол) –блок–ПС–блок–П(БА–со–стирол)–блок–ПС
ПАК–блок–ПС–блок–П(БМА–со–стирол) –блок–ПС–блок–П(БМА–со–стирол)–блок–ПС
Хорошо известно, что в ОПЦ-полимеризации в массе или в растворе порядок введения мономеров в синтез важен с точки зрения контролируемого синтеза блок-сополимеров. Этот порядок определяется тем, какой из полимерных заместителей является лучшей уходящей группой в структуре радикального интермедиата [2]. Однако данную проблему удается решить, если к полимеризации “плохого” мономера добавить некоторое количество “хорошего” мономера, который поможет сдвинуть равновесие при распаде интермедиата в нужную сторону. Такой прием известен в сополимеризации с участием нитроксилов и ОПЦ-сополимеризации, однако в эмульсионной полимеризации для синтеза мультиблок-сополимеров был применен впервые, что дало возможность произвольным образом конструировать блок-сополимеры на основе стирола и метакрилатов [301].
В заключение этого раздела стоит привести пример бессерной ОПЦ-полимеризации [303, 304]. На самом деле речь идет о каталитической передаче цепи с участием кобалоксимов в эмульсионной полимеризации метакриловых мономеров (метил-, этил, бутил- и бензилметакрилат). В результате образуется макромолекула с концевой двойной связью, способная участвовать в полимеризации. Обычно такие процессы приводят к образованию олигомеров. Однако выбранные условия позволяют осуществить синтез гекса- и декаблок-сополимеров с длиной блока 10–50 звеньев, высоким выходом и низкой дисперсностью Đ ∼ 1.2.
В отличие от градиентных сополимеров мультиблок-сополимеры являются достаточно новыми объектами и их свойства только начинают изучать. Очевидно, что области применения градиентных сополимеров будут зависеть не только от химического состава блоков, но и от их длины. Анализ литературы показывает, что короткие мультиблок-сополимеры можно использовать для создания различных биоконъюгатов и носителей, в то время как длинные мультиблок-сополимеры, способные к микрофазному разделению, могут представлять интерес для компатибилизации сложных смесей полимеров.
ЗАКЛЮЧЕНИЕ
В обзоре рассмотрены разные подходы к контролируемому синтезу макромолекул сополимеров заданной архитектуры в условиях полимеризации с обратимой деактивации цепи. Наиболее востребованными из них являются полимеризация с переносом электрона в присутствии Cu(0) и ОПЦ-полимеризация. При этом достаточно перспективными, по-видимому, становятся процессы под действием УФ-облучения, позволяющие мгновенно включать и выключать инициирование полимеризации.
Очевидно, что в руках химиков появился достаточно мощный набор инструментов для создания сополимеров требуемого строения и функционала. Если градиентные сополимеры уже стали достаточно известным классом сополимеров с понятным комплексом свойств, зависящим от химического состава, степени градиентности и в ряде случаев от ММ, то мультиблок-сополимеры только начинают свое активное продвижение. С точки зрения практического применения оба класса сополимеров являются привлекательными, но пока еще требуют разработки методов простого технического воплощения.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 19-33-90096).
Список литературы
Controlled and Living Polymerizations: From Mechanisms to Applicatons / Ed. by A.H.E. Muller, K. Matyjaszewski. Weinheim: Wiley-VCH, 2009.
RAFT Polymerization: Methods, Synthesis, Applications / Ed. by G. Moad, E. Rizzardo. Weinheim: Wiley-VCH, 2022.
Higashihara T., Nagura M., Inoue K., Haraguchi N., Hirao A. // Macromolecules. 2005. V. 38. P. 4577.
Zhao Y., Higashihara T., Sugiyama K., Hirao A. // Macromolecules. 2007. V. 40. P. 228.
Block copolymers / Ed. by N. Hadjichristidis, S. Pispas, G. Floudas. Hoboken: Wiley-Interscience, 2003.
Chernikova E.V., Sivtsov E.V. // Polymer Science B. 2017. V. 59. № 2. P. 117.
Corrigan N., Jung K., Moad G., Hawker C.J., Matyjaszewski K., Boyer C. // Progr. Polym. Sci. 2020. Art.101311.
Zaremski M.Yu., Melik-Nubarov N.S. // Polymer Science C. 2021. V. 63. № 2. P. 126.
Meleshko T.K., Razina A.B., Bogorad N.N., Kurlykin M.P., Kashina A.V., Gofman I.V., Ten’kovtsev A.V., Yakimansky A.V. // Polymer Science B. 2021. V. 63. № 5. P. 385.
Gofman I.V., Nikolaeva A.L., Abalov I.V., Ivan’kova E.M., Kuntzman I.V., Goikhman M.Ya., Yakimansky A.V., Kozlova T.O., Baranchikov A.E., Ivanov V.K. // Polymer Science B. 2020. V. 62. № 6. P. 196.
Voit B.I., Lederer A. // Chem. Rev. 2009. V. 109. P. 5924.
Yokozawa T., Ohta Y. // Chem. Rev. 2016. V. 116. № 4. P. 1950.
Anastasaki A., Nikolaou V., Nurumbetov G., Wilson P., Kempe K., Quinn J.F., Davis T.P., Whittaker M.R., Haddleton D.M. // Chem. Rev. 2016. V. 116. P. 835.
Chernikova E.V., Lysenko E.A., Serkhacheva N.S., Prokopov N.I. // Polymer Science C. 2018. V. 6. № 2. P. 192.
d’Agosto F., Rieger J., Lansalot M. // Angew. Chem. Int. Ed. 2020. V. 59. № 22. P. 8368.
Mansfeld U., Pietsch C., Hoogenboom R., Becer C.R., Schubert U.S. // Polym. Chem. 2010. V. 1. P.1560.
Chernikova E.V., Kudryavtsev Y.V. // Polymers. 2022. V. 14. P. 570.
Huang J., Xiao Z., Liang H., Lu J. // Polym. Int. 2014. V. 63. P. 1122.
Islam M.R., Bach L.G., Park J.M., Hong S.-S., Lim K.T. // J. Appl. Polym. Sci. 2013. V. 127. P. 1569.
Shi Y., Sun J.Z., Qin A. // J. Polym. Sci., Polym. Chem. 2017. V. 55. P. 616.
Alam M.M., Jack K.S., Hill D.J.T., Whittaker A.K., Peng H. // Eur. Polym. J. 2019. V. 116. P. 394.
Chalykh A.E., Nikulova U.V., Gerasimov V.K., Khasbiullin R.R. // Polymer Science A. 2020. V. 62. № 2. P. 85.
Mineeva K.O., Medentseva E.I., Plutalova A.V., Serkhacheva N.S., Bol’shakova A.V., Lysenko E.A., Chernikova E.V. // Polymer Science B. 2021. V. 63. № 6. P. 456.
Kalugin D.I., Zaremski M.Yu., Golubev V.B., Garina E.S. // Polymer Science B. 2011. V. 53. № P. 307.
Huang Y., Liu X., Zhang F., Dong J., Luo Y., Huang C. // Polym. J. 2013. V. 45. P. 125.
Kelley D.J., Tobolsky A.V. // J. Am. Chem. Soc. 1959. V. 81. № 7. P.1597.
Zaremski M.Y. // Polymer Science C. 2015. V. 57. № 1. P. 65.
Mühlebach A., Gaynor S.G., Matyjaszewski K. // Macromolecules. 1998. V. 31. № 18. P. 6046.
Ribaut T., Lacroix-Desmazes P., Fournel B., Sarrade S. // J. Polym. Sci., Polym. Chem. 2009. V. 47. P. 5448.
Otsu T., Ogawa T., Yamamoto T. // Macromolecules. 1986. V. 19. № 7. P. 2087.
Paraskeva S., Hadjichristidis N. // J. Polym. Sci., Polym. Chem. 2000. V. 38. P. 931.
Huang W., Ma H.W., Han L., Liu P.B., Yang L.C., Shen H.Y, Hao X., Li Y. // Macromolecules. 2018. V. 51. P. 3746.
Hutchings L.R., Brooks P.P., Parker D., Mosely J.A., Sevinc S. // Macromolecules. 2015. V. 48. P. 610.
O’Driscoll K., Patsiga R. // J. Polym. Sci. Gen. Pap. 1965. V. 3. P. 1037.
Кабанов В.А., Зубов В.П., Семчиков Ю.Д. Комплексно-радикальная полимеризация. М.: Химия, 1987.
Jenkins A.D., Jones R.G., Moad G. // Pure Applied Chem. 2010. V. 82. № 2. P. 483.
Handbook of Radical Polymerization / Ed. by K. Matyjaszewski, T.P. Davis. Hoboken: Wiley-Interscience, 2002.
Handbook of RAFT Polymerization / Ed. by C. Barner-Kowollik. Weinheim: Wiley-VCH Verlag, 2008.
Lamontagne H.R., Lessard B.H. // ACS Appl. Polym. Mater. 2020. V. 2. № 12. P. 5327.
Yamago S. // J. Polym. Sci., Polym. Chem. 2006. V. 44. № 1. P. 1.
Magenau A.J., Strandwitz N.S., Gennaro A., Matyjaszewski K. // Science. 2011. V. 332. P. 81.
Zaremski M.Yu., Plutalova A.V., Lachinov M.B., Golubev V.B. // Macromolecules. 2000. V. 33. P. 4365.
Zaremski M.Yu., Plutalova A.V., Eremeev I. // Macromol. Theory Simul. 2016. V. 25. № 4. P. 413.
Charleux B., Nicolas J., Guerret O. // Macromolecules. 2005. V. 38. P. 5485.
Moad G., Rizzardo E., Thang S.H. // Austral. J. Chem. 2012. V. 65. № 8. P. 985.
Chernikova E.V., Terpugova P.S., Trifilov M.Y., Garina E.S., Golubev V.B., Sivtsov E.V. // Polymer Science A. 2009. V. 51. № 6. P. 658.
Lee S.B., Russell A.J., Matyjaszewski K. // Biomacromolecules. 2003. V. 4. № 5. P. 1386.
Couvereur L., Charleux B., Guerret O., Magnet S. // Macromol. Chem. Phys. 2003. V. 204. P. 2055.
Borisova O., Billon L., Zaremski M., Grassl B., Bakaeva Z., Lapp A., Stepanek P., Borisov O. // Soft Matter. 2012. V. 8. P. 3649.
Zaremski M.Y., Reznichenko A.L., Grinevich Y.V., Garina E.S., Lachinov M.B., Golubev V.B. // Polymer Science A. 2005. V. 47. № 6. P. 536.
Chernikova E.V., Yulusov V.V., Mineeva K.O., Garina E.S., Sivtsov E.V. // Polymer Science B. 2012. V. 54. № 7–8. P. 349.
Chernikova E.V., Tarasenko A.V., Yulusov V.V., Garina E.S., Golubev V.B. // Polymer Science A. 2009. V. 51. № 6. P. 667.
Zaremski M.Y., Zhaksylykov A.B., Orlova A.P., Garina E.S., Badun G.A., Lachinov M.B., Golubev V.B. // Polymer Science. A. 2005. V. 47. № 6. P. 526.
Zaremski M.Y., Kozhunova E.Yu., Abramchuk S.S., Glavatskaya M.E., Chertovich A.V. // Mendeleev Commun. 2021. V. 31. № 2. P. 277.
Zaremski M.Yu., Kalugin D.I., Golubev V.B. // Polymer Science A. 2009. V. 51. № 1. P. 103.
Matyjaszewski K., Ziegler M.J., Arehart S.V., Greszta D., Pakula T. // J. Phys. Org. Chem. 2000. V. 13. P. 775.
Matyjaszewski K., Xia J. // Chem. Rev. 2001. V. 101. P. 2921.
Rizzardo E., Chiefari J., Chong B.Y.K., Ercole F., Krstina J., Jeffery J., Le T.P.T., Mayadunne R.T.A., Meijs G.F., Moad C.L., Moad G., Thang S.H. // Macromol. Symp. 1999. V. 143. P. 291.
Farcet C., Charleux B., Pirri R. // Macromol. Symp. 2002. V. 182. P. 249.
Kotani Y., Kamigaito M., Sawamoto M. // Macromolecules. 1998. V. 31. P. 5582.
Polymer Handbook / Ed. by J. Brandrup, E.H. Immergut, E.A. Grulke. New York: Wiley, 1999.
Toms R.V., Balashov M.S., Gervald A.Yu., Prokopov N.I., Plutalova A.V., Berkovich A.K., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 6. P. 660.
Toms R.V., Prokopov N.I., Mineeva K.O., Plutalova A.V., Chernikova E.V. // Mendeleev Commun. 2022. V. 32. № 2. P. 238.
Gray M.K., Zhou H., Nguyen S.T., Torkelson J.M. // Macromolecules. 2004. V. 37. № 15. P. 5586.
Gray M.K., Zhou H., Nguyen S.T., Torkelson J.M. // Polymer. 2004. V. 45. № 14. P. 4777.
Kim J., Zhou H., Nguyen S.T., Torkelson J.M. // Polymer. 2006. V. 47. № 16. P. 5799.
Kim J., Mok M.M., Sandoval R.W., Woo D.J., Torkelson J.M. // Macromolecules. 2006. V. 39. № 18. P. 6152.
Mok M.M., Kim J., Torkelson J.M. // J. Polym. Sci., Polym. Phys. 2008. V. 46. № 1. P. 48.
Mok M.M., Pujari S., Burghardt W.R., Dettmer C.M., Nguyen S.T., Ellison C.J., Torkelson J.M. // Macromolecules. 2008. V. 41. № 15. P. 5818.
Mok M.M., Kim J., Wong C.L.H., Marrou S.R., Woo D.J., Dettmer C.M., Nguyen S.T., Ellison C.J., Shull K.R., Torkelson J.M. // Macromolecules. 2009. V. 42. № 20. P. 7863.
Sandoval R.W., Williams D.E., Kim J., Roth C.B., Torkelson J.M. // J. Polym. Sci., Polym. Phys. 2008. V. 46. № 24. P. 2672.
Wylie K., Bennett I., Maric M. // Soft Matter. 2017. V. 13. № 15. P. 2836.
Borisova O., Billon L., Zaremski M., Grassl B., Bakaeva Z., Lapp A., Stepanek P., Borisov O. // Soft Matter. 2012. V. 8. № 29. P. 7649.
Wong C.L.H., Kim J., Torkelson J.M. // J. Polym. Sci., Polym. Phys. 2007. V. 45. № 20. P. 2842.
Cherifi N., Issoulie A., Khoukh A., Benaboura A., Save M., Derail C., Billon L. // Polym. Chem. 2011. V. 2. № 8. P. 1769.
Karaky K., Pere E., Pouchan C., Desbrieres J., Derail C., Billon L. // Soft Matter. 2006. V. 2. № 9. P. 770.
Mok M.M., Masser K.A., Runt J., Torkelson J.M. // Macromolecules. 2010. V. 43. № 13. P. 5740.
Karaky K., Billon L., Pouchan C., Desbrieres J. // Macromolecules. 2007. V. 40. № 3. P. 458.
Karaky K., Derail C., Reiter G., Billon L. // Macromol. Symp. 2008. V. 267. P. 31.
Karaky K., Clisson G., Reiter G., Billon L. // Macromol. Chem. Phys. 2008. V. 209. № 7. P. 715.
Chen J., Li J.-J., Luo Z.-H. // J. Polym. Sci., Polym. Chem. 2013. V. 51. № 5. P. 1107.
Fu Y., Cunningham M.F., Hutchinson R.A. // Macromol. Symp. 2007. V. 259. P. 151.
Jakubowski W., Juhari A., Best A., Koynov K., Pakula T., Matyjaszewski K. // Polymer. 2008. V. 49. № 6. P. 1567.
Zhao Y., Luo Y.-W., Ye C., Li B.-G., Zhu S. // J. Polym. Sci., Polym. Chem. 2009. V. 47. № 1. P. 69.
Zhou Y.-N., Luo Z.-H. // Polym. Chem. 2013. V. 4. № 1. P. 76.
Inoue Y., Watanabe J., Takai M., Yusa S.-I., Ishihara K. // J. Polym. Sci., Polym. Chem. 2005. V. 43. № 23. P. 6073.
Chen Y., Luo W., Wang Y., Sun C., Han M., Zhang C. // J. Colloid Interface Sci. 2012. V. 369. № 1. P. 46.
Guo Y., Zhang J., Xie P., Gao X., Luo Y. // Polym. Chem. 2014. V. 5. № 10. P. 3363.
Li X., Wang W.-J., Weng F., Li B.-G., Zhu S. // Industr. Eng. Chem. Res. 2014. V. 53. № 18. P. 7321.
Sun X., Luo Y., Wang R., Li B.-G., Zhu S. // AIChE J. 2008. V. 54. № 4. P. 1073.
Zheng Z., Gao X., Luo Y., Zhu S. // Macromolecules. 2016. V. 49. № 6. P. 2179.
Wang H., Zhou H., Chen Y., Zhang C. // Colloid Polym. Sci. 2014. V. 292. № 11. P. 2803.
Zhang G., Zhang Q., Wang Q., Zhan X., Chen F. // J. Appl. Polym. Sci. 2016. V. 133. № 5. Art.42936.
Zhang G., Jiang J., Zhang Q., Gao F., Zhan X., Chen F. // Langmuir. 2016. V. 32. № 5. P. 1380.
Moskowitz J.D., Wiggins J.S. // Polymer. 2016. V. 84. P. 311.
Skvortsov I.Y., Chernikova E.V., Kulichikhin V.G., Varfolomeeva L.A., Kuzin M.S., Toms R.V., Prokopov N.I. // Materials. 2020. V. 13. № 16. P. 3454.
Saubern S., Nguyen X., Nguyen V., Gardiner J., Tsanaktsidis J., Chiefari J. // Macromol. React. Eng. 2017. V. 11. № 5. Art.1600065.
Seiner J.A., Litt M. // Macromolecules. 1971. V. 4. P. 308.
Cais R.E., Farmer R.G., Hill D.T.J., O’Donnell J.H. // Macromolecules. 1979. V. 12. P. 835.
Зубов В.П., Кабанов В.А. // Высокомолек. соед. А. 1971. Т. 13. № 6. С.1305.
Rzaev Z.M.O. // Prog. Polym. Sci. 2000. V. 25. P. 163.
Кабанов В.А., Топчиев Д.А. // Высокомолек. соед. А. 1971. Т. 13. № 6. С. 1324.
Saini G., Leoni A., Franco S. // Makromol. Chem. 1971. V. 144. P. 235.
Minsk L.M., Kotlarchik C., Darlak R.S. // J. Polym. Sci., Polym. Chem. Ed. 1973. V. 11. № 2. P. 353.
Семчиков Ю.Д., Рябов А.В., Кашаева В.Н. // Высокомолек. соед. Б. 1972. Т. 14. № 2. С. 138.
Семчиков Ю.Д., Рябов А.В., Кашаева В.Н. // Высокомолек. соед. Б. 1970. Т. 12. № 8. С. 567.
Vasilyeva E.V., Kopylova N.A., Zaitsev S.D., Semchikov Yu.D. // Polymer Science B. 2012. V. 54. № 1–2. P. 15.
Zaitsev S.D., Semchikov Yu.D., Chernikova E.V. // Polymer Science B. 2009. V. 51. № 3–4. P. 84.
Vishnevetskii D.V., Plutalova A.V., Yulusov V.V., Zotova O.S., Chernikova E.V., Zaitsev S.D. // Polymer Science B. 2015. V. 57. № 3. P. 197.
Palmiero U.C., Chovancová A., Cuccato D., Storti G., Lacik I., Moscatelli D. // Polymer. 2016. V. 98. P. 156.
Wang W., Xie W.-Y., Wang G-X., Xu W., Liang E. // Iran. Polym. J. 2021. V. 30. P. 1.
Kozhunova E.Yu., Plutalova A.V., Chernikova E.V. // Polymers. 2022. V. 14. P. 555.
Ito T., Otsu T. // J. Macromol. Sci., Chem. 1969. V. 3. P. 197.
Chapiro A., Dulieu J. // Eur. Polym. J. 1977. V. 13. P. 563.
Saini G., Leoni A., Franco S. // Die Makromol. Chem. 1971. V. 144. № 1. P. 235.
Liang K., Hutchinson R.A. // Macromolecules. 2010. V. 43. № 15. P. 6311.
Fernández-García M., Fernández-Sanz M., Madruga L., Cuervo-Rodriguez R., Hernández-Gordo V., Fernández-Monreal M.C. // J. Polym. Sci., Polym. Chem. 2000. V. 38. № 1. P. 60.
Fernández-Monreal M.C., Martinez G., Sánchez-Chaves M., Madruga L. // J. Polym. Sci., Polym. Chem. 2001. V. 39. P. 2043.
Idowu L.A., Hutchinson R.A. // Polymers. 2019. V. 11. P. 487.
Madruga E.L. // Prog. Polym. Sci. 2002. V. 27. P. 1879.
Plochocka K. // J. Macromol. Sci. C. 1981. V. 20. P. 67.
Harwood H.J. // Makromol. Chem., Macromol. Symp. 1987. V. 10–11. P. 331.
Semchikov Y.D., Smirnova L.A., Knyazewa T.Y., Bulgakova S.A., Sherstyanykh V.I. // Eur. Polym. J. 1990. V. 26. P. 883.
Egorochkin G.A., Semchikov Yu.D., Smirnova L.A., Karyakin N.V., Kut’in A.M. // Eur. Polym. J. 1992. V. 28. № 6. P. 681.
Semchikov Yu.D., Smirnova L.A., Kopylova N.A., Izvolenskii V.V. // Eur. Polym. J. 1996. V. 32. № 10. P. 1213.
Semchikov Yu.D. // Makromol. Chem., Macromol. Symp. 1996. V. 111. P. 317.
Ludin D.V., Kuznetsova Yu.L., Zamyshlyaeva O.G., Zaitsev S.D. // Polymer Science B. 2017. V. 59. № 1. P. 7.
Ludin D.V., Zaitsev S.D. // Rus. Chem. Bull., Int. Ed., 2017. V. 66. № 6. P. 1109.
Mella M., La Rocca M.V., Miele Y., Izzo L. // J. Polym. Sci., Polym. Chem. 2018. V. 56. № 13. P. 1366.
Pai T.S.C., Barner-Kowollik C., Davis T.P., Stenzel M.H. // Polymer. 2004. V. 45. P. 4383.
Borisova O.V., Zaremski M.Yu., Borisov O.V., Billon L. // Polymer Science B. 2013. V. 55. № 11–12. P. 573.
Chernikova E.V., Zaitsev S.D., Plutalova A.V., Mineeva K.O., Zotova O.S., Vishnevetsky D.V. // RSC Adv. 2018. V. 8. Art. 14300.
Mineeva K.O., Osipova N.I., Zaitsev S.D., Plutalova A.V., Medentseva E.I., Serkhacheva N.S., Lysenko E.A., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 6. P. 630.
Sequence Controlled Polymers / Ed. by J.-F. Lutz. Weinheim: Wiley VCH, 2017.
Neve J.De, Haven J.J., Maes L., Junkers T. // Polym. Chem. 2018. V. 9. P. 4692.
Tsuchida E., Tomono T. // Makromol. Chem. 1971. V. 141. P. 265.
Moore E., Pickelman D. // Ind. Eng. Chem. Prod. Res. Dev. 1986. V. 25. № 4. P. 603.
Matsumoto A., Kubota T., Otsu T. // Macromolecules. 1990. V. 23. P. 4508.
Tsuchida E., Tomono T., Sano H. // Makromol. Chem. 1972. V. 151. P. 242.
Chernikova E., Terpugova P., Bui C., Charleux B. // Polymer. 2003. V. 44. № 15. P. 4101.
De Brouwer H., Schellekens M.A.J., Klumperman B., Monteiro M.J., German A.L. // J. Polym. Sci., Polym. Chem. 2000. V. 38. P. 3596.
Lutz J.-F., Kirci B., Matyjaszewski K. // Macromolecules. 2003. V. 36. P. 3136.
Benoit D., Hawker C.J., Huang E.E., Lin Z., Russell T.P. // Macromolecules. 2000. V. 33. P. 1505.
Feng X.-S., Pan C.-Y. // Macromolecules. 2002. V. 35. P. 4888.
Pfeifer S., Lutz J.-F. // J. Am. Chem. Soc. 2007. V. 129. P. 9542.
Pfeifer S., Lutz J.-F. // Chem. – Eur. J. 2008. V. 14. P. 10949.
Moriceau G., Gody G., Hartlieb M., Winn J., Kim H., Mastrangelo A., Smith T., Perrier S. // Polym. Chem. 2017. V. 8. P. 4152.
Tang D., Jiang X., Liu H., Li C., Zhao Y. // Polym. Chem. 2014. V. 5. P. 4679.
Gody G., Zetterlund P., Perrier S., Harrisson S. // Nat. Commun. 2016. V. 7. Art.10514.
Henry S.M., Convertine A.J., Benoit D.S.W., Hoffman A.S., Stayton P.S. // Bioconj. Chem. 2009. V. 20. P. 1122.
Cunningham R.D., Kopf A.H., Elenbaas B.O.W., Staal B.B.P., Pfukwa R., Killian J.A., Klumperman B. // Biomacromolecules. 2020. V. 21. P. 3287.
Delduc P., Tailhan C., Zard S.Z. // J. Chem. Soc., Chem. Commun. 1988. P. 308.
Quiclet-Sire B., Zard S.Z. // Pure Appl. Chem. 2011. V. 83. P. 519.
Quiclet-Sire B., Revol G., Zard S.Z. // Tetrahedron. 2010. V. 66. P. 6656.
Chen M., Ghiggino K.P., Mau A.W.H., Rizzardo E., Sasse W.H.F., Thang S.H., Wilson G.J. // Macromolecules. 2004. V. 37. P. 5479.
Chen M., Ghiggino K.P., Rizzardo E., Thang S.H., Wilson G.J. // Chem. Commun. 2008. P. 1112.
Chen M., Haeussler M., Moad G., Rizzardo E. // Org. Biomol. Chem. 2011. V. 9. P. 6111.
Tanaka S., Nishida H., Endo T. // Macromolecules. 2009. V. 42. P. 293.
Houshyar S., Keddie D.J., Moad G., Mulder R.J., Saubern S., Tsanaktsidis J. // Polym. Chem. 2012. V. 3. P. 1879.
Moad G., Guerrero-Sanchez C., Haven J.J., Keddie D.J., Postma A., Rizzardo E., Thang S.H. // ACS Symp. Ser. 2014. V. 1170. P. 133.
Xu J., Shanmugam S., Fu C., Aguey-Zinsou K.-F., Boyer C. // J. Am. Chem. Soc. 2016. V. 138. P. 3094.
Xu J.T., Jung K., Atme A., Shanmugam S., Boyer C. // J. Am. Chem. Soc. 2014. V. 136. P. 5508.
Shanmugam S., Xu J., Boyer C. // J. Am. Chem. Soc. 2015. V. 137. P. 9174.
Xu J., Fu C., Shanmugam S., Hawker C.J., Moad G., Boyer C. // Angew. Chem., Int. Ed. 2017. V. 56. P. 8376.
Moad G. // Polym. Chem. 2017. V. 8. P. 177.
Xia L., Zhang Z., You Y.-Z. // Polym. J. 2020. V. 52. P. 33.
Nagai A., Koike N., Kudo H., Nishikubo T. // Macromolecules. 2007. V. 40. P. 8129.
Zhang Z., Xia L., Zeng T.-Y., Wu D.-C., Zhang W.-J., Hong C.-Y., You Y-Z. // Polym. Chem. 2019. V. 10. P. 2117.
Zhang Z., Zeng T.Y., Xia L., Hong C.Y., Wu D.C., You Y.Z. // Nat. Commun. 2018. V. 9. 2577.
Aoshima H., Uchiyama M., Satoh K., Kamigaito M. // Angew. Chem. Int. Ed. 2014. V. 53. № 41. P. 10932.
Satoh K., Hashimoto H., Kumagai S., Aoshima H., Uchiyama M., Ishibashi R., Fujiki Y., Kamigaito M. // Polym. Chem. 2017. V. 8. № 34. P. 5002.
Kottisch V., Michaudel Q., Fors B.P. // J. Am. Chem. Soc. 2017. V. 139. № 31. P. 10665.
Peterson B.M., Kottisch V., Supej M.J., Fors B.P. // ACS Cent. Sci. 2018. V. 4. № 9. P. 1228.
Dong H., Zhu Y.J., Li Z.J., Xu J.X., Liu J.J., Xu S.Q., Wang H.X., Gao Y., Guo K. // Macromolecules. 2017. V. 50. P. 9295.
Kamigaito M., Ando T., Sawamoto M. // Chem. Rev. 2001. V. 101. P. 3689.
Davis K.A., Matyjaszewski K. // Adv. Polym. Sci. 2002. V. 159. P. 1.
Matyjaszewski K., Xia J. // Chem. Rev. 2001. V. 101. P. 2921.
Zhang J., Farias-Mancilla B., Destarac M., Schubert U.S., Keddie D.J., Guerrero-Sanchez C., Harrisson S. // Macromol. Rapid Commun. 2018. Art.1800357.
Cho A.S., Broadbelt L.J. // Molec. Simul. 2010. V. 36. № 15. P. 1219.
Chen Y., Chen H., Feng M., Dong Y. // Eur. Polym. J. 2016. V. 85. P. 489.
Lefay C., Save M., Charleux B., Magnet S. // Aust. J. Chem. 2006. V. 59. № 8. P. 544.
Lefay C., Charleux B., Save M., Chassenieux C., Guerret O., Magnet S. // Polymer. 2006. V. 47. № 6. P. 1935.
Borisova O.V., Billon L., Cernochova Z., Lapp A., Stepanek P., Borisov O.V. // Macromol. Symp. 2015. V. 348. № 1. P. 25.
Yuan W., Mok M.M., Kim J.-K., Wong C.L.H., Dettmer C.M., Nguyen S.-B.T., Torkelson J.M., Shull K.R. // Langmuir. 2010. V. 26. № 5. P. 3261.
Mok M.M., Torkelson J.M. // J. Polym. Sci., Polym. Phys. 2012. V. 50. № 3. P. 189.
Farcet C., Charleux B. // Macromol. Symp. 2002. V. 182. P. 249.
Mignard E., Leblanc T., Bertin D., Guerret O., Reed W.F. // Macromolecules. 2004. V. 37. P. 966.
Ziegler M.J., Matyjaszewski K. // Macromolecules. 2001. V. 34. № 3. P. 415.
D'hooge R.D., Van Steenberge H.P., Reyniers M.-F., Marin B.G. // Polymers. 2014. V. 6. № 4. P. 1074.
Li J.-J., Luo Z.-H. // Ind. Eng. Chem. Res. 2014. V. 53. № 5. P. 1900.
Kim B.-S., Lee H.-K., Jeong S., Lee J.-O., Paik H.-J. // Macromol. Res. 2011. V. 19. № 12. P. 1257.
Zhou Y.-N., Li J.-J., Luo Z.-H. // J. Polym. Sci., Polym. Chem. 2012. V. 50. № 15. P. 3052.
Farias-Mancilla B., Zhang J., Kulai I., Destarac M., Schubert U.S., Guerrero-Sanchez C., Harrisson S., Colombani O. // Polym. Chem. 2020. V. 11. P. 7562.
Gu B., Sen A. // Macromolecules. 2002. V. 35. P. 8913.
Borner H.G., Duran D., Matyjazzewski K., da Silva M., Sheiko S.S. // Macromolecules. 2002. V. 35. P. 3387.
Lutz J.F., Pakula T., Matyjaszewski K. // ACS Symp. Ser. 2003. V. 854. P. 268.
Qin S.H., Saget J., Pyun J.R., Jia S.J., Kowalewski T., Matyjaszewski K. // Macromolecules. 2003. V. 36. P. 8969.
Chen Y., Zhang Y., Wang Y., Sun C., Zhang C. // J. Appl. Polym. Sci. 2013. V. 127. № 3. P. 1485.
Alam M.M., Peng H., Jack K.S., Hill D.J.T., Whittaker A.K. // J. Polym. Sci., Polym. Chem. 2017. V. 55. № 5. P. 919.
Guo Y., Gao X., Luo Y. // J. Polym. Sci., Polym. Phys. 2015. V. 53. № 12. P. 860.
Harrisson S., Ercole F., Muir B.W. // Polym. Chem. 2010. V. 1. P. 326.
Zhang J., Farias-Mancilla B., Kulai I., Hoeppener S., Lonetti B., Prévost S., Ulbrich J., Destarac M., Colombani O., Schubert U.S., Guerrero-Sanchez C., Harrisson S. // Angew. Chem. Int. Ed. 2021. V. 60. P. 4925.
Chernikova E.V., Yulusov V.V., Mineeva K.O., Garina E.S., Sivtsov E.V. // Polymer Science B. 2012. V. 54. № 7–8. P. 349.
Wong C.L.H., Kim J., Roth C.B., Torkelson J.M. // Macromolecules. 2007. V. 40. № 16. P. 5631.
Kim J., Torkelson J.M. // Polym. Prepr. Am. Chem. Soc., Div. Polym. Chem. 2007. V. 48. № 1. P. 221.
Tao Y., Kim J., Torkelson J.M. // Polymer. 2006. V. 47. № 19. P. 6773.
Mok M.M., Ellison C.J., Torkelson J.M. // Macromolecules. 2011. V. 44. № 15. P. 6220.
Fu C., Yang B., Zhu C., Wang S., Zhang Y., Wei Y., Tao L. // Polym. Chem. 2013. V. 4. № 24. P. 5720.
Zhang M., Ray W.H. // J. Appl. Polym. Sci. 2002. V. 86. № 7. P. 1630.
Al-Harthi M., Soares J.B.P., Simon L.C. // Macromol. React. Eng. 2007. V. 1. № 4. P. 468.
Al-Harthi M.A., Masihullah J.K., Abbasi S.H., Soares J.B.P. // Macromol. Theory Simul. 2009. V. 18. № 6. P. 307.
Wang W., Zhou Y.-N., Luo Z.-H. // Ind. Eng. Chem. Res. 2014. V. 53. № 30. P. 11873.
Zapata-Gonzalez I., Hutchinson R.A., Matyjaszewski K., Saldivar-Guerra E., Ortiz-Cisneros J. // Macromol. Theory Simul. 2014. V. 23. № 4. P. 245.
Fortunatti C., Sarmoria C., Brandolin A., Asteasuain M. // Macromol. React. Eng. 2014. V. 8. № 4. P. 260.
Zhao Y., Luo Y.-W., Li B.-G., Zhu S. // Langmuir. 2011. V. 27. № 18. P. 11306.
Sun X., Luo Y., Wang R., Li B.-G., Liu B., Zhu S. // Macromolecules. 2007. V. 40. № 4. P. 849.
Wang R., Luo Y., Li B., Sun X., Zhu S. // Macromol. Theory Simul., 2006. V. 15. № 4. P. 356.
Wang R., Luo Y., Li B.-G., Zhu S. // AIChE J. 2007. V. 53. № 1. P. 174.
Dworak A., Utrata-Wesolek A., Oleszko N., Walach W., Trzebicka B., Aniol J., Sieron A.L., Klama-Baryla A., Kawecki M. // J. Mater. Sci., Mater. Med. 2014. V. 25. № 4. P. 1149.
Wang L., Broadbelt L.J. // Macromolecules. 2009. V. 42. № 21. P. 8118.
Wang L., Broadbelt L.J. // Macromol. Theory Simul. 2011. V. 20. № 3. P. 191.
Wang L., Broadbelt L.J. // Macromolecules. 2009. V. 42. № 20. P. 7961.
Wang L., Broadbelt L.J. // Macromolecules. 2010. V. 43. № 5. P. 2228.
Zargar A., Schork F.J. // Macromol. React. Eng. 2009. V. 3. № 2–3. P. 118.
Zargar A., Schork F.J. // Ind. Eng. Chem. Res. 2009. V. 48. № 9. P. 4245.
Ye Y., Schork F.J. // Ind. Eng. Chem. Res. 2009. V. 48. № 24. P. 10827.
Borisova O., Billon L., Zaremski M., Grassl B., Bakaeva Z., Lapp A., Stepanek P., Borisov O. // Soft Matter. 2011. V. 7. Art. 10824.
Cernochova Z., Bogomolova A., Borisova O.V., Filippov S.K., Cernoch P., Billon L., Borisov O.V., Stepanek P. // Soft Matter. 2016. V. 12. P. 6788.
Pham Q.T., Russel W.B., Thibeault J.C., Lau W. // Macromolecules. 1999. V. 32. P. 5139.
Charbonneau C., Chassenieux C., Colombani O., Nicolai T. // Macromolecules. 2012. V. 45. P. 1025.
Charbonneau C., Chassenieux C., Colombani O., Nicolai T. // Phys. Rev. E. 2013. V. 87. Art. 062302.
Shedge A., Colombani O., Nicolai T., Chassenieux C. // Macromolecules. 2014. V. 47. P. 2439.
Charbonneau C., De Souza Lima M.M., Chassenieux C., Colombani O., Nicolai T. // Phys. Chem. Chem. Phys. 2013. V. 15. P. 3955.
Colombani O., Lejeune E., Charbonneau C., Chassenieux C., Nicolai T. // J. Phys. Chem. B. 2012. V. 116. P. 7560.
Colombani O., Ruppel M., Burkhardt M., Drechsler M., Schumacher M., Gradzielski M., Schweins R., Muller A.H.E. // Macromolecules. 2007. V. 40. P. 4351.
Levina Yu.V., Plutalova A.V., Zaitsev S.D., Toms R.V., Serkhacheva N.S., Lysenko E.A., Chernikova E.V. // Polymer Science B. 2020. V. 62. № 3. P. 225.
Zheng C., Huang H., He T. // Macromol. Rapid Commun. 2014. V. 35. P. 309.
Gil E.S., Hudson S.M. // Prog. Polym. Sci. 2004. V. 29. P. 1173.
D’Agosto F., Rieger J., Lansalot M. // Angew. Chem. Int. Ed. 2020. V. 59. P. 8368.
Yañez-Macias R., Kulai I., Ulbrich J., Yildirim T., Sungur P., Hoeppener S., Guerrero-Santos R., Schubert U.S., Destarac M., Guerrero-Sanchez C. // Polym. Chem. 2017. V. 8. P. 5023.
Xu S., Zhang T., Kuchel R.P., Yeow J., Boyer C. // Macromol. Rapid Commun. 2019. V. 41. Art. 1900493.
Xu S., Corrigan N., Boyer C. // Polym. Chem. 2021. V. 12. P. 57.
Zhang J., Farias-Mancilla B., Destarac M., Schubert U.S., Keddie D.J., Guerrero-Sanchez C., Harrisson S. // Macromol. Rapid Commun. 2018. V. 39. Art. 1800357.
Kozhunova E.Y., Gavrilov A.A., Zaremski M.Y., Chertovich A.V. // Langmuir. 2017. V. 33. P. 3548.
Grigoreva A., Polozov E., Zaitsev S. // Colloid Polym. Sci. 2019. V. 297. P. 1423.
París R., De la Fuente J.L. // J. Polym. Sci. Polym. Phys. 2007. V. 45. P. 1845.
Kalugin D., Borisova O., Zaremski M., Garina E., Kolesov D., Bulgakov BAvdeev V. // Eur. Polym. J. 2014. V. 60. P. 213.
Chalykh A.E., Nikulova U.V., Shcherbina A.A. // Polymer Science A. 2020. V. 62. № 4. P. 368.
Chalykh A.E., Nikulova U.V., Gerasimov V.K., Khasbiullin R.R. // Polymer Science A. 2020. V. 62. № 2. P. 85.
Chalykh A.E., Nikulova U.V., Shcherbina A.A., Chernikova E.V. // Polymer Science A. 2019. V. 61. № 2. P. 175.
Pakula T., Matyjaszewski K. // Macromol. Theory Simul. 1996. V. 5. P. 987.
Mok M.M., Torkelson J.M. // J. Polym. Sci, Polym. Phys. 2012. V. 50. P. 189.
Kim J., Sandoval R.W., Dettmer C.M., Nguyen S.T., Torkelson J.M. // Polymer. 2008. V. 49. № 11. P. 2686.
Kim J., Gray M.K., Zhou H., Nguyen S.T., Torkelson J.M. // Macromolecules. 2005. V. 38. P. 1037.
Palermo E.F., van der Laan H.L., McNeil A.J. // Polym. Chem. 2013. V. 4. P. 4606.
Palermo E.F., Darling S.B., McNeil A.J. // J. Mater. Chem. C. 2014. V. 2. P. 3401.
Penfold H.V., Holder S.J., Mkenzie B.E. // Polymer. 2010. V. 51. P. 1904.
Luo Y., Guo Y., Gao X., Li B.-G., Xie T. // Adv. Mater. 2013. V. 25. P. 743.
Milonaki Y., Kaditi E., Pispas S., Demetzos C. // J. Polym. Sci., Polym. Chem. 2012. V. 50. P. 1226.
Vlassi E., Pispas S. // Macromol. Chem. Phys. 2015. V. 216. P. 873.
Park J.-S., Kataoka K. // Macromolecules. 2006. V. 39. P. 6622.
Shull K.R. // Macromolecules. 2002. V. 35. P. 8631.
Yuan W., Laprade E.J., Henderson K.J., Shull K.R. // Soft Matter. 2014. V. 10. P. 1142.
Amonoo J.A., Li A., Purdum G.E., Sykes M.E., Huang B., Palermo E.F., McNeil A.J., Shtein M., Loo Y.-L., Green P.F. // J. Mater. Chem. A. 2015. V. 3. P. 20174.
Williams B.M., Barone V., Pate B.D., Peralta J.E. // Comput. Mater. Sci. 2015. V. 96. P. 69.
Lee E., Kim D., Yoon J. // ACS Appl. Mater. Interfaces. 2016. V. 8. P. 26359.
Matyjaszewski K., Acar M.H., Beers K.L., Coca S., Davis K.A., Gaynor S.G., Miller P.J., Paik H., Shipp D.A., Teodorescu M., Xia J., Zhang X. // Polym. Prepr. 1999. V. 40. P. 966.
Eastwood E.A., Dadmun M.D. // Macromolecules. 2001. V. 34. P. 740.
Hadjiantoniou N.A., Krasia-Christoforou T., Loizou E., Porcar L., Patrickios C.S. // Macromolecules. 2010. V. 43. P. 2713.
Vandenbergh J., Junkers T. // Macromolecules. 2014. V. 47. P. 5051.
Chernikova E.V., Vishnevetskii D.V., Garina E.S., Plutalova A.V., Litmanovich E.A., Korolev B.A., Shlyakhtin A.V., Kostina Yu.V., Bondarenko G.N. // Polymer Science B. 2012. V. 54. № 3–4. P. 127.
Ebeling B., Vana P. // Polymers. 2011. V. 3. P. 719.
Soeriyadi A.H., Boyer C., Nyström F., Zetterlund P.B., Whittaker M.R. // J. Am. Chem. Soc. 2011. V. 133. P. 11128.
Boyer C., Derveaux A., Zetterlund P.B., Whittaker M.R. // Polym. Chem. 2012. V. 3. P. 117.
Zhang Q., Anastasaki A., Li G.Z., Haddleton A.J., Wilson P., Haddleton D.M. // Polym. Chem. 2014. V. 5. P. 3876.
Anastasaki A., Waldron C., Wilson P., Boyer C., Zetterlund P.B., Whittaker M.R., Haddleton D. // ACS Macro Lett. 2013. V. 2. P. 896.
Zhang Q., Wilson P., Li Z., McHale R., Godfrey J., Anastasaki A., Waldron C., Haddleton D.M. // J. Am. Chem. Soc. 2013. V. 135. P. 7355.
Simula A., Nikolaou V., Anastasaki A., Alsubaie F., Nurumbetov G., Wilson P., Kempe K., Haddleton D.M. // Polym. Chem. 2015. V. 6. P. 2226.
Alsubaie F., Anastasaki A., Wilson P., Haddleton D.M. // Polym. Chem. 2015. V. 6. P. 406.
Aksakal R., Resmini M., Becer C.R. // Polym. Chem. 2016. V. 7. P. 171.
Anastasaki A., Nikolaou V., Pappas G.S., Zhang Q., Wan C., Wilson P., Davis T.P., Whittaker M.R., Haddleton D.M. // Chem. Sci. 2014. V. 5. № 9. P. 3536.
Chuang Y.M., Ethirajan A., Junkers T. // ACS Macro Lett. 2014. V. 3. P. 732.
Anastasaki A., Nikolaou V., McCaul N.W., Simula A., Godfrey J., Waldron C., Wilson P., Kempe K., Haddleton D.M. // Macromolecules. 2015. V. 48. P. 1404.
Wenn B., Martens A.C., Chuang Y.M., Gruber J., Junkers T. // Polym. Chem. 2016. V. 7. P. 2720.
Anastasaki A., Oschmann B., Willenbacher J., Melker A., Van Son M.H.C., Truong N.P., Schulze M.W., Discekici E.H., McGrath A.J., Davis T.P., Bates C.M., Hawker C.J. // Angew. Chem., Int. Ed. 2017. V. 56. P. 14483.
Gody G., Maschmeyer T., Zetterlund P.B., Perrier S. // Nat. Commun. 2013. V. 4. P. 2505.
Gody G., Maschmeyer T., Zetterlund P.B., Perrier S. // Macromolecules. 2014. V. 47. P. 639.
Gody G., Maschmeyer T., Zetterlund P.B., Perrier S. // Macromolecules. 2014. V. 47. P. 3451.
Zetterlund P.B., Gody G., Perrier S. // Macromol. Theory Simul. 2014. V. 23. P. 331.
Radzevicius P., Steponaviciute M., Krivorotova T., Makuska R. // Polym. Chem. 2017. V. 8. P. 7217.
Gody G., Barbey R., Danial M., Perrier S. // Polym. Chem. 2015. V. 6. P. 1502.
Tanaka J., Gurnani P., Cook A.B., Häkkinen S., Zhang J., Yang J., Kerr A., M. Haddleton D., Perrier S., Wilson P. // Polym. Chem. 2019. V. 10. P. 1186.
Baeten E., Haven J.J., Junkers T. // Polym. Chem. 2017. V. 8. P. 3815.
Kuroki A., Martinez-Botella I., Hornung C.H., Martin L., Williams E.G.L., Locock K.E.S., Hartlieb M., Perrier S. // Polym. Chem. 2017. V. 8. P. 3249.
Fu C., Xu J., Tao L., Boyer C. // ACS Macro Lett. 2014. V. 3. P. 633.
Liu Z., Lv Y., An Z. // Angew. Chem., Int. Ed. 2017. V. 56. P. 13852.
Ng G., Yeow J., Chapman R., Isahak N., Wolvetang E., Cooper-White J.J., Boyer C. // Macromolecules. 2018. V. 51. P. 7600.
Judzewitsch P.R., Nguyen T.K., Shanmugam S., Wong E.H.H., Boyer C. // Angew. Chem., Int. Ed. 2018. V. 57. P. 4559.
Kerr A., Hartlieb M., Sanchis J., Smith T., Perrier S. // Chem. Commun. 2017. V. 53. P. 11901.
Guimarães T.R., Khan M., Kuchel R.P., Morrow I.C., Minami H., Moad G., Perrier S., Zetterlund P.B. // Macromolecules. 2019. V. 52. P. 2965.
Khan M., Guimarães T.R., Choong K., Moad G., Perrier S., Zetterlund P.B. // Macromolecules, 2021. V. 54. № 2. P. 736.
Clothier G.K.K., Guimarães T.R., Khan M., Moad G., Perrier S., Zetterlund P. B. // ACS Macro Lett. 2019. V. 8. P. 989.
Engelis N.G., Anastasaki A., Nurumbetov G., Truong N.P., Nikolaou V., Shegiwal A., Whittaker M.R., Davis T.P., Haddleton D.M. // Nat. Chem. 2017. V. 9. P. 171.
Engelis N.G., Anastasaki A., Whitfield R., Jones G.R., Liarou E., Nikolaou V., Nurumbetov G., Haddleton D.M. // Macromolecules. 2018. V. 51. P. 336.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия С)