

Химия высоких энергий, 2019, T. 53, № 2, стр. 161-163
Самотушение флуоресценции Hoechst 33258 и SYBR Green I на матрице холестерической жидкокристаллической ДНК
В. Н. Морозов a, *, В. А. Кузьмин a
a Институт биохимической физики им. Н.М. Эмануэля Российской Академии наук
117997 Москва, ул. Косыгина, 4, Россия
* E-mail: morozov.v.n@mail.ru
Поступила в редакцию 16.08.2018
После доработки 26.10.2018
Принята к публикации 24.10.2018
ДНК-специфичные лиганды демонстрируют явление самотушения флуоресценции при взаимодействии с биомакромолекулами [1, 2]. Это явление наблюдается в случае близкого (менее 10 нм) расположения хромофоров при высоких концентрациях лигандов и происходит за счет образования нефлуоресцирующего комплекса, безызлучательного переноса энергии или переноса электрона между флуорофорами [3]. Самотушение флуоресценции также наблюдается в условиях плотной упаковки ДНК в биологических системах [4]. В некотором приближении упаковку ДНК in vivo способны моделировать молекулярно-организованные ансамбли, например, холестерические жидкокристаллические дисперсии (ХЖКД) ДНК [5, 6]. При взаимодействии с такими пространственно-упорядоченными модельными системами большое значение приобретают размерные и структурные особенности лигандов, а также строение их комплексов с ДНК. Известно несколько типов нековалентного взаимодействия низкомолекулярных лигандов с ДНК: внешнее связывание, интеркаляция между парами оснований и связывание в бороздке [7]. В настоящем сообщении приведены результаты исследования самотушения флуоресценции интеркалятора SYBR Green I (SG) и АТ-специфичного узкобороздочного лиганда Hoechst 33258 (Ht58) на матрицах ДНК и ХЖКД ДНК.
ХЖКД ДНК получали смешением в пропорции 1 : 1 водно-солевого раствора низкомолекулярной ДНК с 34% раствором полиэтиленгликоля (ПЭГ) [6]. Водно-солевой раствор ДНК готовили на основе 10–2 М фосфатного (NaH2PO4) буфера, содержащего 0.3 M NaCl (pH ~ 7.4) с использованием 7.5 × 10–5 М (ε260 ≈ 6600 М–1 cм–1) коммерческого препарата ДНК (Деринат®, Россия). ПЭГ с молекулярной массой 4000 Да (Me-rck, Германия) использовали без дополнительной очистки. Использовали коммерческие красители SYBR Green I и Hoechst 33258 (Sigma, США). Концентрации красителей определяли спектрофотометрически, используя коэффициенты экстинкции: ε498 ≈ 73 000 М–1 см–1 (SG) [8], ε343 ≈ ≈ 42 000 М–1 см–1 (Ht58) [9]. Спектры поглощения были получены на спектрофотометре UV-Vis 3101 PC (Shimadzu, Япония). Спектры флуоресценции регистрировали на спектрофлуориметре RF5301PC (Shimadzu, Япония). Времена жизни флуоресценции измеряли на спектрофлуориметре FluoTime 300 (PicoQuant, Германия). Все измерения проводили в кварцевых кюветах (0.2 × × 1.0 см). Оптический путь возбуждающего света составлял 0.2 см.
В органических растворах концентрационная зависимость интенсивности флуоресценции красителей имеет линейный характер, поскольку взаимодействие хромофоров отсутствует из-за больших расстояний между ними. При наличии в растворе молекул субстрата, белков или нуклеиновых кислот, связывающих флуоресцирующие лиганды, реализуется микрофазная модель, согласно которой локализация флуорофоров происходит в основном на макромолекулярной матрице, в то время как их концентрация в растворе пренебрежимо мала [10]. В настоящем сообщении матрицей, связывающей Ht58 и SG, являлись молекулы ДНК в растворе, а также низкомолекулярные ДНК конденсированные ПЭГ в частицы ЖКД с холестерической закруткой слоев, обеспечивающие взаимодействие хромофоров на расстояниях ~3.4 нм [6]. Такое близкое расположение молекул Ht58 и SG в микрофазе ХЖКД ДНК приводит к самотушению флуоресценции, которое сопровождается изменением времен жизни возбужденного состояния Ht58 (τ1 = 2 нс, τ2 = 4.1 нс (6 × 10–7 M), τ1 = 1.7 нс, τ2 = 3.6 нс (6 × × 10–5 М)); SG (τ1 = 4.5 нс (6 × 10–7 M), τ1 = 3.1 нс (1.2 × 10–5 М)). Уменьшение времени жизни флуоресценции красителей при увеличении их концентрации согласуется с полученными ранее результатами [2] и свидетельствует о переносе энергии по механизму homo-FRET. Кривые самотушения флуоресценции красителей (рис. 1) имеют характерный для этого процесса вид [11], однако характер зависимостей в растворе ДНК и молекулярно-организованном ансамбле ХЖКД ДНК значительно отличается для SG и Ht58. Структура комплекса SG одинакова для раствора ДНК и конденсированной мезофазы, где флуорофор защищен от взаимодействия с другими молекулами ДНК. Структура частиц ХЖКД ДНК способствует сближению хромофоров, интеркалированных между парами оснований соседних двойных спиралей, что приводит к самотушению флуоресценции при меньших концентрациях SG по сравнению с ДНК в растворе.
Рис. 1.
Зависимость нормированной интенсивности флуоресценции SYBR Green I (а) и Hoechst 33258 (б) в ДНК и ХЖКД ДНК от концентрации красителей.
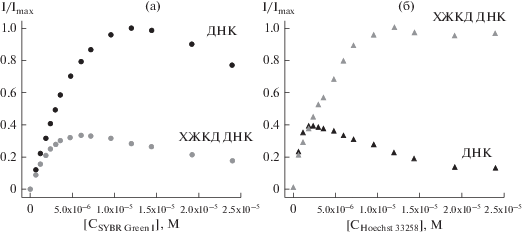
Концентрационные зависимости интенсивности флуоресценции Ht58 имеют иной характер. При малых концентрациях молекулы Ht58 занимают наиболее выгодные сайты связывания с ДНК, однако с ростом концентрации красителя таких сайтов становится меньше, что приводит к димерному связыванию и образованию комплексов с более сложной стехиометрией [12]. В растворе ДНК эффективное самотушение флуоресценции Ht58 происходит в первую очередь за счет димеров, образование которых при малых концентрациях залегателя ограничено в ХЖКД ДНК стерическим фактором. При возрастании концентрации Ht58 происходит изменение шага холестерической закрутки слоев в частицах дисперсии, наблюдаемое по уменьшению амплитуды аномальной отрицательной полосы в области поглощения азотистых оснований (~280 нм) на спектре кругового дихроизма ХЖКД ДНК (данные не приведены). Переход холестерической упаковки ДНК в более упорядоченную мезофазу маловероятен, поскольку сопровождался бы значительно более эффективным самотушением флуоресценции [2] за счет уменьшения расстояния между соседними слоями до ~2.6 нм [13, 14]. Известно, что интеркалирующие соединения способны препятствовать конденсации высокомолекулярной ДНК за счет увеличения персистентной длины молекулы [15], однако изменение длины и увеличение жесткости ДНК при связывании SG в исследуемых концентрациях не влияет на упаковку низкомолекулярных ДНК в частицах ХЖКД. В отличие от интеркаляторов связывание залегателей не приводит к изменениям структуры ДНК [7], однако если интеркалированный краситель экранирован азотистыми основаниями от взаимодействия с окружающими молекулами [16], в случае образования комплекса залегания стерические препятствия для взаимодействия Ht58 с соседними сайтами значительно меньше [17–19]. Таким образом, плотная упаковка ДНК управляет связыванием Ht58, а узкобороздочный комплекс сильнее влияет на пространственную организацию компактизованной ДНК, чем интеркалирующий, что существенно отличает поведение Ht58 в молекулярно-организованных системах с высокой плотностью упаковки от поведения в растворе ДНК.
Исследования выполнены с использованием оборудования ЦКП “Новые материалы и технологии” ИБХФ РАН.
Список литературы
Gudnason H., Dufva M., Bang D.D., Wolff A. // Nucleic acids res. 2007. V. 35. P. e127.
Лисицына Е.С., Дурандин Н.А., Калюжный Д.Н., Кузьмин В.А. // Химия высоких энергий. 2013. Т. 47. № 1. С. 39.
Chen R.F., Knutson J.R. // Anal. Biochem. 1988. V. 172. P. 61.
Estandarte A.K., Botchway S., Lynch C., Yusuf M., Robinson I. // Sci. rep. 2016. V. 6. 31417.
Livolant F. // Physica A. 1991. V. 176. P. 117.
Евдокимов Ю.М., Салянов В.И., Семенов С.В., Скуридин С.Г. Жидкокристаллические дисперсии и наноконструкции ДНК. М.: Радиотехника, 2008.
Blackburn G.M., Gait M.J., Loakes D., Williams D.M. Nucleic Acids in Chemistry and Biology. Cambridge: The Royale Society of Chemistry, 2006.
Zipper H., Brunner H., Bernhagen J., Vitzthum F. // Nucleic acids res. 2004. V. 32. P. e103.
Latt S.A., Wohlleb J.C. // Chromosoma. 1975. V. 52. P. 297.
Kuzmin M.G., Soboleva I.V., Durandin N.A., Lisitsyna E.S., Kuzmin V.A. // J. Phys. Chem. B. 2014. V. 118. P. 4245.
Hamann S., Kiilgaard J.F., Litman T., Alvarez-Leef-mans F.J., Winther B.R., Zeuthen T. // J. Fluoresc. 2002. V. 12. P. 139.
Bazhulina N.P., Nikitin A.M., Rodin S.A., Surovaya A.N., Kravatsky Y.V., Pismensky V.F., Archipova V.S., Martin R., Gursky G.V. // J. Biomol. Struct. Dyn. 2009. V. 26. P. 701.
Livolant F., Leforestier A. // Prog. Polym. Sci. 1996. V. 21. P. 1115.
De Frutos M., Leforestier A., Livolant F. // Biophys. Rev. and Lett., 2014. V. 9. P. 81.
Rocha M.S., Cavalcante A.G., Silva R., Ramos E.B. // J. Phys. Chem. B. 2014. V. 118. P. 4832.
Geacintov N.E., Waldmeyer J., Kuzmin V.A., Kolubayev T. // J. Phys. Chem. 1981. V. 85. P. 3608.
Saito M., Kobayashi M., Iwabuchi S., Morita Y., Takamura Y., Tamiya E. // J. Biochem. 2004. V. 136. P. 813.
Saito M., Takamura Y., Tamiya E. // NanoBiotechnology 2005. V. 1. P. 361.
Silva E.F., Ramos E.B., Rocha M.S. // J. Phys. Chem. B. 2013. V. 117. P. 7292.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий