

Химия высоких энергий, 2019, T. 53, № 3, стр. 178-185
Сопровождаемая золь-гель переходом радикальная полимеризация, инициируемая двумя первичными радикалами
И. П. Ким a, *, В. А. Бендерский a
a Институт проблем химической физики РАН
142432 Московская обл., Черноголовка, просп. Академика Семенова 1, Россия
* E-mail: ipkim@icp.ac.ru
Поступила в редакцию 29.11.2018
После доработки 10.12.2018
Принята к публикации 25.12.2018
Аннотация
Когда при радиационном воздействии образуются два радикала, cпособных инициировать рост цепи с различными константами скорости ее передачи, возникают два канала полимеризации с различной средней длиной цепи. Каналы не перемешиваются на начальном участке баллистического роста. Их перемешивание в актах передачи цепи сопровождается переходом от баллистического роста к диффузионному. В области золь-гель перехода каналы пространственно разделяются, если передача цепи зависит от вращательной подвижности реагентов и уменьшается с ростом вязкости по мере накопления образующихся олигомеров. В качестве примера рассмотрена полимеризация ТФЭ в растворах силанов и оксисиланов.
ВВЕДЕНИЕ
Жидкофазная радикальная полимеризация (ЖРП) приводит к образованию линейных цепей, изменяющих свойства раствора, в котором она происходит. По мере накопления достаточно длинных олигомеров вязкость раствора возрастает, что изменяет константны скорости роста и передачи цепи. Когда концентрация образовавшихся олигомеров достигает критического значения, гомогенный раствор переходит в коллоидный, затем переходящий в гель. Эта картина взаимно связанных цепной химической реакции и фазового перехода наблюдается при полимеризации тетрафторэтилена (ТФЭ) в различных органических растворителях [1–5], и по сравнению с гомогенной полимеризацией обладает следующими особенностями: золь-гель переход превращает одномодовое молекулярно-массовое распределение (ММР) в двухмодовое, которое является пространственно неоднородным: более короткие олигомеры остаются в гомогенном растворе, а длинные сосредоточены в составе коллоидных частиц и геля. Плотная фаза состоит из агрегатов, в состав которых помимо длинных олигомеров входят молекулы растворителя, образующие сольватные оболочки звеньев цепи. Разделение фаз сопровождается резким уменьшением эффективной константы скорости роста цепи.
Традиционное рассмотрение кинетики радикальной полимеризации предполагает, что рост цепи инициирует один первичный радикал ${{R}_{0}},$ который отличается более высоким квантовым выходом, отсутствием рекомбинации в клеточных реакциях, где, в частности, гибнут Н-атомы и ОН-радикалы, и последующей регенерацией в реакции передачи цепи
где Rn – ведущий цепь макрорадикал, S = R0H – молекула растворителя, Pn – олигомер, содержащий n звеньев мономера. Из (1) следует, что перенос цепи является реакцией переноса атома водорода от S к Rn и ее энтальпия равна разности стандартных энтальпий разрыва СН-связи в образующемся олигомере и в молекуле растворителяСогласно эмпирическому правилу Поляни–Семенова–Эванса (ПСЭ) (см. напр. [6]), энергия активации передачи цепи пропорциональна $\Delta {{H}_{t}}.$ Для быстрого роста длинных олигомеров в процессе ЖРП необходимо, чтобы ${{k}_{t}}$ удовлетворяла двум неравенствам
где $k$ – константа скорости роста, ${{k}_{r}}$ – константа скорости обрыва цепи, обусловленного гибелью макрорадикалов в совокупности реакций их рекомбинации, ${{M}_{0}},$ $S,$ $R \approx {{R}_{0}}$ – концентрации ТФЭ, растворителя, первичного радикала и всех макрорадикалов соответственно, обычно равные ${{M}_{0}}$ = 0.1–1, $S$ ≈ 10 и ${{R}_{0}}$ ~ 10–4 моль/л. С ростом ${{k}_{t}},$ длина цепи уменьшается ${{\sim k} \mathord{\left/ {\vphantom {{\sim k} {{{k}_{t}}}}} \right. \kern-0em} {{{k}_{t}}}}{\text{,}}$ а при ${{k}_{t}}\sim {{k}_{r}}$ цепь вырождается. Поскольку рост цепи, как правило, сильно экзотермичен (для присоединения ТФЭ к макрорадикалу $\Delta {{H}^{0}}$ = –36 Ккал/моль), он ограничен скоростью диффузионного потока мономера к макрорадикалу и его константа скорости обратно пропорциональна вязкости раствора $\eta $(4)
$k \approx 2{{k}_{B}}T\,{{{{R}_{g}}} \mathord{\left/ {\vphantom {{{{R}_{g}}} 3}} \right. \kern-0em} 3}{{r}_{0}}\eta ,$(5)
${{k{{M}_{0}}} \mathord{\left/ {\vphantom {{k{{M}_{0}}} S}} \right. \kern-0em} S} \gg {{k}_{t}} \gg k{{{{R}_{0}}} \mathord{\left/ {\vphantom {{{{R}_{0}}} S}} \right. \kern-0em} S}.$Интервал энтальпий, разделяющий безактивационные (энергия активации${{E}^{\# }} = 0$) и безбарьерные (${{E}^{\# }} = \Delta {{H}^{0}}$) реакции переноса водорода, в котором реализуется кинетический режим, обычно составляет ~10 Ккал/моль [7]. Этот интервал энтальпии соответствует изменению ${{k}_{t}}$ в интервале (5). Оптимальные значения ${{k}_{t}}$ соответствуют энергетически нейтральным реакциям, для которых
(6)
$\left| {\Delta {{H}_{t}}} \right| \leqslant 5{\text{ }}{{{\text{К к а л }}} \mathord{\left/ {\vphantom {{{\text{К к а л }}} {{\text{м о л ь }}}}} \right. \kern-0em} {{\text{м о л ь }}}}.$При выполнении (6) ожидаемая средняя длина цепи $\bar {n}$ ~ 102. Зависимость $\bar {n}$ от $\Delta H_{s}^{0}$ качественно согласуется с экспериментальными данными о полимеризации ТФЭ в различных растворителях. Энергия разрыва связи СН в концевой группе олигомеров CHF2$\Delta H_{P}^{0}$ = 104 Ккал/моль. Например, $\Delta H_{s}^{0}$ в ацетоне – 102 Ккал/моль и $\bar {n}$ достигает 20–25, тогда как в метаноле, где $\Delta H_{s}^{0}$ = 87 Ккал/моль, $\bar {n} \leqslant 6$ [2]. Отмеченная в [8, 9] особенность ЖРП в триалкилсиланах HSiR3 и триалкоксисиланах HSi(OR)3 состоит в возможности образования двух первичных радикалов при разрыве в S двух связей СН ($\Delta H_{s}^{0}$ = 100–104 Ккал/моль) и связи SiH, энергия которой на 14–18 Ккал/моль меньше, чем CH. Согласно правилу ПСЭ, в ЖРП должен доминировать канал, связанный с быстрым образованием коротких олигомеров ${{{\text{R}}}_{{\text{3}}}}{\text{Si(}}{{{\text{C}}}_{{\text{2}}}}{{{\text{F}}}_{{\text{4}}}}{{{\text{)}}}_{n}}{\text{H}}$ и ${{({\text{RO)}}}_{{\text{3}}}}{\text{Si(}}{{{\text{C}}}_{{\text{2}}}}{{{\text{F}}}_{4}}{{)}_{n}}{\text{H}},$ как и в метаноле, а образование длинных олигомеров HR2SiCH2(C2F4)nH и ${\text{H(RO}}{{{\text{)}}}_{{\text{2}}}}{\text{SiOC}}{{{\text{H}}}_{{\text{2}}}}{{{\text{(}}{{{\text{C}}}_{{\text{2}}}}{{{\text{F}}}_{4}})}_{n}}{\text{H}}$ должно быть подавлено. Однако, экспериментальные данные [8, 9], где R = CH3 не подтверждают это предположение. Средняя длина цепи и ММР оказались приблизительно такими же, как и в алкил- и алкоксисиланах, не имеющих слабой связи SiH. В обоих случаях образуются олигомеры, в которых длинная фторалкильная цепь соединена с атомом кремния фрагментами СН2 или ОСН2, образующимися при разрыве более прочной CH связи, а не связи SiH.
Цель настоящей статьи – установление причин обнаруженного несоответствия экспериментальных данных правилу ПСЭ. Объяснение основано на указанном во Введении разделении фаз, которое уменьшает скорость не только диффузионно-ограниченного роста цепи, но и кинетической передачи цепи, связанной с разрывом связи SiH, в которой образование реактивного начального состояния для передачи цепи связано с преодолением пространственного барьера, создаваемого тремя метильными или метоксильными группами, экранирующими связь SiH [10–12].
Передача цепи возможна в узкой области телесных углов и связана с вращательной диффузией молекулы растворителя, которая уменьшается в плотной фазе с ростом вязкости сильнее, чем трансляционная диффузия, ограничивающая рост цепи [13–16]. Резкий рост вращательного барьера в вязкой среде идейно связан с рептильным механизмом Эдвардса–Дои [17–19], определяющим динамику полимеров в полуразбавленных растворах. Подавление вращательной подвижности в структурированных вязких средах происходит, когда поперечный размер эффективной трубки, не содержащей других олигомеров, становится сравним с радиусом инерции молекулы растворителя. По сравнению с ЖРП, инициируемой одним первичным радикалом, рассматриваемый в этой статье процесс обладает рядом кинетических особенностей, связанных с перемешиванием и конкуренцией двух каналов роста неразветвленных цепей.
КИНЕТИКА ГОМОГЕННОЙ ПОЛИМЕРИЗАЦИИ, ИНИЦИИРУЕМОЙ ДВУМЯ ПЕРВИЧНЫМИ РАДИКАЛАМИ
Рассмотрим схему ЖРП по двум каналам А и В, создаваемым двумя первичными радикалами $R_{0}^{A}$ и $R_{0}^{B}.$ Скорость роста цепи в обоих каналах обусловлена присоединением мономера к макрорадикалу, не зависит от первичного радикала и в обоих каналах одинакова
Передача цепи (1) происходит с различными константами скорости $k_{t}^{A} < k_{t}^{B}$ и приводит к регенерации обоих первичных радикалов и перемешиванию каналов в каждом акте передачи цепи
(8)
$\begin{gathered} R_{n}^{A} + S\left\{ {\begin{array}{*{20}{c}} {\xrightarrow{{k_{t}^{A}}}R_{0}^{A} + P_{n}^{A}} \\ {\xrightarrow{{k_{t}^{B}}}R_{0}^{B} + P_{n}^{A}} \end{array}} \right., \\ R_{n}^{B} + S\left\{ {\begin{array}{*{20}{c}} {\xrightarrow{{k_{t}^{A}}}R_{0}^{A} + P_{n}^{B}} \\ {\xrightarrow{{k_{t}^{B}}}R_{0}^{B} + P_{n}^{B}} \end{array}.} \right. \\ \end{gathered} $Уравнения Смолуховского для реакций (7) и (8) имеют вид
(9)
$\begin{gathered} \dot {M} = - kM\sum\limits_{n = 0}^N {\left( {R_{n}^{A} + R_{n}^{B}} \right)} , \\ \dot {R}_{0}^{A} = - kMR_{0}^{A} + \sum\limits_{n = 1}^N {k_{t}^{A}(R_{n}^{A} + R_{n}^{B})} , \\ \dot {R}_{0}^{B} = - kMR_{0}^{B} + \sum\limits_{n = 1}^N {k_{t}^{B}(R_{n}^{A} + R_{n}^{B})} , \\ \dot {R}_{n}^{A} = kMR_{{n - 1}}^{A} - \left( {kM + k_{t}^{A} + k_{t}^{B}} \right)R_{n}^{A}, \\ \dot {R}_{n}^{B} = kMR_{{n - 1}}^{B} - \left( {kM + k_{t}^{A} + k_{t}^{B}} \right)R_{n}^{B}, \\ \dot {P}_{n}^{A} = \left( {k_{t}^{A} + k_{t}^{B}} \right)R_{n}^{A},\,\,\,\,\dot {P}_{n}^{B} = \left( {k_{t}^{A} + k_{t}^{B}} \right)R_{n}^{B}. \\ \end{gathered} $Первое из уравнений (9) определяет расход мономера по двум каналам, пропорциональный суммарной концентрации А- и В-макрорадикалов. Четыре следующих уравнения определяют концентрации первичных радикалов и макрорадикалов в обоих каналах, которые также пропорциональны суммарной концентрации макрорадикалов, но происходят с различными константами скорости передачи цепи. Два последних уравнения относятся к росту концентраций А- и В-олигомеров, пропорциональных соответствующим концентрациям макрорадикалов и суммарной скорости передачи цепи. Стехиометрические коэффициенты в (9) выбраны так, что начальное число мономеров ${{M}_{0}}$ сохраняется в процессе ЖРП
(10)
$M + \sum\limits_{n = 1}^N {n\left( {R_{n}^{A} + R_{n}^{B} + P_{n}^{A} + P_{n}^{B}} \right) = {{M}_{0}}} .$При численном решении уравнений (9) точность расчета концентраций определяется выбранным значением максимальной длины цепи $N.$ Расчеты, выполненные в настоящей работе, показали, что и при $N \geqslant {k \mathord{\left/ {\vphantom {k {k_{t}^{A}}}} \right. \kern-0em} {k_{t}^{A}}}$ результаты не зависят от $N$ с точностью, лучшей 3%. Из (10) следует, что макроскопические характеристики ЖРП (степень превращения, суммарные концентрации олигомеров, средняя длина цепи) зависят только от суммарной константы скорости передачи в обоих каналах. От отношения констант ${{k_{t}^{A}} \mathord{\left/ {\vphantom {{k_{t}^{A}} {k_{t}^{B}}}} \right. \kern-0em} {k_{t}^{B}}}$ зависят только ММР и средние длины цепи А- и В-олигомеров. Типичные кинетические зависимости представлены на рис. 1. Когда ${{k_{t}^{A}} \mathord{\left/ {\vphantom {{k_{t}^{A}} k}} \right. \kern-0em} k}$ ~ 10–2–10–3, но ${{k_{t}^{B}} \mathord{\left/ {\vphantom {{k_{t}^{B}} k}} \right. \kern-0em} k}$ ~ 1, рост олигомеров в обоих каналах происходит в том же масштабе времени ${{\sim 1} \mathord{\left/ {\vphantom {{\sim 1} {k_{t}^{B}}}} \right. \kern-0em} {k_{t}^{B}}},$ что и расход мономеров и приводит к образованию только коротких олигомеров, вне зависимости от $k_{t}^{A}.$ Когда обе константы передачи цепи на порядки меньше константы роста, ${{k_{t}^{A}} \mathord{\left/ {\vphantom {{k_{t}^{A}} k}} \right. \kern-0em} k}\sim {{10}^{{ - 2}}},$ ${{k_{t}^{A}} \mathord{\left/ {\vphantom {{k_{t}^{A}} k}} \right. \kern-0em} k}\sim {{10}^{{ - 1}}},$ рост длинных А-олигомеров и коротких В-олигомеров происходит в различных масштабах времени (рис. 2): рост А-олигомеров с длиной цепи $n \leqslant 10$ происходит за время ${{\sim 1} \mathord{\left/ {\vphantom {{\sim 1} k}} \right. \kern-0em} k},$ когда передача цепи незначительна и мало влияет на концентрацию радикалов (верхняя панель).
Рис. 1.
Кинетика макроскопических характеристик ЖРП с участием двух первичных радикалов с различными константами скорости передачи цепи. ${{k_{t}^{A}} \mathord{\left/ {\vphantom {{k_{t}^{A}} k}} \right. \kern-0em} k}$ = 0.2, ${{k_{t}^{B}} \mathord{\left/ {\vphantom {{k_{t}^{B}} k}} \right. \kern-0em} k}$ = 0.9. Начальные концентрации первичных радикалов $R_{0}^{A}(0) = R_{0}^{B}(0) = 0.01{{M}_{0}}.$ Верхняя панель: зависящие от времени концентрации мономеров ${{M(t)} \mathord{\left/ {\vphantom {{M(t)} {{{M}_{0}}}}} \right. \kern-0em} {{{M}_{0}}}}$ и мономерных фрагментов в составе всех А- и В-олигомеров, ${{P}^{{A(B)}}}(t) = \sum\nolimits_{n = 1}^N {nP_{n}^{{A(B)}}(t)} $ – кривые 1–3. Пунктиром показано сохранение полного числа мономеров (10) в процессе ЖРП. Вторая и третья панели: зависящие от времени концентрации мономерных фрагментов в составе А- и В-олигомеров, $P_{n}^{{A(B)}}(t) = nP_{n}^{{A(B)}}(t)$, $n$ = 1, 2, 4, 6 – кривые (1–4). Нижняя панель: зависящая от времени средняя длина цепи А- и В-олигомеров, $\left\langle {{{n}^{A}}} \right\rangle ,$ $\left\langle {{{n}^{B}}} \right\rangle $ – кривые 1 и 2 соответственно. Масштаб времени одинаков для всех панелей.
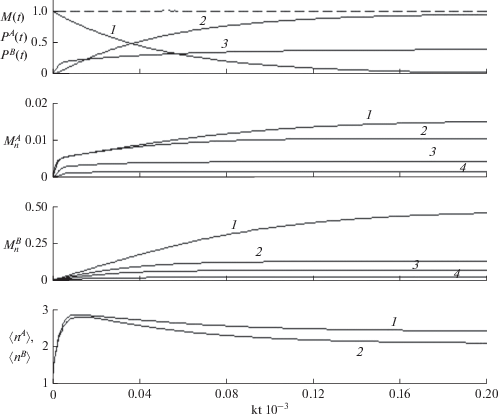
Рис. 2.
Зависящие от времени концентрации макрорадикалов $R_{n}^{{(A)}}$ и $R_{n}^{{(B)}}$ – верхняя и нижняя панель; $n$ = 1, 3, 5, 7, 10 – кривые 1–5 соответственно; $R_{0}^{{(A)}}(0) = R_{0}^{{(B)}}(0) = 0.01\,{{M}_{0}};$ $k_{t}^{A}$ = 0.01$k,$ 0.1 $k_{t}^{B}$ = 0.1$k.$ Масштаб времени одинаков для обеих панелей.
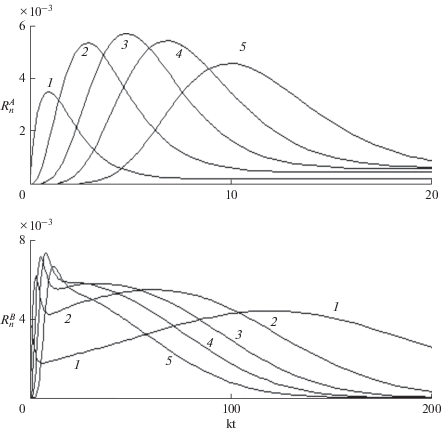
Режим роста радикалов в отсутствие передачи цепи является аналогом баллистического распространения, когда мономеры присоединяются к радикалам первого поколения и пропорциональный времени рост длины не сопровождается актами возврата (регенерации первичных радикалов) и повторного роста. В кинетике В-радикалов (нижняя панель) наблюдаются два участка. Начальный баллистический рост происходит в том же масштабе времени, что и А-радикалов, но в дальнейшем он переходит в диффузионный, когда доминирует рост макрорадикалов следующих поколений после многократной регенерации первичных В-радикалов. В этом процессе передача цепи от А-макрорадикалов и вызывает рост длинных В-олигомеров в масштабе времени ${{\sim 1} \mathord{\left/ {\vphantom {{\sim 1} {k_{t}^{A}}}} \right. \kern-0em} {k_{t}^{A}}}.$ Максимальные концентрации А- и В-макрорадикалов одного порядка, но высокая концентрация последних существует в течение более длительного времени, что приводит к более высокой концентрации В-, чем А-олигомеров. Если время существования А-олигомеров растет с увеличением их длины, для В-олигомеров оно, напротив, уменьшается (сравни кривые 1–5 на верхней и нижней панелях). В результате, средняя длина А-олигомеров оказывается больше, чем В-олигомеров. Обнаруженный переход от баллистического к диффузионному режиму характерен для процессов слабо диссипативной передачи энергии, когда первое прохождение остается баллистическим, а последующие многократные возвраты (в данном случае передача цепи) приводят к диффузионному распространению [20–22]. Рассмотренная сложная кинетика макрорадикалов приводит к тому, что средняя длина цепи А- и В-олигомеров, несмотря на перемешивание каналов, оказывается различной (рис. 3). Хотя при двухканальной ЖРП длина цепи более чем вдвое меньше, чем при одноканальной (кривые 1 и 2 по сравнению с кривой 4), с уменьшением $k_{t}^{A} \ll k_{t}^{B}$ средняя длина А-олигомеров в несколько раз больше, чем В-олигомеров. Таким образом, даже в условиях перемешивания каналов в гомогенном растворе распределение олигомеров не описывается одним ММР с усредненной средней длиной цепи.
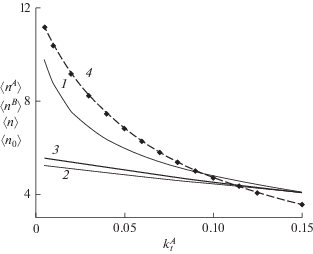
Рис. 3.
Зависимость средней длины цепи А- и В-олигомеров $\left\langle {{{n}^{{A(B)}}}} \right\rangle $ (кривые 1 и 2), средней длины цепи обоих каналов $\left\langle n \right\rangle $ (кривая 3) от константы скорости передачи цепи ${{k_{t}^{A}} \mathord{\left/ {\vphantom {{k_{t}^{A}} k}} \right. \kern-0em} k}$ при ${{k_{t}^{B}} \mathord{\left/ {\vphantom {{k_{t}^{B}} k}} \right. \kern-0em} k}$ = 0.15, $R_{0}^{{(A)}}(0) = R_{0}^{{(B)}}(0) = 0.01\,{{M}_{0}}.$ Пунктиром показана уменьшенная вдвое средняя длина цепи одноканальной ЖРП $\bar {n}{\text{/}}2$ с такими же константами роста и передачи цепи (кривая 4).
Следует подчеркнуть, что рассмотренная сложная кинетика роста цепей в двух режимах не описывается в квазистационарном приближении (КСП), традиционно применяемом для расчета ММР радикальной полимеризации. Поскольку точность КСП увеличивается с ростом длины цепи, оно надежно применимо для высокомолекулярных полимеров, когда рост цепи происходит в диффузионном режиме. Однако в кинетике ЖРП, где образуются олигомеры, применимость КСП требует специального рассмотрения, связанного с решением уравнений (9). Обнаруженный в настоящей работе начальный баллистический режим присущ не только олигомерам, но и многим процессам полимеризации, в которых константы скорости зависят от длины цепи. Интерес к таким процессам связан с возможностями получения материалов с контролируемой наноразмерной структурой [23–25].
ДВУХКАНАЛЬНАЯ РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ В ОБЛАСТИ РАЗДЕЛЕНИЯ ФАЗ
Имеется широкая область длин фторалкильной ФА цепи, разделяющая гомогенный раствор и гель, где фторалкил-силановые (ФАС) олигомеры образуют коллоидные растворы. Эта особенность первоначально связывалась с тем, что ФА цепи в отличие от алкильных цепей имеют форму жестких стержней, состоящих из углеродной цепи в транс-конфигурации и обматывающей ее спирали из атомов фтора в составе фрагментов CF2 [26–28]. Золь-гель переход в системе жестких стержней обусловлен нематическим (ориентационным) упорядочением. Подробно изученная модель Онзагера [29, 30], описывающая этот переход, предсказывает, что коллоидный раствор существует в узкой области отношений длины стержней $L$ к их диаметру $d{\text{:}}$ ${{\Phi }_{1}} < {L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d} < {{\Phi }_{2}}.$ При ${L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d} < {{\Phi }_{1}}$ раствор изотропен, а при ${L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d} > {{\Phi }_{2}}$ образуется гель с ориентированным расположением стержней. Для жестких стержней ${{\Phi }_{1}}$ = 3.34, ${{\Phi }_{2}}$ = = 4.49. Поскольку ${L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d} \approx n,$ эта модель не согласуется с экспериментальными данными для растворов ФАС олигомеров.
Чтобы согласовать теоретическую модель с экспериментом, необходимо учесть, что в коллоидных частицах и гелях ФА цепи окружены оболочками из молекул растворителя. Описание этой модели в рамках приближения среднего поля учитывает взаимодействие мономеров (элементов стержня) друг с другом ${{V}_{{pp}}}$ и с молекулами растворителя ${{V}_{{ps}}}$ [31, 32]. Взаимодействие сольватных оболочек создает эффективное притяжение между соседними олигомерами. Когда ${{V}_{{pp}}} > {{V}_{{ps}}}$ растворитель с ростом концентрации и длины олигомеров выжимается из плотной фазы, а при ${{V}_{{pp}}} < {{V}_{{ps}}}$ олигомер сильнее набухает. Уменьшение числа сольватирующих молекул растворителя при увеличении длины цепи, наблюдаемое экспериментально [1], показывает, что в растворах олигомеров ФАС доминирует притяжение ФА цепей.
Особенности анизотропной трансляционной и вращательной диффузии реагентов в форме вытянутых эллипсоидов рассмотрены в [14–16].
Расслоение фаз изменяет кинетику ЖРП [33, 34]. Поскольку рост цепи ограничен потоком подвижных молекул мономера к малоподвижному радикалу, скорость роста пропорциональна коэффициенту диффузии мономера, который обратно пропорционален вязкости раствора (см. (4)). Поток образующихся олигомеров, уходящих в объем, пропорционален их коэффициенту диффузии, который уменьшается с ростом длины цепи и вязкости.
В разбавленном растворе олигомеров, когда занимаемый ими исключенный объем мал, относительный рост вязкости также мал и незначительно уменьшает скорость роста цепи. Дальнейший рост концентрации длинных олигомеров вызывает резкий рост характеристической вязкости, обусловленный образованием цепей. В образующемся полуразбавленном растворе коэффициент диффузии мономеров и константа роста цепи экспоненциально уменьшаются. Вновь образующиеся длинные олигомеры не способны покинуть реакционный объем, где их локальная концентрация возрастает по сравнению со средней по всему объему. Напротив, короткие олигомеры уходят в объем. ММР становится пространственно неоднородным. В области расслоения (разделения фаз) в составе коллоидных частиц преобладают длинные олигомеры, а в гомогенной фазе – короткие. В силу пространственной неоднородности суммарные ММР бимодальны.
Теоретическое описание ЖРП возможно на основе кинетических уравнений (9) при учете изменений констант скорости роста и передачи цепи, учитывающих изменения свойств среды при образовании олигомеров. Эти изменения, приводящие к золь-переходам, описываются в динамической (в частности, скейлинговой) теории растворов полимеров [21, 33–36]. Когда доля объема, занятого сферами с диаметром, равным длине олигомера L = $\bar {n}$l0 (l0 – длина фрагмента цепи), мала, раствор олигомеров считается разбавленным, а движение олигомеров независимым, если средняя длина цепи остается меньше определенного значения N1
(11)
${{M}_{0}}{{V}_{0}} < {{\left( {\sum\limits_n {{{n}^{3}}{{P}_{n}}} } \right)}^{{ - 1}}},\,\,\,\,\bar {n} < {{N}_{1}}.$С ростом длины и концентрации олигомеров траектории их движения запутываются, и раствор превращается в полуразбавленный и затем в плотный. При постоянной концентрации мономера, раствор можно считать полуразбавленным в области длин цепи, меньшей N2
(12)
${{\left( {\sum\limits_n {{{n}^{3}}{{P}_{n}}} } \right)}^{{ - 1}}} < {{M}_{0}}{{V}_{0}} < {{\left( {\sum\limits_n {{{n}^{2}}{{P}_{n}}} } \right)}^{{ - 1}}},\,\,\,\,{{N}_{1}} < \bar {n} < {{N}_{2}}.$Пользуясь соотношениями подобия для изменений коэффициентов диффузии в областях (11) и (12), оценим изменения констант скорости роста и передачи цепи. Коэффициенты продольной (D||), поперечной (${{D}_{ \bot }}$) поступательной и вращательной $({{D}_{R}})$ диффузии для вытянутых эллипсоидов (${L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d} > 1$) пропорциональны ${d \mathord{\left/ {\vphantom {d L}} \right. \kern-0em} L}$ и ${{({d \mathord{\left/ {\vphantom {d L}} \right. \kern-0em} L})}^{3}}$ соответственно
(13)
$\begin{gathered} {{D}_{{||}}} = {{k}_{B}}T\ln {{(1 + {L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d})} \mathord{\left/ {\vphantom {{(1 + {L \mathord{\left/ {\vphantom {L d}} \right. \kern-0em} d})} 2}} \right. \kern-0em} 2}\pi {{\eta }_{0}}L, \\ {{D}_{ \bot }} = {{{{D}_{{||}}}} \mathord{\left/ {\vphantom {{{{D}_{{||}}}} 2}} \right. \kern-0em} 2},\,\,\,\,{{D}_{r}} = {{6{{D}_{{||}}}} \mathord{\left/ {\vphantom {{6{{D}_{{||}}}} {{{L}^{2}}}}} \right. \kern-0em} {{{L}^{2}}}}. \\ \end{gathered} $Уменьшение вращательной диффузии создает вращательный барьер для реакций, требующих определенной относительной ориентации реагентов. Рост вращательного барьера означает, что реакция переноса цепи возможна только в ограниченном интервале углов, который резко сужается с ростом характеристической вязкости раствора $\left[ \eta \right] = {{(\eta - {{\eta }_{0}})} \mathord{\left/ {\vphantom {{(\eta - {{\eta }_{0}})} {{{\eta }_{0}}}}} \right. \kern-0em} {{{\eta }_{0}}}} \gg 1$ в области золь-гель перехода. В той же области поступательная диффузия мономера сменяется рептильным движением вдоль эффективной трубки, образующейся в сетке, создаваемой олигомерами и свободной от их непроницаемых фрагментов. Согласно модели Дои–Эдвардса [17–19] коэффициент рептильной диффузии связан с вязкостью соотношением
(14)
${{D(\eta )} \mathord{\left/ {\vphantom {{D(\eta )} D}} \right. \kern-0em} D}\left( {{{\eta }_{0}}} \right) \approx {{({\eta \mathord{\left/ {\vphantom {\eta {{{\eta }_{0}}}}} \right. \kern-0em} {{{\eta }_{0}}}})}^{\gamma }},$Соотношение (15) справедливо для масштаба смещений, превышающих длину олигомера $\bar {n}$l0. Поскольку телесный угол Ω ≈ (drep/Ln)2~ ${{\bar {n}}^{{ - 4}}},$ поток мономера к макрорадикалу, находящемуся в эффективной трубке, ограничен этим углом.
Пока в разбавленном растворе вращательный барьер отсутствует, и поступательная диффузия мономеров удовлетворяет условию (5), можно считать, что механизм ЖРП остается таким же, как в гомогенном растворе. Однако с ростом вязкости уменьшается не только константа скорости роста цепи, пропорциональная D||, но и константа скорости передачи цепи, зависящая от Ω. Как следует из (5), основным кинетическим параметром является зависящее от вязкости (т.е. N1 и N2) отношение скоростей передачи и роста цепи
(16)
$X(\bar {n}) = {{{{k}_{t}}S} \mathord{\left/ {\vphantom {{{{k}_{t}}S} {k{{M}_{0}}}}} \right. \kern-0em} {k{{M}_{0}}}},$поведение которого в областях (11) и (12) для А- и В-радикалов различно. Считая, что для А-радикалов вращательный барьер мал, изменение X(n–) определяет уменьшение коэффициента поступательной диффузии при постоянстве kt
(17)
$X(\bar {n})\sim \left\{ {\begin{array}{*{20}{c}} {{{{(1 + \beta )}}^{{ - 1}}}\left( {1 + \beta {{\bar {n}} \mathord{\left/ {\vphantom {{\bar {n}} {{{N}_{1}}}}} \right. \kern-0em} {{{N}_{1}}}}} \right),\,\,\,\,\bar {n} \leqslant {{N}_{1}}} \\ {{{{\left( {{{\bar {n}} \mathord{\left/ {\vphantom {{\bar {n}} {{{N}_{1}}}}} \right. \kern-0em} {{{N}_{1}}}}} \right)}}^{\alpha }},\,\,\,\,\bar {n} \geqslant {{N}_{1}}} \end{array}} \right.,$Рис. 4.
Переход от одно- к бимодальному ММР, обусловленный изменением константы скорости роста цепи при золь-гель переходе. Значения индексов в соотношении (16): $\alpha = 4,$ $\beta = 0.4,$ ${{N}_{1}}$ = 3, 4, 6, 10, (кривые 1–4 соответственно) ${{k}_{t}}{\text{/}}k = 0.03.$
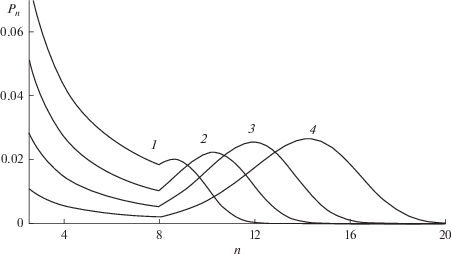
Следует указать, что соотношения (14) и (17), основанные на представлениях подобия, требуют, как и КСП, проверки с помощью решения кинетических уравнений (9) и анализа зависящих от времени ММР. Насколько известно авторам, экспериментальные и теоретические исследования таких процессов еще не проводились.
ВЫВОДЫ
1. Решение уравнений Смолуховского позволяет выявить сложную кинетику радикальной полимеризации, в которой начальный баллистический режим роста цепи первого поколения (в отсутствие передачи цепи) переходит в диффузионный режим с участием макрорадикалов многих предыдущих поколений.
2. В области перехода от баллистического к диффузионному росту цепи радикальная полимеризация с участием двух первичных радикалов не приводит к единому распределению олигомеров, характерному для ЖРП, инициируемому одним первичным радикалом. Несмотря на перемешивание обоих каналов в каждом акте передачи, образуются два распределения олигомеров с различной средней длиной цепи, зависящей от констант скорости и начальных концентраций обоих первичных радикалов.
3. В области золь-гель перехода каналы ЖРП, инициируемые двумя первичными радикалами, пространственно разделяются, если кинетически ограниченные скорости передачи цепи зависят от вращательной подвижности реагентов и уменьшаются с ростом характеристической вязкости среды в полуразбавленных растворах олигомеров, образующихся в плотной фазе.
Работа выполнена при финансовой поддержке Программы Фундаментальных Исследований Президиума РАН ПФИ I.55П и по теме Государственного задания № 0089-2014-0025.
Список литературы
Ким И.П., Шестаков А.Ф. // Химия высоких энергий. 2009. Т. 43. № 6. С. 516.
Ким И.П., Мартыненко И.М., Шульга Ю.М., Шестаков А.Ф. // Химия высоких энергий. 2010. Т. 44. № 6. С. 483.
Ким И.П. // Химия высоких энергий. 2011. Т. 45. № 5. 399.
Ким И.П., Перепелицина Е.О., Шестаков А.Ф., Шульга Ю.М., Куница А.А. // Химия высоких энергий. 2011. Т. 45. № 6. С. 511.
Ким И.П. // Изв. АН Сер. хим. 2013. № 9. С. 2065.
Куница А.А., Шестаков А.Ф., Ким И.П. // Изв. АН Сер. хим. 2011. № 9. С. 1761.
Benderskii V.A., Makarov D.E., White C.A. Chemical Dynamics at low Temperatures. 1994. Wiley. N.Y. Ch. 6.
Ким И.П., Бендерский В.А., Мартыненко В.М., Черняк А.В. // Химия высоких энергий. 2017. Т. 51. № 4. С. 292.
Ким И.П., Мартыненко В.М., Черняк А.В., Бендерский В.А. // Химия высоких энергий. 2017. Т. 51. № 4. С. 300.
Brinker C.J., Scherer G.W. // Sol-Gel Science. 1990. Academic Press. Boston. Ch. 3.
Lopez M.C., Demitras I., Maskill H., Mishima M. // J. Phys. Org. Chem. 2008. 21. P. 614.
Ignatyev I.S., Montenjo M., Gonzalez J.L. // Dalton Trans. 2010. 39. P. 6967.
Agarwal U.S., Khakhar D.V. // J. Chem. Phys. 1993. 99. P. 1382.
Gupta J.S., Khakhar D.V. // J. Chem. Phys. 1998. 108. P. 5626.
Baldo M., Grass A., Raudino A. // Phys. Rev. A. 1989. 40. P. 1017.
Vasanthi R., Bhattacharyya S., Bagchi B. // J. Chem. Phys. 2002. 116. P. 1092.
Doi M., Edwards S.F. // J. Chem. Soc. Farad.Trans II. 1978. 74. P. 918.
Doi M., Edvards S.F. // J. Chem. Soc. Farad. Trans II. 1978. 74. P. 1802. P. 1818.
Дой М., Эдвардс С. Динамическая теория полимеров. 1998. М.: Мир. [Doi M., Edvards S.F. The Theory of Polymer Dynamics. 1986. Oxford Univ. Press. N.Y.]
Rubtsov I.V. // Acc.Chem. Res. 2009. 42. P. 1385.
Бендерский В.А., Кац Е.И. // ЖЭТФ. 2013. Т. 143. № 1. С. 5.
Бендерский В.А., Коткин А.С., Кац Е.И., Рубцов И.В. // Письма в ЖЭТФ. 2013. Т. 98. № 4. С. 219.
Lynd N.A., Hillmayer M.A. // Macromolecules. 2007. 40. P. 8050.
Listak J., Jakubowski W., Mueller L., Plichta A., Matyjaszwski K., Bockstaller M.R. // Macromolecules. 2008. 41. P. 5919.
Gentekos D.T., Dupuis L.N., Fors B.P. // J. Am. Chem. Soc. 2016. 138. P. 18548.
D-Amorem M., Talarico G., Barone V. // J. Am. Chem. Soc. 2006. 128. P. 1099.
Tsige M., Curro J.G., Grest G.S. // J. Chem. Phys. 2008. 129. P. 214901.
Dalvi V.H., Rossky P.J. // Proc.Nat. Acad.Sci. USA. 2010. 107. P. 13603.
Чандрасекар С. Жидкие кристаллы. 1980. М.: Мир. [Chandrasekhar S. Liquid Crystals. 1977. Cambridge Univ. Press. Cambridge. Ch.2.]
Kayser R.F., Raveche H.J. // Phys. Rev. A.1978. 17. P. 2067.
Zilman A., Kiefier J., Molino F., Porte G., Safran S.A. // Phys. Rev. Lett. 2003. 91. P. 015901.
Ким И.П., Бендерский В.А. // Химия высоких энергий. 2010. Т. 44. № 5. С. 387.
Ким И.П., Бендерский В.А. // Химия высоких энергий. 2011. Т. 45. № 5. С. 406.
Ким И.П., Бендерский В.А. // Химия высоких энергий. 2015. Т. 49. № 1. С. 3.
де Жен П. Идеи скейлинга в физике полимеров. 1982. М.: Мир. [De Gennes P.G. Scaling Concept in Polymer Physics. 1979. Cornell Univ. Press. Ithaca.]
Гросберг А.Ю., Хохлов А.Р. Статистическая физика макромолекул. 1989. М.: Наука. [Grosberg A.Yu., Khokhlov A.R. Statistical Physics of Macromolecules. 1994. Springer. N.Y. Ch.4.]
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий