

Химия высоких энергий, 2021, T. 55, № 3, стр. 181-194
ТУШЕНИЕ ФЛУОРЕСЦЕНЦИИ 3,5-ДИФЕНИЛ-8-CF3-BODIPY ЛЮМИНОФОРОВ С АМИНОФЕНИЛЬНЫМИ ЗАМЕСТИТЕЛЯМИ АРОМАТИЧЕСКИМИ МОЛЕКУЛАМИ
А. В. Гадомская a, b, *, А. В. Невидимов a, b, С. А. Товстун a, b, О. В. Петрова c, Л. Н. Собенина c, Б. А. Трофимов c, В. Ф. Разумов a, b, d
a Институт проблем химической физики РАН
142432 Черноголовка, просп. акад. Семенова, 1, Россия
b Московский физико-технический институт (национальный исследовательский университет)
141701 Долгопрудный, Институтский пер., 9, Россия
c Иркутский институт химии им. А.Е. Фаворского Сибирского отделения РАН
664033 Иркутск, ул. Фаворского, 1, Россия
d Центр фотохимии РАН, ФНИЦ “Кристаллография и фотоника” РАН
119421 Москва, ул. Новаторов, 7а, Россия
* E-mail: ann.gadomsky@gmail.com
Поступила в редакцию 28.12.2020
После доработки 14.01.2021
Принята к публикации 15.01.2021
Аннотация
Обнаружено, что квантовый выход флуоресценции 3,5-дифенил-8-CF3-BODIPY люминофоров с аминофенильными заместителями в ароматических растворителях сильно зависит от того, в каком положении находится аминогруппа анилина по отношению к положению присоединения к ядру молекулы BODIPY. Было выдвинуто и обосновано предположение о том, что причиной тушения мета-изомеров BODIPY является специфическое взаимодействие молекул люминофоров с ароматическими молекулами.
ВВЕДЕНИЕ
Известно, что флуоресценция люминофоров на основе производных 4‑бора‑3а,4а‑диаза‑s‑индацена (BODIPY) может сильно зависеть от полярности растворителя. Это связано с тем, что процессы излучения и поглощения света в этих молекулах сопровождаются внутримолекулярным переносом заряда и при этом может сильно изменяться дипольный момент молекулы.
Примером таких соединений являются исследованные в данной работе аминофенильные производные BODIPY, структуры которых приведены на схеме 1 . В этих соединениях два атома фтора в положении 4 и группа CF3 в положении 8 являются сильными акцепторами электрона, а аминофенильные заместители в положениях 3 и 5 проявляют значительные электронодонорные свойства, при этом внутримолекулярный перенос заряда для представленных на схемах 1 структур наиболее выражен для соединений P1 и P2, в которых аминогруппа находится в пара‑положении по отношению к месту замещения по положениям 3 и 5.
В неполярных растворах алифатических углеводородов соединения M1, M2, P1, P2 обладают высоким квантовым выходом флуоресценции, а вот в полярных растворителях эффективность люминесценции значительно уменьшается. Внутримолекулярное пространственное разделение зарядов, возникающее при фотовозбуждении красителей BODIPY, стабилизируется полярными апротонными растворителями, такими как 1,4-диоксан, этилацетат, ацетонитрил, диметилсульфоксид (ДМСО), и делает эти соединения практически нелюминесцирующими [1].
Резкое уменьшение квантового выхода люминесценции в полярных растворителях связывают с тем, что в результате сильного взаимодействия с дипольными молекулами растворителя происходит резкое понижение электронного уровня возбужденного состояния и это влечет за собой значительное увеличение константы безызлучательного перехода в основное состояние [2].
Вместе с тем квантовый выход люминесценции соединений M1 и M2, в которых аминогруппа находится в мета‑положении, в растворе ДМСО и подобных полярных растворителях уменьшается, но не так значительно, как это наблюдается для соединений Р1 и Р2. Это, по‑видимому, связано с тем, что в силу меньшего взаимодействие соединений M1 и M2 с дипольными молекулами растворителя уровень возбужденного электронного состояния соединений M1 и M2 не так значительно понижается, как это имеет место для молекул Р1 и Р2.
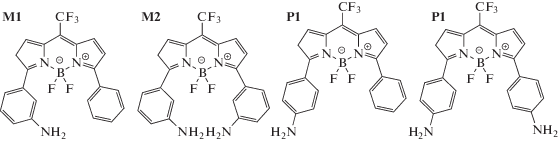
Схема 1 . Производные 8-CF3-BODIPY, исследованные в работе.
В данной работе было обнаружено, что в ароматических растворителях (бензол, толуол и о‑ксилол) люминесцентные характеристики люминофоров очень сильно зависят от того, в каком положении находится аминогруппа анилина по отношению к положению присоединения к ядру молекулы BODIPY. В работе [3] было проведено специальное исследование влияния полярности, поляризуемости и рН растворителей на положение электронных спектров молекул с внутримолекулярным переносом заряда. Для объяснения особой роли ароматических растворителей был введен новый эмпирический показатель, так называемая величина диполярности (dipolarity, SdP), которая наряду с такими величинами как полярность и поляризуемость растворителя позволяет найти эмпирическое соответствие сдвигов полос поглощения и люминесценции молекул, в которых электронные переходы связаны с внутренним разделением зарядов. Не вдаваясь в детали, можно констатировать, что влияние ароматических растворителей тем больше, чем больше изменяется дипольный момент молекулы люминофора в результате перехода в возбужденное состояние. Однако в случае молекул BODIPY, соединения M1, M2, P1, P2, в ароматических растворителях (бензол, толуол и о‑ксилол) наблюдается обратный эффект: люминесценция мета‑изомеров (соединения M1 и M2) тушится значительно сильнее, чем люминесценция пара‑изомеров (соединения P1 и P2), хотя внутримолекулярный перенос заряда в мета‑изомерах гораздо меньше, чем в пара‑изомерах.
Таким образом, это аномальное поведение мета‑ и пара‑изомеров в ароматических растворителях нельзя объяснить внутримолекулярным переносом заряда. В частности, бесплодными будут попытки использовать понятие параметра диполярности, согласно которому бензол и толуол можно условно рассматривать как некоторый промежуточный случай между неполярным н‑гексаном и полярными растворителями. В таком случае, тушение люминесценции пара‑изомера должно быть существенно больше, чем для мета‑изомера, чего не наблюдается в эксперименте, подробности которого обсуждаются далее. Именно это противоречие и послужило мотивацией для постановки данной работы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы и материалы
В работе были использованы следующие растворители: н‑гексан (99%, HPLC, Scharlau), циклогексан (99%, Sigma-Aldrich), бензол (99%, Sigma‑Aldrich), толуол (99.99%, HPLC, Fisher Scientific), о‑ксилол (99%, Sigma‑Aldrich). Все используемые реагенты и органические растворители дополнительной очистке не подвергались.
Спектральные измерения
Регистрацию спектров поглощения проводили на спектрофотометре “Shimadzu UV 3101PC”, спектров люминесценции – на спектрофлуориметре “Shimadzu RF‑6000”. Кинетики затухания люминесценции измерялись на времяразрешенном спектрофлюориметре “PicoQuant FluoTime 200”, в качестве источника возбуждения использовался полупроводниковый лазер с длиной волны 467 нм.
Квантовые выходы люминесценции определяли при оптической плотности образцов на длине волны возбуждения не более чем 0.1. В качестве стандарта для измерения квантового выхода люминесценции использовали этанольный раствор родамина 6Ж (квантовый выход в этанольном растворе равен 0.95% [4]). При вычислении квантового выхода люминесценции ККТ учитывались показатели преломления соответствующих растворителей [5].
Все спектральные и спектрально-кинетические измерения проводились при комнатной температуре в кварцевых флуоресцентных кюветах с длиной оптического пути 10 мм. Спектральное разрешение при измерении люминесценции составляло 3 нм, а для образцов с низким квантовым выходом – 5 нм.
Спектры ЯМР 1Н (400 МГц), 13С (100 МГц), 15N (41 МГц) и 19F (377 МГц) регистрировали на приборе “Bruker Avance 400” в СDСl3 и DMCO-d6. Химические сдвиги приведены в м.д. Отнесение сигналов в спектрах ЯМР 1Н осуществляли с помощью экспериментов COSY и NOESY. Резонансные сигналы атомов углерода установлены на основе экспериментов 1H−13C HSQC и 1H−13C HMBC, значения δ 15N − на основе эксперимента 2D1H−15NHMBC. Химические сдвиги (δ) 1H приведены относительно сигналов остаточных протонов дейтерированных растворителей (7.27 м.д. для CDCl3, 2.50 м.д. для DMSO-d6), химические сдвиги в спектрах 13C − относительно пиков дейтерированных растворителей (77.1 м.д. для CDCl3, 39.5 м.д. для DMSO-d6), в спектрах 15N – относительно MeNO2 (0.0 м.д.) и в спектрах 19F – относительно CFCl3 (0.0 м.д.). Константы спин-спинового взаимодействия измеряли в герцах (Гц) по одномерным спектрам 1H. ИК-спектры получали на спектрометре “Bruker IFS-25” (400–4000 см–1, KBr). Микроанализ (C, H, N) проводили на приборе FlashEA 1112 СHNS-O/MAS (CHNAnalyzer). Содержание фтора определяли на спектрометре SPECOL 11 (CarlZeissJena, Germany). Температуры плавления (без коррекции) определяли с помощью цифрового аппарата StuartScientificSMP3.
Квантово‑химические расчеты для оптимизации геометрии структур в основном и возбужденном состояниях проводили с помощью свободно распространяемой программы ORCA [6, 7]. Для основного состояния применяли теорию функционала электронной плотности (DFT), для возбужденного состояния TD‑DFT, с функционалом PBE0 и базисом 6-311G**. Для найденной геометрии проводили расчет матрицы вторых производных, чтобы удостовериться в истинности найденного минимума. Поиск геометрии возбужденного состояния начинали с найденной оптимальной геометрии основного состояния. Для найденных оптимальных геометрий рассчитывали энергию вертикальных переходов: из оптимизированного состояния S0 в S1 без изменения конфигурации ядер и из неоптимизированного состояния S0 в оптимизированное состояние S1 (также без изменения конфигурации ядер).
Молекулярно-динамические расчеты в программе NAMD [8] использовали для исследования геометрической структуры возможных комплексов BODIPY и бензола. Для этого рассматривали ячейку с периодическими граничными условиями, в центре которой располагали молекулу красителя, а остальное пространство заполняли бензолом. Заряды атомов молекулы красителя заимствовали из результатов квантово‑химических расчетов [9 ] . Интегрирование классических уравнений движения производили по схеме Верле со скоростями с шагом интегрирования 1 фс, общая длина траектории молекулярной динамики составила 1.1 нс, из которых первые 0.1 нс проводили минимизацию энергии. Через каждые 100 шагов сохраняли текущие координаты всех атомов в файл траектории, из анализа которого отыскивали возможные структуры комплексов BODIPY с бензолом. Найденные структуры оптимизировали в программе ORCA, как описано выше.
Синтез люминофоров M1, M2, P1 и P2
Синтез люминофоров M1 и P1 подробно описан в статье [1].
Люминофоры M2 и P2 были получены впервые по методике [10] из 2‑(3‑ и 4‑аминофенил)пирролов 5a,b, которые были синтезированы из соответствующих ацетофеноноксимов и ацетилена по реакции Трофимова [11].
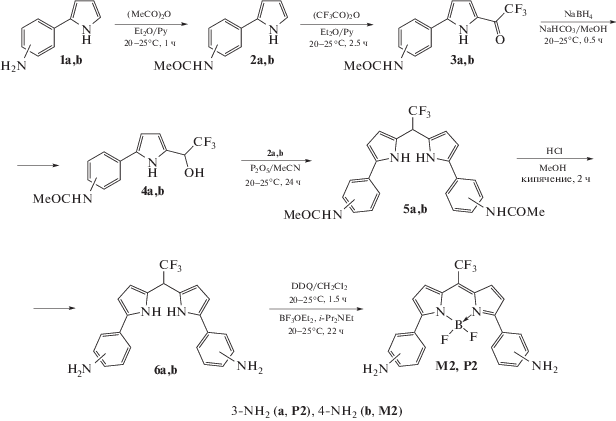
Схема 2 . Схема синтеза люминофоров M2 и P2.
На схеме 2 представлены следующие стадии синтеза люминофоров M2 и P2: (i) защита аминогруппы 2‑(3‑ и 4‑аминофенил)пирролах 1a,b уксусным ангидридом; (ii) трифторацетилирование 2‑(3‑ и 4‑ацетамидофенил)пирролов 2a,b трифторуксусным ангидридом; (iii) восстановление 2‑трифторацетилпирролов 3a,b с помощью NaBH4; (iv) конденсация трифторметил(пиррол)этанолов 4a,b с 2‑(3‑ и 4‑ацетамидофенил)пирролами 2a,b; (v) снятие ацетамидной защиты в дипиррометанах 5a,b и (vi) окисление/комплексообразование дипиррометанов 6a,b в люминофоры М2 и P2.
Синтез и спектральные свойства пирролов 1a,b–4a,b описаны в статье [1].
Синтез дипиррометанов 5a,b
Смесь пирролов 2а,b (0.200 г, 1.0 ммоль), 4а,b (0.298 г, 1.0 ммоль) и P2O5 (0.142 г, 1.0 ммоль) в MeCN (30 мл) перемешивали при комнатной температуре в течение 24 ч. Затем добавляли NaHCO3 (0.101 г, 1.2 ммоль) и реакционную смесь перемешивали в течение 1.5 ч. Осадок отфильтровали и промывали MeCN (3 × 5 мл). Растворитель удаляли из фильтрата. Дипиррометан 5a,b выделили из осадка колоночной хроматографией (SiO2, элюент MeCN/н-гексан/CH2Cl2 в соотношении 20 : 1 : 1).
N,N'-[(2,2,2-трифторэтан-1,1-диил)бис(1H-пиррол-5,2-диил-3,1-фенилен)]диацетамид (5a). Выход 0.226 г (47%), Тпл. 148–150°C.
ЯМР 1Н (400 МГц, DMSO-d6), δ: 11.35 (br s, 2 H, NH), 9.90 (br s, 2 H, NHCO), 7.79 (m, 2 H, H-2 Ph), 7.36 (m, 2 H, H-4 Ph), 7.28 (m, 4 H, H-5,6 Ph), 6.37 (m, 2 H, H-4), 6.22 (m, 2 H, H-3), 5.11 (q, J = 9.5 Гц, 1 H, CH), 2.06 (s, 6 H, Me).
ЯМР 13С (100 МГц, DMSO-d6), δ: 168.2, 139.5, 133.0, 131.5, 129.5, 125.4, 125.4 (q, J = 279.5 Гц, CF3), 118.6, 117.0, 114.7, 109.3, 105.9, 42.1 (q, J = 30.1 Гц, CH), 23.9.
ЯМР 19F (377 МГц, CDCl3), δ: –67.8 d (J = 9.6 Гц).
ЯМР 15N (41 МГц, DMSO-d6), δ: –245.5 (J = = 90.8 Гц, NHCO), –226.5 (1JNH = 95.5 Гц, NH).
ИК (KBr), ν: 3406, 3303, 3110, 2930, 1672, 1614, 1594, 1540, 1500, 1429, 1384, 1372, 1256, 1189, 1158, 1108, 1075, 1046, 882, 866, 775, 713, 690, 538 см–1.
Вычислено для C26H23F3N4O2 (480.49): C, 64.99; H, 4.83; F, 11.86; N, 11.66. Найдено: C, 64.72; H, 4.95; F, 11.68; N, 11.58.
N,N'-[(2,2,2-трифторэтан-1,1-диил)бис(1H-пиррол-5,2-диил-4,1-фенилен)]диацетамид (5b). Выход 0.207 г (43%), Тпл. 154–156°C.
ЯМР 1Н (400 МГц, DMSO-d6), δ: 11.23 (br s, 2 Н, NH), 9.91 (br s, 2 Н, NHCO), 7.55 (m, 8 Н, H-2,3,5,6 Ph), 6.39 (m, 2 H, H-4), 6.18 (m, 2 Н, Н-3), 5.03 (q, J = 9.7 Гц, 1 Н, СН), 2.04 (s, 6 Н, Me).
ЯМР 13С (100 МГц, DMSO-d6), δ: 168.2, 137.3, 131.5, 127.7, 125.4 (q, J = 279.3 Гц, CF3), 124.9, 123.9, 119.3, 109.3, 105.4, 42.2 (q, J = 30.3 Гц, СН), 24.0.
ЯМР 19F (377 МГц, CDCl3), δ: –67.9 (d, J = 9.7 Гц).
ЯМР 15N (41 МГц, DMSO-d6), δ: –246.5 (J = = 90.4 Гц, NHCOMe), –226.9 (J = 95.3 Гц, NH).
ИК (KBr), ν: 3402, 3304, 2930, 1673, 1600, 1524, 1426, 1370, 1318, 1254, 1158, 1107, 831, 776 см-1.
Вычислено для C26H23F3N4O2 (480.49): C, 64.99; H, 4.83; F, 11.86; N, 11.66. Найдено: C, 64.78; H, 5.09; F, 11.63; N, 11.85.
Синтез дипиррометанов 6a,b
Раствор дипиррометана 5a,b (1.201 г, 2.5 ммоль) в MeOH (80 мл) кипятили в присутствии 15% HCl в течение 2 ч. После охлаждения к смеси добавляли воду (40 мл) и раствор NH4OH (до pH ~ 7–8). Затем смесь экстрагировали CH2Cl2 (4 × 30 мл), осадки промывали водой (3 × 20 мл) и сушили над Na2SO4. Растворитель удалили и осадок очистили с помощью колоночной хроматографией (SiO2, эфир), получив дипиррометан 6a,b.
3,3'-[(2,2,2-трифторэтан-1,1-диил)бис(1H-пирролe-5,2-диил)]дианилин (6a). Выход 0.888 г (90%), Тпл. 126–127°C.
ЯМР 1Н (400 МГц, DMSO-d6), δ: 11.19 (br s, 2 Н, NH), 7.00 (m, 2 Н, H-5 Ph), 6.79 (m, 4 Н, H-2,6 Ph), 6.41 (m, 2 H, H-4 Ph), 6.29 (m, 2 Н, Н-4), 6.16 (m, 2 H, H-3), 5.07 (q, J = 9.8 Гц, 1 Н, СН), 5.01 (br s, 4 Н, NH2).
ЯМР 13С (100 МГц, DMSO-d6), δ: 148.7, 133.2, 132.3, 129.1, 125.5 (q, J = 279.8 Гц, CF3), 124.8, 112.1, 111.9, 109.4, 109.1, 105.3, 42.0 (q, J = 30.2 Гц, CH).
ИК (KBr), ν: 3426, 3396, 2957, 2925, 2864, 1615, 1506, 1471, 1330, 1253, 1194, 1158, 1106, 864, 778, 691 см–1.
Вычислено для C22H19F3N4 (396.42): C, 66.66; H, 4.83; F, 14.38; N, 14.13. Найдено: C, 66.39; H, 4.99; F, 14.27; N, 14.35.
4,4'-[(2,2,2-трифторэтан-1,1-диил)бис(1H-пиррол-5,2-диил)]дианилин (6b). Выход 0.829 (84%), Тпл. 182–183°C.
ЯМР 1Н (400 МГц, DMSO-d6), δ: 10.93 (br s, 2 Н, NH), 7.29 (m, 4 Н, H-3,5 Ph), 6.56 (m, 4 Н, H-2,6 Ph), 6.15 (m, 2 H, H-4), 6.09 (m, 2 Н, Н-3), 5.06 (br s, 4 H, NH2), 4.98 (q, J = 9.9 Гц, 1 Н, СН).
ЯМР 13С (100 МГц, DMSO-d6), δ: 146.9, 132.6, 125.6 (q, J = 280.3 Гц, CF3), 124.8, 123.7, 121.1, 114.1, 108.8, 103.1, 42.2 (q, J = 30.3 Гц, СН).
ИК (KBr), ν: 3430, 2925, 2825, 1622, 1600, 1524, 1479, 1333, 1258, 1158, 1105, 832, 778 см–1.
Вычислено для C22H19F3N4 (396.42): C, 66.66; H, 4.83; F, 14.38; N, 14.13. Найдено: C, 66.58; H, 4.94; F, 14.25; N, 14.23.
Синтез BODIPY M2 и P2
Смесь дипиррометана 6a,b (0.595 г, 1.50 ммоль) и DDQ (2,3-дихлор-5,6-дицианобензохинона, 0.341 г, 1.50 ммоль) в CH2Cl2 (45 мл) перемешивали при комнатной температуре в течение 1.5 ч. Затем добавляли (i-Pr)2NEt (1.939 г, 15.0 ммоль), полученный раствор перемешивали в течение 10 мин и затем в него по каплям добавляли BF3.OEt2 (3.193 г, 22.5 ммоль) в течение 30 мин. Реакционную смесь перемешивали при комнатной температуре в течение 22 ч и разбавляли насыщенным раствором NaHCО3, органический слой отделяли, водный слой экстрагировали CH2Cl2 (3 × 20 мл). Объединенные органические соли промывали водой (3 × × 30 мл) и высушивали над K2CO3. После удаления растворителя из остатка, BODIPY M2 и P2 выделяли колоночной хроматографией (SiO2, элюент н‑гексан/эфир, градиент от 1 : 1 до 0 : 1 для 2a; н‑гексан/CH2Cl2, градиент от 1 : 1 до 0 : 1 для 4b).
4,4-дифтор-3,5-бис(3-анилин)-8-трифторметил-4-бор-3a,4a-диаза-s-индацен (М2). Выход 0.061 г (9%), Тпл 164–166°C.
ЯМР 1Н (400 МГц, СDCl3), δ: 7.42 (m, 2 H, H-2 Ph), 7.21 (m, 4 H, H-5,6 Ph), 7.17 (m, 2 H, H-1,7), 6.75 (m, 2 H, H-2,6), 6.70 (m, 2 H, H-4 Ph), 3.75 (br s, 4 H, NH2).
ЯМР 13С (100 МГц, СDCl3), δ: 162.2, 146.4, 133.4, 132.9, 130.1, 129.3, 126.5 (q, J = 31.5 Гц, C-8), 123.1, 122.8 (q, J = 276.0 Гц, CF3), 120.2 (t, J = 2.8 Гц), 117.3, 115.9 (t, J = 4.0 Гц).
ИК (KBr), ν: 3442, 3371, 2924, 2859, 1620, 1566, 1509, 1474, 1421, 1306, 1269, 1225, 1135, 1085, 978, 902, 783 см–1.
Вычислено для C22H16BF5N4 (442.20); C, 59.76; H, 3.65; B, 2.44; F, 21.48; N, 12.67. Найдено: C, 59.84; H, 3.48; F, 21.68; N, 12.79.
4,4-дифтор-3,5-бис(4-анилин)-8-трифторметил-4-бор-3a,4a-диаза-s-индацен (P2). Выход 0.093 г (14%), Тпл 165–167°C.
ЯМР 1Н (400 МГц, СDCl3), δ: 7.81 (m, 4 H, H-2,6 Ph), 7.34 (m, 2 H, H-1,7), 6.71 (m, 2 H, H-2,6), 6.68 (m, 4 H, H-3,5 Ph), 4.00 (br s, 4 H, NH2).
ЯМР 13С (100 МГц, СDCl3), δ: 160.5, 148.9, 133.4, 131.8, 131.7, 128.9, 123.2 (q, J = 276.0 Гц, CF3), 123.0 (q, J = 32.0 Гц, C-8), 122.0, 114.6.
ИК (KBr), ν: 3480, 3390, 3216, 2922, 2854, 1623, 1563, 1470, 1446, 1304, 1276, 1226, 1133, 1080, 907, 730 см–1.
Вычислено для C22H16BF5N4 (442.20); C, 59.76; H, 3.65; B, 2.44; F, 21.48; N, 12.67. Найдено: C, 59.94; H, 3.38; F, 21.25; N, 12.87.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
В табл. 1 сведены экспериментальные данные положения максимумов спектров поглощения и люминесценции, стоксовых сдвигов, квантовых выходов и времен жизни люминесценции. Кроме того, в таблице для сравнения приведены эти же характеристики для 3,5‑дифенил‑8‑CF3‑BODIPY (F2) в различных растворителях и для P1, M1 в полярном растворителе ДМСО, взятые из работы [1].
Таблица 1.
Спектральные характеристики BODIPY в разных растворителях: положение пика поглощения (λпогл) и люминесценции (λлюм), стоксов сдвиг (Δν), квантовый выход люминесценции (φ) и время жизни возбужденного состояния (τ)
| № | Растворитель | λпогл, нм | λлюм, нм | Δν, см–1 | φ | τ, нс |
|---|---|---|---|---|---|---|
| M1 | н-гексан | 598 | 632 | 895 | 0.53 | 5.16 |
| циклогексан | 601 | 634 | 878 | 0.52 | 5.16 | |
| бензол | 605 | 636 | 810 | 0.011 | 5.34 | |
| толуол | 606 | 636 | 797 | 0.017 | 5.42 | |
| ДМСО* | 600 | 648 | 1180 | 0.0004 | <0.1 | |
| M2 | н-гексан | 605 | 638 | 863 | 0.42 | 5.41 |
| циклогексан | 607 | 642 | 920 | 0.43 | 5.41 | |
| бензол | 610 | 646 | 893 | 0.003 | 5.41 | |
| толуол | 612 | 652 | 991 | 0.002 | 5.35 | |
| P1 | н-гексан | 632 | 660 | 735 | 0.425 | 4.3 |
| циклогексан | 635 | 665 | 729 | 0.40 | 4.3 | |
| бензол | 647 | 692 | 1044 | 0.19 | 2.1 | |
| толуол | 648 | 693 | 1056 | 0.21 | 2.2 | |
| ДМСО* | 676 | 754 | 1530 | n.d. | < 0.1 | |
| P2 | н-гексан | 662 | 692 | 685 | 0.26 | 2.35 |
| циклогексан | 665 | 696 | 691 | 0.24 | 2.35 | |
| бензол | 682 | 730 | 977 | 0.15 | 2.35 | |
| толуол | 685 | 730 | 961 | 0.19 | 2.3 | |
| F2* | н-гексан | 592 | 622 | 820 | 0.8 | 6.29 |
| 1,4-диоксан | 594 | 630 | 950 | 0.83 | 5.89 | |
| этилацетат, | 592 | 626 | 940 | 0.82 | 6.13 | |
| ацетонитрил | 586 | 626 | 1090 | 0.84 | 6.43 | |
| ДМСО | 599 | 639 | 1030 | 0.71 | 5.08 |
* Данные взяты из работы [1].
Во-первых, следует отметить, что электронодонорный характер аминофенильных заместителей хорошо проявляется в батохромном сдвиге спектров поглощения и люминесценции исследуемых соединений, измеряемых в неполярном растворителе (н‑гексан или циклогексан), по сравнению с соединением F2. Видно, что для мета‑изомеров сдвиг значительно меньше (Δλлюм = 10 нм и Δλлюм = 16 нм для M1 и M2, соответственно), чем для пара-изомеров (Δλлюм = 38 нм и Δλлюм = 70 нм для P1 и P2 соответственно), при этом наличие двух аминофенильных заместителя смещает спектр люминесценции сильнее, чем один.
Стоксов сдвиг в неполярных растворителях, таких как н‑гексан, примерно одинаков для всех этих соединений. То же относится и к величине квантового выхода люминесценции: в целом он достаточно высокий и находится в пределах 80–25%, при этом снижение наблюдается при сдвиге максимума люминесценции в длинноволновую область. В полярных растворителях, как это уже обсуждалось во введении, резкое падение люминесценции (в 103–104 раз) наблюдается для всех исследованных аминофенильных производных BODIPY. Важно отметить, что для пара‑изомеров тушение люминесценции больше, чем для мета‑изомеров, при этом квантовый выход люминесценции F2 остается практически неизменным даже в наиболее полярном растворителе ДМСО.
Во-вторых, обращает на себя внимание необычное влияние ароматических растворителей бензола и толуола на исследуемые аминофенильные производные BODIPY, о котором говорилось во введении. Если для пара‑изомеров P1 и P2 в растворах бензола и толуола квантовый выход и время жизни фотолюминесценции незначительно меняются, то для мета‑изомеров M1 и M2 тушение люминесценции происходит более чем в 100 раз, однако время жизни – не меняется. Как уже было сказано во введении, необычность этого влияния заключается в том, что мета‑изомеры M1 и M2 характеризуются меньшей величиной внутримолекулярного переноса заряда при поглощении кванта света, чем пара‑изомеры P1 и P2. Это подтверждают данные табл. 1. Действительно, стоксов сдвиг и тушение люминесценции в полярном растворителе ДМСО значительно больше для пара‑изомеров, чем для мета‑изомеров, в то время как в ароматических растворителях (бензол и толуол) все ровно наоборот. Поэтому механизм тушения люминесценции в ароматических растворителях следует, видимо, искать в некотором специфическом взаимодействии, контролируемом структурным соответствием молекул растворителя и люминофора. Для более детального исследования механизма этого процесса были проведены измерения квантового выхода люминесценции растворов M1, M2, P1, P2 в н‑гексане в зависимости от концентрации добавляемых в раствор ароматических молекул: бензола, толуола, о‑ксилола.
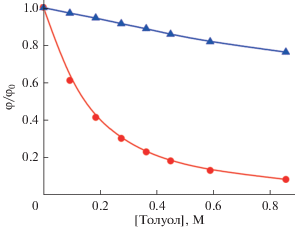
На рис. 1 для сравнения показано изменение относительного квантового выхода люминесценции мета‑ M1 и пара‑изомеров P1 в зависимости от концентрации толуола. Добавление в раствор н‑гексана всего 1 мольного процента толуола приводит к тушению люминесценции соединения M1 на 40%, в то время как P1 тушится всего лишь на 3.5%. Возможно, что специфическое взаимодействие толуола происходит для обоих соединений M1 и P1. Однако, для соединения P1 такое взаимодействие намного слабее. Измерение таких же концентрационных зависимостей для соединений M1 и P1 в случае добавления бензола и о‑ксилола, а также и для соединений M2 и P2 при добавлении толуола, бензола и о‑ксилола привели к качественно аналогичным результатам. Другими словами, люминесценция мета‑изомеров при увеличении концентрации ароматических добавок тушится значительно сильнее, чем это имеет место в случае пара‑изомеров.
Рис. 1.
Зависимость квантового выхода люминесценции соединений M1 (кружки) и P1 (треугольники) от концентрации толуола в н-гексане. Здесь φ0 и φ – квантовые выходы люминесценции в отсутствие и в присутствии тушителя соответственно.
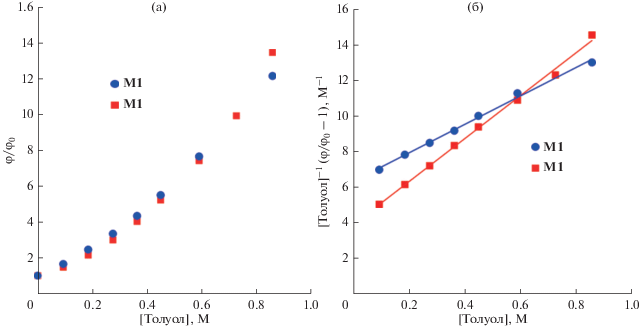
На рис. 2 результаты двух серий экспериментов тушения люминесценции соединений M1 и M2 в растворах н‑гексана в зависимости от мольной доли толуола представлены в координатах Штерна–Фольмера в предположении только динамического механизма тушения (рис. 2а) и смешанного тушения (рис. 2б), т.е. динамического и статического одновременно.
Рис. 2.
Обработка экспериментальных данных соединений M1 и M2 по уравнению Штерна–Фольмера: (а) динамическое тушение, (б) смешанное тушение: динамическое и статическое.
Динамическое тушение люминесценции описывается уравнением:
(1)
$\frac{{{{\varphi }_{0}}}}{\varphi } = \left( {1 + {{K}_{{\text{S}}}}\left[ {\text{Q}} \right]} \right)\left( {1 + {{K}_{{\text{D}}}}\left[ {\text{Q}} \right]} \right).$В этом случае концентрационная зависимость должна спрямляться в координатах:
(2)
$\frac{1}{{{\text{[Q}}]}}\left( {\frac{{{{\varphi }_{0}}}}{\varphi } - 1} \right) \to {\text{[Q}}].$Из рис. 2б видно, что экспериментальные данные неплохо спрямляются в координатах (2), т.е. для смешанного механизма тушения. Эти экспериментальные данные описываются уравнением (1) по методу наименьших квадратов с критерием достоверности R2 = 0.9994 для соединения М1 и R2 = 0.9991 для М2. Нетрудно заметить, что тангенс угла наклона линейных функций на рис. 2б определяет произведение констант динамического и статического тушения, а точка пересечения с осью ординат – сумму этих констант. Для М1 решение соответствующего квадратного уравнения позволяет определить значения констант с точностью до перестановки: KD = 5.5, KS = 1.3 [M–1] (KS = = 5.5, KD = 1.3 [M–1]). Для М2 вещественных решений для констант нет. Следовательно, для люминофора М2 с двумя аминофенильными группами механизм тушения является более сложным.
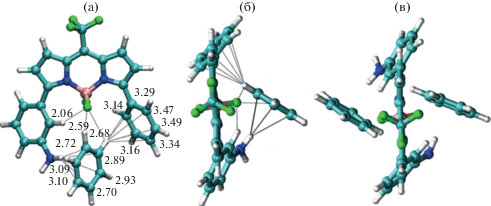
В качестве рабочей гипотезы было сделано предположение, что тушение люминесценции молекулы люминофора М2 с двумя аминофенильными группами может происходит в результате образования двух комплексов состава 1 : 1 и 1 : 2, т.е. молекула люминофора может образовывать комплекс как с одной, так и с двумя молекулами тушителя. На рис. 3 показана структура таких комплексов. Комплексы образуются в результате одновременного взаимодействия бензольного кольца с аминогруппой и атомами фтора.
Рис. 3.
Структура комплексов бензола с М1 состава 1 : 1 фронтальный вид (a) и вид сверху (б); комплекс М2 с бензолом состава 1 : 2 (в).
Если принять эту гипотезу о наличии двух комплексов, то зависимость квантового выхода люминесценции от концентрации тушителя будет иметь вид:
(3)
$\frac{{{{\varphi }_{0}}}}{\varphi } = \left( {1 + {{K}_{{\text{D}}}}[{\text{Q}}]} \right)\left( {1 + {{K}_{{\text{S}}}}[{\text{Q}}] + {{K}_{{\text{S}}}}{{K}_{{{\text{S2}}}}}{{{[{\text{Q}}]}}^{2}}} \right),$Таким образом, казалось бы, гипотеза о возможности образования двух комплексов позволяет интерпретировать полученные экспериментальные данные, однако при этом возникает другая проблема. Дело в том, что независимо от механизма статического тушения люминесценции, экспериментальные данные по сокращению времени жизни возбужденного состояния от концентрации тушителя должны спрямляться в штерн-фольмеровских координатах, но как будет показано далее, это не наблюдается. Поэтому мы обратились к более детальному исследованию кинетики затухания люминесценции в зависимости от концентрации толуола и длины волны наблюдения.
В результате проведенных исследований было обнаружено, что для пара‑изомеров P1 и P2 кинетика затухания люминесценции имеет моноэкспоненциальных характер и с уменьшением квантового выхода люминесценции при переходе от н‑гексана к ароматическому растворителю наблюдается закономерное сокращение времени жизни люминесценции (см. табл. 1), в то время как для мета‑изомеров M1 и M2 наблюдается сложная биэкспоненциальная кинетика затухания люминесценции. Рассмотрим полученные данные для этих соединений более подробно.
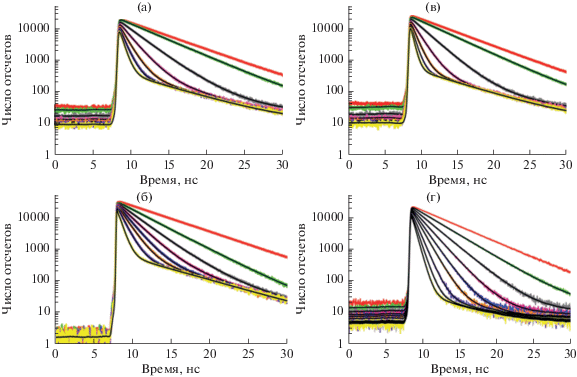
На рис. 4 показано влияние добавок бензола, толуола и о‑ксилола на вид кривых кинетики затухания люминесценции соединений М1 и М2 в н‑гексане. Видно, что в чистом н‑гексане кривые для обоих соединений являются почти моноэкспоненциальными, в то время как добавки толуола значительно усложняют их вид. Такой вид кривых, полученных в присутствии толуола, может быть связан с тем, что каждое из исследуемых соединений М1 и М2 представляет из себя смесь двух люминофоров, а ароматические молекулы (бензол, толуол и о-ксилол) проявляет свойства тушителя только по отношению к одному из них. В случае с M1 и M2 проявление подобного рода “двухкомпонентности” может быть связано, например, с тем, что оба соединения присутствуют в растворе в виде по крайней мере двух различных конформеров.
Рис. 4.
Экспериментальные данные кинетики затухания люминесценции для соединения М1 в зависимости от добавки бензола (a), толуола (б), о-ксилола (в) и для M2 в зависимости от добавки толуола (г). Верхняя (красная) кривая на каждом рисунке относится к кинетике затухания люминесценции в чистом н-гексане. Длина волны возбуждения 470 нм, кинетики регистрировались в максимуме люминесценции на длине волны 632 нм. Сплошные черные кривые – обработка экспериментальных данных в соответствии с формулой (4).
Другую альтернативу, связанную с возможным наличием неконтролируемой примеси люминесцирующих молекул близкого строения, мы рассмотрим отдельно в конце работы.
Для упрощения дальнейшей обработки экспериментальных данных было принято допущение, что каждое из соединений M1 и M2 присутствует только в двух конформеров, одна из которых взаимодействует с тушителем, а другая – нет. В этом случае для обработки кинетических кривых спада люминесценции можно использовать следующее уравнение:
(4)
${\text{PL}}(t) = {{A}_{1}}{{e}^{{\left( { - {t \mathord{\left/ {\vphantom {t {{{\tau }_{{_{1}}}}}}} \right. \kern-0em} {{{\tau }_{{_{1}}}}}}} \right)}}} + {{A}_{2}}{{e}^{{\left( { - {t \mathord{\left/ {\vphantom {t {{{\tau }_{{_{2}}}}}}} \right. \kern-0em} {{{\tau }_{{_{2}}}}}}} \right)}}},$Численные значения времен жизни и предэкспоненциальных множителей, полученные путем обработки кинетических кривых затухания люминесценции по уравнению (4), представлены в табл. 2. Видно, что концентрационные зависимости для соединений M1 и M2 хорошо описываются уравнением (4). Однако, для соединения M1 с одной аминогруппой мы получаем с хорошей достоверностью два времени жизни τ1 и τ2 уже при первой же добавкe толуола, в то время как для соединения M2 это наблюдается начиная с концентрации толуола 0.45 М. Это связано по-видимому с тем, что в соединении M2 второй компоненты, которая не тушится толуолом, гораздо меньше по отношению к первой компоненте (которая тушится), чем это имеет место для соединения М1.
Таблица 2.
Численные значения времен жизни и предэкспоненциальных множителей, полученные в результате обработки кинетик затухания люминесценции соединений М1 и М2 по уравнению (4)
| М1 | Конц. бензола | τ1, нс | τ2, нс | A1 | A2 | A1τ1/(A1τ1 + A2τ2) | A2τ2/(A1τ1 + A2τ2) |
| 0 | 5.14 ± 0.1 | – | 20 126 | – | 1 | – | |
| 0.04 | 4.3 ± 0.8 | – | 19 905 | – | 1 | – | |
| 0.22 | 2.4 ± 0.03 | 6.1 ± 1.5 | 17 274 | 446 | 0.97 | 0.03 | |
| 0.45 | 1.5 ± 0.04 | 5.5 ± 0.7 | 15 074 | 559 | 0.96 | 0.04 | |
| 0.67 | 1.0 ± 0.01 | 5.8 ± 0.5 | 13 257 | 462 | 0.97 | 0.03 | |
| 0.90 | 0.7 ± 0.02 | 6.0 ± 0.3 | 11 911 | 426 | 0.97 | 0.03 | |
| 1.12 | 0.5 ± 0.01 | 5.9 ± 0.2 | 10 975 | 426 | 0.96 | 0.04 | |
| М1 | Конц. толуола | τ1, нс | τ2, нс | A1 | A2 | A1τ1/(A1τ1 + A2τ2) | A2τ2/(A1τ1 + A2τ2) |
| 0 | 5.30 ± 0.2 | – | 35 647 | 0 | 1 | 0 | |
| 0.09 | 3.4 ± 0.2 | 7.0 ± 5.1 | 33 292 | 460 | 0.99 | 0.01 | |
| 0.18 | 2.4 ± 0.1 | 6.8 ± 2.4 | 30 901 | 613 | 0.98 | 0.02 | |
| 0.27 | 1.8 ± 0.1 | 6.3 ± 1.4 | 28 821 | 750 | 0.97 | 0.03 | |
| 0.36 | 1.4 ± 0.04 | 6.3 ± 1.1 | 26 955 | 747 | 0.97 | 0.03 | |
| 0.45 | 1.2 ± 0.1 | 6.2 ± 1.1 | 25 173 | 754 | 0.97 | 0.03 | |
| 0.59 | 0.9 ± 0.04 | 6.2 ± 0.8 | 23 164 | 732 | 0.97 | 0.03 | |
| 0.86 | 0.6 ± 0.1 | 6.2 ± 0.9 | 18 812 | 729 | 0.96 | 0.04 | |
| М1 | Конц. о-ксилола | τ1, нс | τ2, нс | A1 | A2 | A1τ1/(A1τ1 + A2τ2) | A2τ2/(A1τ1 + A2τ2) |
| 0 | 5.05 ± 0.2 | – | 27 726 | – | 1 | – | |
| 0.03 | 4.1 ± 0.1 | – | 25 570 | – | 1 | – | |
| 0.17 | 2.1 ± 0.03 | 6.6 ± 1.4 | 23 160 | 415 | 0.98 | 0.02 | |
| 0.33 | 1.6 ± 0.01 | 6.0 ± 0.4 | 20 839 | 541 | 0.97 | 0.03 | |
| 0.50 | 0.7 ± 0.01 | 6.1 ± 0.2 | 19 060 | 565 | 0.97 | 0.03 | |
| 0.66 | 0.5 ± 0.01 | 6.0 ± 0.3 | 17 442 | 552 | 0.97 | 0.03 | |
| 0.83 | 0.4 ± 0.01 | 6.0 ± 0.2 | 16 236 | 562 | 0.97 | 0.03 | |
| М2 | Конц. толуола | τ1, нс | τ2, нс | A1 | A2 | A1τ1/(A1τ1 + A2τ2) | A2τ2/(A1τ1 + A2τ2) |
| 0 | 4.58 ± 0.2 | – | 14 486 | 0 | 1 | 0 | |
| 0.09 | 3.3 ± 1.6 | 3.3 ± 3.7 | 14 031 | 335 | 0.98 | 0.02 | |
| 0.18 | 2.4 ± 1.3 | 3.4 ± 7.5 | 13 052 | 112 | 0.99 | 0.008 | |
| 0.27 | 1.8 ± 0.04 | 3.9 ± 4.6 | 12 565 | 213 | 0.98 | 0.02 | |
| 0.36 | 1.3 ± 0.03 | 2.9 ± 7.7 | 11 944 | 52 | 0.996 | 0.004 | |
| 0.45 | 1.1 ± 0.02 | 4.4 ± 5.9 | 11 535 | 40 | 0.996 | 0.003 | |
| 0.59 | 0.8 ± 0.01 | 5.2 ± 3.1 | 10 691 | 37 | 0.996 | 0.003 | |
| 0.86 | 0.5 ± 0.02 | 5.2 ± 2.5 | 9438 | 22 | 0.998 | 0.002 |
Для того чтобы получить дополнительную информации об этих формах люминофора, были проведены измерения время‑разрешенных спектров люминесценции (TRES – time‑resolved emission spectroscopy) при концентрации толуола в н‑гексане равной 0.86 М.
Данные TRES получают путем измерения набора кинетик спада люминесценции, снятых на разных длинах волн наблюдения, которые представляют собой функцию двух переменных PL(λ, t), где λ – длина волны наблюдения люминесценции. В случае смеси двух различных люминофоров функция PL(λ, t) представляется в виде:
(5)
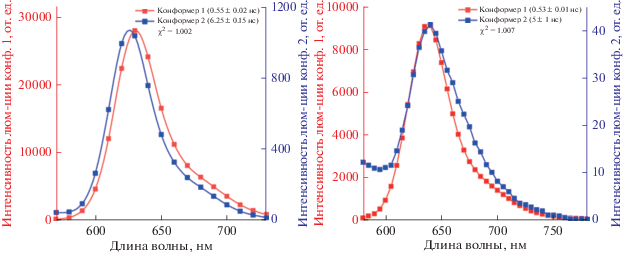
${\text{PL}}(\lambda ,t){\text{ }} = {{\xi }_{1}}{{g}_{1}}(\lambda ){{e}^{{({{ - t} \mathord{\left/ {\vphantom {{ - t} {{{\tau }_{1}}}}} \right. \kern-0em} {{{\tau }_{1}}}}){\text{ }}}}} + {{\xi }_{1}}{{g}_{2}}(\lambda ){{e}^{{({{ - t} \mathord{\left/ {\vphantom {{ - t} {{{\tau }_{2}}}}} \right. \kern-0em} {{{\tau }_{2}}}})}}},$На рис. 5 представлены результаты разложения экспериментальных данных по формуле (5). Видно, что для соединения M1 и M2 при концентрации толуола в н‑гексане равной 0.85 М имеются две люминесцирующие формы люминофора, спектры которых незначительно отличаются, однако время жизни возбужденных состояний этих форм отличается на порядок.
Рис. 5.
Спектры люминесценции двух форм для соединений M1 (слева) и M2 (справа), полученные из разложения матрицы TRES на две компоненты по формуле (5).
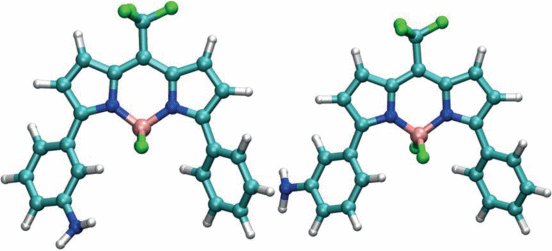
Можно предположить, что две формы люминофора – это два конформера молекулы люминофора, показанные на рис. 5. Конформеры отличаются друг от друга взаимным расположением аминогруппы и BF2‑группы ядра BODIPY. Переход одного конформера в другой происходит в результате вращения аминофенильной группы вокруг одинарной связи, по которой она присоединена к ядру BODIPY в положении 3 или 5. Из рис. 6 видно, что комплекс молекулы BODIPY с ароматической молекулой может образовываться только для мета‑изомеров А‑конформера.
Рис. 6.
Два конформера молекулы М1: А-конформер (слева) и В-конформер (справа), отличающиеся разной ориентацией аминогруппы.
Если предположить, что В‑конформер не тушится ароматической молекулой (то есть не образует соответствующий комплекс), а А‑конформер тушится и статически и динамически, то зависимость квантового выхода люминесценции от концентрации тушителя будет определяться следующим уравнением:
(6)
$\frac{{{{\varphi }_{0}}}}{\varphi } = 1 + \frac{{\left( {{{K}_{{\text{D}}}} + {{K}_{{\text{S}}}}} \right)[{\text{Q}}] + {{K}_{{\text{D}}}}{{K}_{{\text{S}}}}{{{[{\text{Q}}]}}^{2}}}}{{1 + {{K}_{{\text{B}}}}\left( {1 + {{K}_{{\text{D}}}}[{\text{Q}}]} \right)}},$При добавлении второго конформера в модель для соединения М2 и выполнении аналогичной процедуры по аппроксимации кинетических кривых (см. рис. 4 и табл. 2) получаем значение константы KB ≈ 0.003. В этом случае введение в модель второго конформера также не приводит к изменению констант тушения KD, KS, KS2.
Были проведены квантово‑химические расчеты электронной структуры исследованных соединений и компьютерное моделирование молекул BODIPY и их комплексов с бензолом.
Чтобы понять, какие вообще могут быть комплексы, сначала использовали метод молекулярной динамики, в котором молекула красителя была окружена ≈ 103 молекулами бензола. В течение 1000 нс наблюдали за системой и отмечали наиболее устойчивые мотивы взаимодействия красителя с бензолом. Далее для найденных претендентов в комплексы проводили оптимизацию геометрии в квантово‑химической программе. В результате для комплексов М1 с бензолом получены несколько стабильных структур, характеризующиеся расположением бензола – в своеобразной полости, образованной группой BF2, с аминофенильным и фенильным кольцами. Взаимодействие происходит с одним или двумя атомами водорода фенильных колец с атомами фторами, а также всех шести атомов углерода с атомом водорода NH2‑группы. Наибольший выигрыш энергии составляет 5 ккал/моль для структуры, показанной на рис. 6. Аналогичное строение имеют и комплексы с одной или двумя молекулами бензола для красителя М2. При этом наибольший выигрыш энергии составляет около 11 ккал/моль, что вполне согласуется с невысокими численными значениями KS, KS2.
Для иных расположений бензола вблизи молекул М1 и М2, а также для молекул Р1 и Р2 образования устойчивых структур “BODIPY-бензол” обнаружить не удалось. Поэтому, если комплексы М1 и М2 с бензолом и образуются, то их строение должно описываться структурами, показанными на рис. 6, и обусловлено специфическим взаимодействием тушителя с красителем.
Из табл. 3 видно, что в результате специфического взаимодействия молекулы бензола с мета‑изомерами уменьшается энергия вертикальных переходов S0 → S1. При этом молекула бензола стабилизирует состояние с большим дипольным моментом. Ниже представлены диаграммы энергетических уровней для индивидуальных красителей и их комплексов с бензолом для М1 (рис. 7) и М2 (рис. 8).
Таблица 3.
Характеристики комплексов с бензолом молекул M1 и M2 в основном и возбужденном (*) состояниях
| M1 + 1бензол | M2 + 1бензол | М2 + 2бензола | ||||
|---|---|---|---|---|---|---|
| $\angle $Ph-ядро | 33 | 46 | 33 | 42 | 35 | 45 |
| $\angle $Ph-ядро* | 71 | 42 | 69 | 37 | 68 | 40 |
| $\angle $NH2 | 22 | 23 | 25 | 24 | 22 | |
| $\angle {\text{NH}}_{2}^{*}$ | 6 | 7 | 30 | 6 | 29 | |
| µопт → µ*, D | 1.9 → 26.1 | 3.0 → 27.2 | 2.7 → 26.5 | |||
| $\mu _{{{\text{опт}}}}^{*}$ → µ, D | 26.3 → 3.3 | 26.9 → 3.5 | 26.8 → 3.2 | |||
| S0,опт → S1, eV | 1.95 | 2.01 | 2.00 | |||
| S0 → S1,опт, eV | 0.89 | 0.95 | 0.88 | |||
| q(N)опт, e– | –0.49 | –0.49 | –0.49 | –0.49 | –0.49 | |
| q(N)*, e– | –0.17 | –0.18 | –0.49 | –0.18 | –0.49 | |
| ${\text{q(N}})_{{{\text{опт}}}}^{*},$ e– | –0.23 | –0.22 | –0.48 | –0.23 | –0.49 | |
| q(N), e– | –0.51 | –0.51 | –0.48 | –0.51 | –0.49 | |
Рис. 7.
Энергетические переходы S0 → S1 и молекулярные орбитали HOMO−LUMO в М1 (слева) и комплексе М1–бензол (справа).
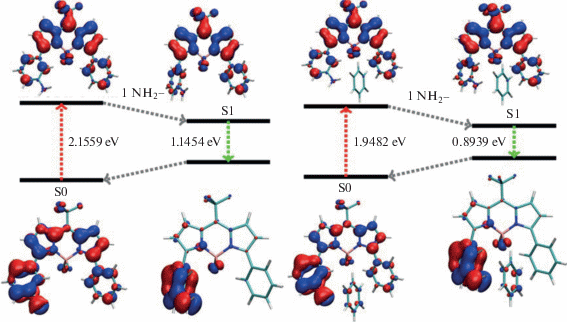
Рис. 8.
Энергетические переходы S0 → S1 и молекулярные орбитали HOMO−LUMO в М2 (слева) и комплексах М2−бензол (1:1 по центру, 1 : 2 справа).
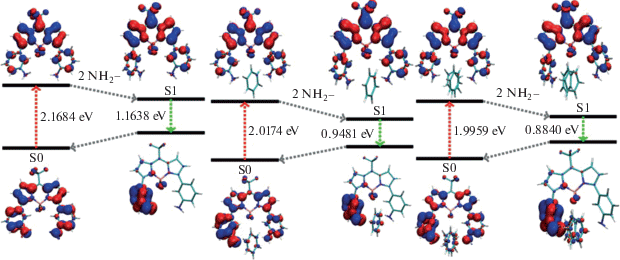
В заключение вернемся к обсуждению упомянутой выше альтернативы объяснения наблюдаемых экспериментальных данных, связанную с возможным наличием неконтролируемой примеси люминесцирующих молекул близкого строения. Действительно, в процессе синтеза соединения М1 возможно не полностью избавились от соединения 3‑(3‑ацетамидофенил)‑5‑фенил‑8‑CF3‑BODIPY, спектрально-люминесцентные свойств которого были ранее описаны в статье [1]. Однако, для соединения М2 прямых свидетельств наличия неконтролируемой люминесцирующей примеси у нас нет. Тем не менее категорически отметать такую альтернативу тоже нельзя. Поэтому мы рассматриваем предложенную трактовку наблюдаемых экспериментальных данных как возможную гипотезу, так как прямых экспериментальных доказательств наличия конформеров соединений М1 и М2 у нас тоже нет.
ВЫВОДЫ
В результате проведенных исследований показано, что люминесценция рассмотренных в работе производных красителя BODIPY с положением аминогруппы в мета‑положении чувствительна к присутствию ароматических неполярных соединений в смеси с другими неполярными соединениями. Заметные изменения спектральных свойств красителя наступают при мольной доле бензола или толуола в их смеси с н‑гексаном ниже 1%. Причиной такого резкого изменения спектральных свойств могут являться комплексы, которые формируются между мета‑изомерами и ароматической молекулой. Энергия формирования таких комплексов близка к энергии обычной водородной связи (3 ккал/моль), что делает их достаточно устойчивыми при комнатной температуре.
Список литературы
Petrushenko K.B., Petrushenko I.K., Petrova O.V., Sobenina L.N., Ushakov I.A., Trofimov B.A. // Asian J. Org. Chem. 2017. V. 6. P. 852.
Grabowski Z.R., Rotkiewicz K. // Chem. Rev. 2003. V. 103. P. 3899.
Catalan J. // J. Phys. Chem. B 2009. V. 113. P. 5951.
Magde D., Wong R., Seybold P.G. // Photochem. Photobiol. 2002. V. 75. P. 327.
Паркер С. Фотолюминесценция растворов. М.: Мир, 1972. (Parker C.A. Photoluminescence of solutions: with applications to photochemistry and analytical chemistry. Amsterdam–London–New York: Elsevier Pub. Co., 1968.)
Neese F. // Comput. Mol. Sci. 2012. V. 2. P. 73.
Neese F. // Comput. Mol. Sci. 2017. V. 8. P. e1327.
Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kale L., Schulten K. // J. Comput. Chem. 2005. V. 26. P. 1781.
Nevidimov A.V. // Rus. J. Phys. Chem. A. 2020 V. 5. P. 951.
Sobenina L.N., Vasil’tsov A.M., Petrova O.V., Petrushenko K.B., Ushakov I.A., Clavier G., Meallet-Renault R., Mikhaleva A.I., Trofimov B.A. // Org. Lett. 2011. V. 13. P. 2524.
Trofimov B.A., Mikhaleva A.I., Schmidt E.Yu., Sobenina L.N. Chemistry of Pyrroles, CRC Press Taylor&Fransis Group, Boca Raton. 2014. 398 p.
Lakowicz J.R. Principles of fluorescence spectroscopy, 3rd edn. Springer, 2006. 954 pp.
Treibs A., Kreuzer F.-H. // Justus Liebigs Ann. Chem. 1968. V. 718. P. 208.
Monsma F.J., Barton A.C., Kang H.C., Brassard D.L., Haughland R.P., Sibley D.R. // J. Neurochem. 1989. V. 52. P. 1641.
Golovkova T.A., Kozlov D.V., Neckers D.C. // J. Org. Chem. 2005. V. 70. P. 5545.
McCusker C., Carroll J.B., Rotello V.M. // Chem. Commun. 2005. V. 8. P. 996.
Hattori S., Ohkubo K., Urano Y., Sunahara H., Nagano T., Wada Y., Tkchanko N.V., Lemmetyinen H., Fukuzumi S. // J. Phys. Chem. B. 2005. V. 109. P. 15368.
Boens N., Verbelen B., Dehaen W. // Eur. J. Org. Chem. 2015. P. 6577.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий