

Химия высоких энергий, 2021, T. 55, № 5, стр. 356-360
Инверсия эффективности дезактивации возбужденных состояний при селективном фотовозбуждении комплекса иона уранила с D‑триптофаном в водных и метанольных растворах
С. С. Остахов a, *, Г. А. Масягутова a, С. Л. Хурсан a
a Уфимский Институт химии УФИЦ РАН
450054 Уфа, просп. Октября, 71, Россия
* E-mail: chemlum@anrb.ru
Поступила в редакцию 13.01.2021
После доработки 09.03.2021
Принята к публикации 12.03.2021
Аннотация
Изучено тушение флюоресценции уранил-иона ${\text{UO}}_{2}^{{2 + }}$ и триптофана Trp при их селективном внутрикомплексном фотовозбуждении в водных и метанольных растворах. Определены константы Штерна−Фольмера для всех исследованных систем. Установлен статический механизм тушения ФЛ. Найдено, что в водных растворах наблюдается только внутрикомплексный фотоиндуцированный перенос электрона (ФПЭ) от Trp к ${\text{UO}}_{2}^{{2 + }}$ независимо от типа фотовозбуждения. При возбуждении ${\text{UO}}_{2}^{{2 + }}$ в комплексе Trp⋅⋅⋅${\text{UO}}_{2}^{{2 + }}$ в среде CH3OH наряду с ФПЭ наблюдается конкурирующий с ним межмолекулярный фотоиндуцированный перенос атома водорода от метанола на электронно-возбужденный ион уранила.
Спектроскопия и фотохимия соединений уранила, обладающего яркой люминесценцией и выраженной способностью к комплексообразованию с различными органическими лигандами, изучается уже более полутора веков [1, 2]. Для фотореакций ${\text{*UO}}_{2}^{{2 + }}$ (* – наличие электронного возбуждения) доказано протекание процессов, определяющих двойственную природу электронно-возбужденного иона уранила [3]:
– внутримолекулярные реакции фотоиндуцированного переноса электрона (ФПЭ) от лиганда (L) на ${\text{UO}}_{2}^{{2 + }}$;
– межмолекулярные реакции фотоиндуцированного переноса атома водорода, которые, как правило, протекают по атомам кислорода уранил-иона и характерны, в частности, для реакций ${\text{*UO}}_{2}^{{2 + }}$ со спиртами.
Если удается селективно возбудить ${\text{UO}}_{2}^{{2 + }}$ и лиганд, которые люминесцируют в разных спектральных областях, то возможно исследование дезактивации возбужденных состояний за счет внутрикомплексного ФПЭ в сопряженных фотофизических процессах [4–8]:
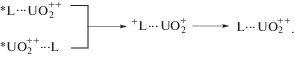 (1)
(1)
Этому условию удовлетворяют прочные комплексы ${\text{UO}}_{2}^{{2 + }}$ с аминокислотами (константы устойчивости 106–109 М–1 [9]), в том числе с триптофаном (Trp) [10]. При селективном возбуждении компонентов комплекса Trp⋅⋅⋅${\text{UO}}_{2}^{{2 + }}$ удачно сочетаются электронно-донорные свойства *Trp с высокой окислительной способностью ${\text{*UO}}_{2}^{{2 + }}$: стандартные окислительно-восстановительные потенциалы E(Trp+/Trp) = 1.13 эВ [11], E(${\text{UO}}_{2}^{{2 + }}$/${\text{UO}}_{2}^{ + }$) = 0.06 эВ [12], E(${\text{*UO}}_{2}^{{2 + }}$) = 2.6 эВ [13], E(*Trp) = 3.9 эВ [14]. Легко увидеть, что при любом способе фотовозбуждения ФПЭ термодинамически вероятен (протекает с уменьшение энергии Гиббса):
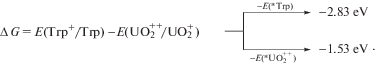
С другой стороны, для фотохимических превращений ${\text{*UO}}_{2}^{{2 + }}$ характерны реакции радикального отрыва атома водорода от органических субстратов с пониженной прочностью C–H связи. В частности, тушение возбужденного состояния ${\text{*UO}}_{2}^{{2 + }}$ алифатическими спиртами, в том числе метанолом, осуществляется в результате межмолекулярного отрыва H‑атома от молекулы спирта атомом кислорода уранил-иона [15]:
с последующим U(IV)/U(VI) диспропорционированием UO2+.
Наблюдать конкуренцию двух каналов дезактивации в фотореакциях ${\text{*UO}}_{2}^{{2 + }}$ удается не часто [3]. Условия для этого можно создать при фотолюминесценции комплекса уранил-иона с триптофаном в среде органического растворителя. Таким образом, для изучения дуализма природы электронно-возбужденного иона уранила в настоящей работе исследовано тушение флюоресценции (ФЛ) ${\text{*UO}}_{2}^{{2 + }}$ и *Trp при их селективном внутрикомплексном фотовозбуждении в кислых водных и метанольных растворах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Электронные спектры поглощения регистрировали на спектрофотометре “Specord M-40”. Скорректированные спектры ФЛ записывали на спектрофлюориметре СМ-2203. UO2(NO3)2 ⋅ 6H2O марки ч. д. а. был использован без предварительной очистки. Интенсивности (I) люминесценции *Trp и ${\text{*UO}}_{2}^{{2 + }}$ измеряли на длинах волн эмиссии λфл = 350 и 510 нм соответственно. Время жизни τ ФЛ ${\text{*UO}}_{2}^{{2 + }}$ измеряли на импульсном лазерном флюориметре LIF-200. D‑Триптофан марки ч.д.а. перекристаллизовывали из бидистиллированной воды и сушили под вакуумом. Для устранения гидролиза ${\text{UO}}_{2}^{{2 + }}$ растворы готовили в 0.01 М HClO4. Концентрированную хлорную кислоту (73.6%) перегоняли под вакуумом, чистоту проверяли спектрофотометрически (А = 1 при 220 нм, l = 5 см).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
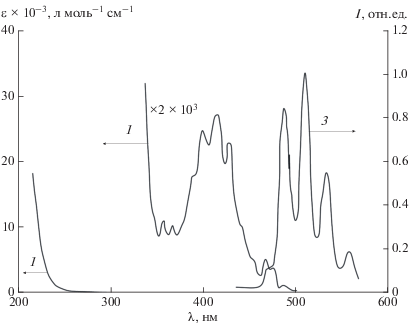
Спектры поглощения и люминесценции Trp (рис. 1) и ${\text{UO}}_{2}^{{2 + }}$ (рис. 2) не перекрываются, что позволяет провести их селективное фотовозбуждение (λвозб = 280 и 415 нм соответственно) и исследовать процессы внутрикомплексной дезактивации ${\text{*UO}}_{2}^{{2 + }}$ и *Trp (1).
Рис. 1.
Спектры поглощения (1) и ФЛ (2) триптофана ([Trp] = 10–4 моль/л, λвозб = 280 нм, 0.01 М HClO4, 298 К).
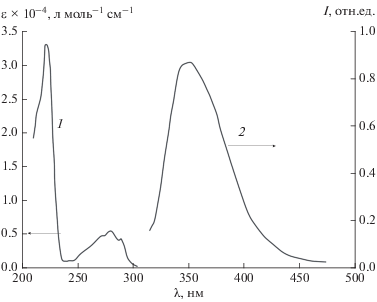
Рис. 2.
Спектры поглощения (1) и ФЛ (2) UO2(NO3)2 · 6H2O ([UO22+] = 10–2 моль/л, λвозб = 415 нм, 0.01 М НClO4, 298 K).
Уменьшение интенсивности (I0/I) в максимумах ФЛ при селективном фотовозбуждении Trp (рис. 3) или ${\text{UO}}_{2}^{{2 + }}$ (рис. 4) в водных и метанольных растворах от концентрации тушителя [Q] (${\text{UO}}_{2}^{{2 + }}$ или Trp соответственно) описывается уравнением Штерна–Фольмера: I0/I – 1 = К [Q], где К – константа тушения.
Рис. 3.
Тушение ФЛ *Trp уранилом в растворах: 1 – CH3OH, 2 – 0.01 М HClO4 (λвозб = 280 нм, [Trp] = = 10–4 моль/л, 298 К).
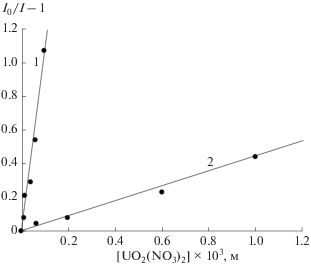
Рис. 4.
Тушение ФЛ $*{\text{UO}}_{2}^{{2 + }}$ триптофаном в растворах: 1 – CH3OH, 2 – 0.01 М HClO4 (λвозб = 415 нм, [UO2(NO3)2 ⋅ 6H2O] = 10–2 моль/л, 298 К).
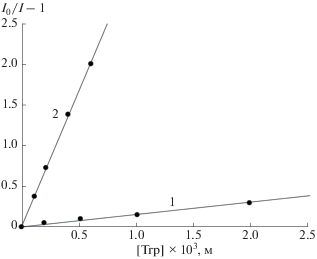
Известно, что устойчивость координационных соединений уменьшается с ростом диэлектрической проницаемости (ε) растворителей, поскольку равновесие определяется конкуренцией процессов сольватации и комплексообразования. Можно ожидать, что следствием перехода от H2O (ε = 78.3 [12]) к менее полярному метанолу (ε = = 32.63 [12]) будет увеличение констант внутрикомплексного тушения ФЛ как ${\text{*UO}}_{2}^{{2 + }}$ триптофаном, так и *Trp уранил-нитратом. Однако как можно видеть из зависимостей на рис. 3 и рис. 4, а также значений констант Штерна–Фольмера, приведенных в табл. 1, ожидаемый эффект наблюдается только для тушения ФЛ *Trp уранил-ионом. Влияние растворителя на тушение ФЛ ${\text{*UO}}_{2}^{{2 + }}$ в CH3OH (зависимость 1) и H2O (зависимость 2) противоположно наблюдаемому для *Trp.
Таблица 1.
Константы тушения ФЛ $*{\text{UO}}_{2}^{{2 + }}$ (λвозб = = 415 нм, λфл = 350 нм) и *Trp (λвозб = 280 нм, λфл = = 510 нм) в водных и метанольных растворах
| Тушение ФЛ | K, М–1 | |
|---|---|---|
| H2O | CH3OH | |
| [${\text{UO}}_{2}^{{2 + }}$···*Trp] | 430 | 1000 |
| [$*{\text{UO}}_{2}^{{2 + }}$···Trp] | 3400 | 150 |
Константа тушения флюоресценции *Trp уранил-ионом в 2.3 раза выше в CH3OH, чем в H2O, что естественно объяснить влиянием полярности растворителя на устойчивость комплексов [${\text{UO}}_{2}^{{2 + }}$···Trp]. Следует отметить, что спектры ФЛ *Trp (рис. 1) и оптического поглощения UO22+ (рис. 2) перекрываются, однако сенсибилизированное свечение последнего при тушении ФЛ *Trp уранил-ионом не наблюдается. Этот факт указывает на то, что дезактивация возбужденного состояния триптофана осуществляется не по механизму безызлучательного переноса энергии, а в результате внутрикомплексного переноса электрона (реакция (1)).
Тушение ФЛ ${\text{*UO}}_{2}^{{2 + }}$ триптофаном может протекать, как по динамическому межмолекулярному, так и по статическому внутрикомплексному механизму переноса электрона. На статический характер тушения ФЛ ${\text{*UO}}_{2}^{2}$ указывает неизменность времени жизни ${\text{*UO}}_{2}^{{2 + }}$ (τ = 21 мкс, 0.01 М HClO4, 298 К) в широком интервале концентраций аминокислоты (10–4–10–3 М). Таким образом, константа тушения Штерна–Фольмера представляет собой эффективную величину, которая включает в себя константу устойчивости Kуст комплекса [${\text{UO}}_{2}^{{2 + }}$⋅⋅⋅Trp] и долю тушения по механизму ФПЭ (α) в общей совокупности процессов дезактивации возбужденных состояний уранила K ∼ αKуст [16, 17].
Как следует из результатов эксперимента, тушение ФЛ ${\text{*UO}}_{2}^{{2 + }}$ триптофаном протекает с существенно большей эффективностью в воде, нежели в метаноле (K(H2O)/K(CH3OH) = 23). В свете вышеизложенного, трудно представить, что этот эффект обусловлен значительным снижением константы устойчивости комплекса. По всей видимости, в метанольных растворах происходит значительное уменьшение величины α. Таким образом, наблюдаемая инверсия эффективности дезактивации возбужденного состояния комплекса [${\text{*UO}}_{2}^{{2 + }}$⋅⋅⋅Trp] по сравнению с [${\text{UO}}_{2}^{{2 + }}$⋅⋅⋅*Trp] в водных и метанольных растворах может быть непротиворечиво интерпретирована с точки зрения двойственной природы электронно-возбужденного иона уранила. В водных растворах в обоих случаях доминирует внутрикомплексный ФПЭ от Trp к ${\text{UO}}_{2}^{{2 + }}$ (реакция (1)). Этот же канал дезактивации сохраняется для [${\text{UO}}_{2}^{{2 + }}$⋅⋅⋅*Trp] и в среде метанола, в то время как для возбужденного состояния ${\text{*UO}}_{2}^{{2 + }}$ в CH3OH имеет место конкуренция внутрикомплексный ФПЭ от Trp к ${\text{UO}}_{2}^{{2 + }}$ (реакция (1)). Этот же канал дезактивации сохраняется для [${\text{UO}}_{2}^{{2 + }}$⋅⋅⋅*Trp] и в среде метанола, в то время как для возбужденного состояния ${\text{*UO}}_{2}^{{2 + }}$ в CH3OH имеет место конкуренция внутрикомплексного ФПЭ и межмолекулярного отрыва атома водорода от растворителя атомом кислорода уранил-иона (реакция (2)). Эффективность последнего процесса, по-видимому, выше, что обусловливает существенное снижение константы Штерна–Фольмера в CH3OH по сравнению с водным раствором.
Несмотря на то, что константа тушения Штерна–Фольмера ФЛ водных растворов ${\text{*UO}}_{2}^{{2 + }}$ метанолом невысока, K = 12 М–1 [15], фотовосстановление ${\text{*UO}}_{2}^{{2 + }}$ в CH3OH (реакция 2) протекает с высоким квантовым выходом (0.15 [15]). Сочетание этих факторов позволяет зарегистрировать конкуренцию ФПЭ и фотоиндуцированного отрыва H‑атома в метанольных растворах ${\text{UO}}_{2}^{{2 + }}$ и Trp, иллюстрирующую дуализм в реакционной способности электронно-возбужденного ${\text{*UO}}_{2}^{{2 + }}$.
Список литературы
Рабинович Е., Белфорд Р. // Спектроскопия и фотохимия соединений уранила. М.: Атомиздат. 1968. С. 342.
Burrows HD., Kemp T.J. // Chemical Society Reviews. 1974. V. 3. № 2. P. 139.
Бучаченко А.Л., Худяков И. В. // Успехи химии. 1991. Т. 60. № 6. С. 1105.
Казаков В.П., Остахов С.С., Осина И.О., Алябьев А.С., Гайнетдинова С.Г., Ахмадеева Г.Х. // Радиохимия. 2006. Т. 48. № 5. С. 399.
Казаков В.П., Остахов С.С., Осина И.О., Алябьев А.С., Гайнетдинова С.Г., Ахмадеева Г.Х. // Радиохимия. 2006. Т. 48. № 5. С. 403.
Казаков В.П., Остахов С.С., Осина И.О., Алябьев А.С., Гайнетдинова С.Г., Ахмадеева Г.Х. // Радиохимия. 2006. Т. 48. № 5. С. 406.
Ostakhov S.S., Kazakov V.P., Osina I.O. // Mendeleev Communication. 2009. V. 19. № 2. P. 113.
Казаков В.П., Остахов С.С., Осина, И.О., Султанбаев М.В. // Химия высоких энергий. 2010. Т. 44. № 4. С. 326.
Сергеев Г.М., Коршунов И.А. // Радиохимия. 1973. Т. 15. № 4. С. 621.
Patil S.S., Thakur G.A., Shaikh M.M. // International Scholarly Research Notices. 2011. V. 2011(Article ID 168539).
Meers P. // Biochemistry. 1990. V. 29. № 13. P. 3325.
CRC Handbook of Chemistry and Physics, 89th edition / Edited by Lide D R. Boka Raton, Florida: CRC Press, 2000.
Balzani V., Bolletta F., Gandolfi M.T., Maestri M. // Bimolecular electron transfer reactions of the excited states of transition metal complexes. In: Organic Chemistry and Theory. Topics in Current Chemistry / Berlin, Heidelberg: Springer, 1978. V. 75.
Horrocks W.D., Bolender J.P., Smith W.D., Supkowski R.M. // J. Am. Chem. Soc. 1997. V. 119. № 25. P. 5972.
Matsushima R., Sakuraba S. // J. Am. Chem. Soc. 1971. V. 93. № 21. P. 5421.
Барлтроп Дж., Койл Дж. // Возбужденные состояния в органической химии. М.: Мир. 1978. С. 446
Паркер С. // Фотолюминесценция растворов / Под ред. Васильев Р.Ф.: М.: Мир, 1972. С. 510.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий