

Химия высоких энергий, 2021, T. 55, № 6, стр. 446-454
Спектральнo-люминесцентные и фотохимические свойства производных гидрированных фуро- и тиенохинолинов
В. В. Шахматов a, Е. Н. Ходот b, Т. Д. Некипелова a, *, В. А. Кузьмин a
a Институт биохимической физики им. Н.М. Эмануэля РАН
119334 Москва, ул. Косыгина, 4, Россия
b Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Ленинский просп., 47, Россия
* E-mail: nekip@sky.chph.ras.ru
Поступила в редакцию 10.06.2021
После доработки 01.07.2021
Принята к публикации 06.07.2021
Аннотация
Синтезированы новые производные гидрированных фуро- и тиенохинолинов и исследованы их спектрально-люминесцентные свойства и фотохимические превращения. С использованием метода импульсного фотолиза доказана триплетная природа короткоживущих состояний при фотовозбуждении и получены спектрально-кинетические характеристики триплетных состояний синтезированных фурогидрохинолинов и образующихся аминильных радикалов. Процессы с участием триплетного состояния исследованных соединений являются важными для использования исследуемых соединений в фотодерматологии.
В последнее время гетероциклические соединения с аннелированной фурогруппой привлекают пристальное внимание в связи с потенциальной возможностью их использования в качестве биологически активных веществ и лекарственных препаратов [1, 2]. Фурокумарины или псоралены широко используются в фотодерматологии при лечении кожных заболеваний [3]. Лечебный эффект фурокумаринов обусловлен образованием триплетного состояния при поглощении света УФА диапазона с последующим взаимодействием их триплетного состояния с тиминовым основанием ДНК с образованием циклобутанового цикла между двойной связью фуранового кольца и связью С(5)=С(6) тимина. Псоралены используются в фотодерматологии более 60 лет, и за это время проявились побочные отрицательные последствия терапевтического применения этих соединений, а именно: образование кросс-сшивок в молекуле ДНК за счет возможности протекания реакции не только по двойной связи фуранового кольца, но и по двойной связи кумарина, что может приводить к развитию локальных злокачественных образований. В этой связи, поиск новых соединений, способных образовывать моноаддукты с тиминовым основанием ДНК и не способных образовывать диаддукты, представляется актуальной задачей. Для замены фурокумаринов в фотодерматологии было предложено использовать фурохинолоны [4, 5] в качестве потенциальной замены псораленам при аналогичном механизме терапевтического действия.
В современной фармакологической и агрохимической индустрии широко и интенсивно используются производные хинолинов [6]. Хинолины представляют интерес в качестве исходных структур для синтеза на их основе новых соединений путем введения в их структуру фуранового цикла, что приводит к увеличению электронного сопряжения в молекуле и способствует получению соединений удовлетворяющим требованиям PUVA-терапии.
Соединения, которые активны в фотодерматологии, должны удовлетворять следующим требованиям: быть нетоксичными без облучения, образовывать при фотовозбуждении с высоким квантовым выходом триплетные состояния, вступать в реакции с тиминовым основанием ДНК, образуя при этом моноаддукты, и не образовывать диаддукты. 1,2-Дигидрохинолины (ДГХ) хорошо известны как эффективные нетоксичные антиоксиданты [7], их фотохимические и фотофизические свойства в зависимости от заместителей в гетероцикле и ароматическом кольце, а также от природы растворителя детально исследованы [8–13]. Известно, что выход триплетного состояния у этих соединений невелик [8]. Ранее на примере 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолина было показано, что введение фуранового кольца в структуру дигидрохинолина увеличивает выход триплетного состояния с сохранением антиоксидантных свойств этих соединений и их низкой токсичности [14–18].
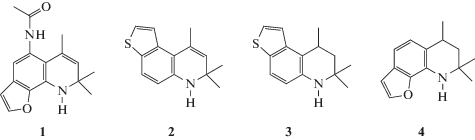
В данной работе продолжено исследование спектрально-люминесцентных и фотохимических свойств новых производных гидрированных хинолинов с аннелированой фуро- или тиено-группой: ацетамидного производного фуродигидрохинолина (1), тиенодигидрохинолина (2) и фуротетрагидрохинолинов (3) и (4).
МЕТОДИКА ЭКСПЕРИМЕНТА
Синтез N-(6,8,8-триметил-8,9-дигидрофуро[3,2-h]хинолин-5-ил)ацетамида (1) описан в [19]. 1H ЯМР (DMSO-d6) δ, м.д.: 9.40 (1H), 7.90 (d, J 8 Hz, 1H), 6.78 (d, J 8 Hz, 1H), 6.60 (s, 1H), 6.04 (s, 1H), 5.23 (s, 1H), 2.03 (s, 3H), 1.97 (s, 3H), 1.24 (s, 6H).
Принципиальнаясхемасинтеза 7,7,9-триметил-6,7-дигидротиено[3,2-f]хинолина (2) включает 4 стадии (схема 1 ). Исходный 1-бензотиофен-5-амин (7) для синтеза (2) по реакции Скраупа был получен по описанной методике [20].
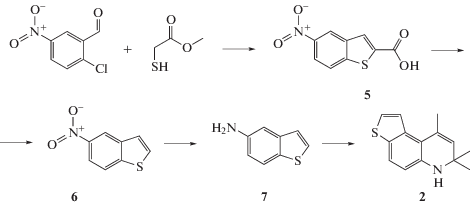
Схема 1 .
Соединение 2 было выделено в виде стеклообразной массы, выход 30%; 1H ЯМР (CDСl3) δ, м.д: 7.65 (уш.с., 1H), 7.53 (д, J 8 Hz, 1H), 7.40 (д, J 8, Hz, 1H), 6.69 (уш.с., 1H), 5.42 (уш.с., 1H), 4.32 (уш.с., 1Н), 2.37 (с, 3Н), 1.31 (с, 6Н); МСВР: (М + Н+)/z 230.0996.
7,7,9-триметил-6,7,8,9-тетрагидрофуро[3,2-f]хинолин (3) получали восстановлением 7,7,9-триметил-6,7-дигидрофуро[3,2-f]хинолина (8), синтез которого был описан ранее [14], с использованием Pd/C катализатора (схема 2 ).
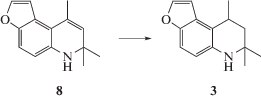
Схема 2 .
Целевой продукт (3) получали в виде зеленоватых кристаллов, Выход 25%. 1H ЯМР (CDСl3) δ, м.д: 7.55 (д, J 2.1 Hz, 1H), 7.17 (дд, J 8.7, 1.0 Hz, 1H), 6.79 (дд, J 2.1, 1.0 Hz, 1H), 6.51 (д, J 8.7 Hz, 1H), 3.27 уш.с., 1H), 3.22 (м, 1H), 1.94 (м, 1H), 1.51 (м, 1H), 1.47 (д, J 8 Hz, 3H), 1.29 (с, 3Н), 1.17 (с, 3Н).
Синтез 6,8,8-триметил-6,7,8,9-тетрагидрофуро[3,2-h]хинолина (4) проводили аналогично восстановлением соответствующего фуродигидрохинолина. Однако этот синтез осложняется тем, что соответствующий фуродигидрохинолин подвергается самопроизвольной олигомеризации. Это приводит к тому, что выход целевого продукта не превышает 16%.
В качестве растворителей использовали метанол (Merck, for spectrophotometry), этанол, гексан, ацетонитрил (Компонент Реактив, для ВЭЖХ).
Спектры поглощения и флуоресценции регистрировали на спектрофотометре “Shimadzu UV‑3101 PC” в области 200-500 нм и спектрофлуориметре “Shimadzu RF-5301 PC” в кварцевых кюветах с длиной оптического пути 1 см. Квантовые выходы флуоресценции (φfl) измеряли методом сравнения со стандартом [20], в качестве которого использовали 1,4-ди-[2-(5-фенилоксазолил)]-бензол (POPOP) с квантовым выходом флуоресценции в гексане 0.97, максимумы поглощения и испускания которого близки к соответствующим максимумам анализируемых веществ. Квантовые выходы флуоресценции (φfl) вычисляли по уравнению (1), которое учитывает изменение свойств растворителя через соотношение их показателей преломления [21].
(1)
$\varphi _{1}^{{{\text{fl}}}} = {{\varphi _{2}^{{{\text{fl}}}}n_{1}^{2}{{I}_{1}}{{A}_{2}}} \mathord{\left/ {\vphantom {{\varphi _{2}^{{{\text{fl}}}}n_{1}^{2}{{I}_{1}}{{A}_{2}}} {n_{2}^{2}}}} \right. \kern-0em} {n_{2}^{2}}}{{I}_{2}}{{A}_{1}},$Стационарный фотолиз соединений 1 и 2 проводили в растворе спирта и ацетонитрила при облучении светодиодным источником (λex = (365 ± ± 10) нм), а соединений 3 и 4 при облучении ртутной лампой ДРШ-1000. Для исследования методом стационарного фотолиза использовали кварцевые кюветы с длиной оптического пути 1 см, эксперименты проводили на воздухе или после продувки аргоном в течение 15 мин, для удаления следов кислорода.
Импульсный фотолиз соединений 1 и 3 осуществляли в кварцевой кювете с длиной оптического пути 20 см с использованием Xe лампы (75 Дж, 20 мкс) на установке импульсного фотолиза, описанной ранее [16]. Изменения в поглощении после импульса света измеряли в диапазоне 400–720 нм системой, состоящей из лампы накаливания (75 вт), монохроматора ЗМР-3, фотоумножителя ФЭУ-38 и осциллографа на базе PCI Bordo 211. Удаление кислорода в экспериментах по импульсному фотолизу проводили путем вакуумирования. Концентрация фурогидрохинолинов 1 и 3 в ацетонитриле составляла 4 × 10–4 мол/л. Обработку экспериментальных данных импульсного фотолиза проводили методом глобального анализа путем аппроксимации экспериментальных кривых гибели промежуточного поглощения мультиэкспоненциальным уравнением (2).
(2)
$\Delta {{A}^{\lambda }} = \Sigma \Delta A_{i}^{\lambda }\exp ( - {{k}_{i}}t) + \Delta A_{{{\text{res}}}}^{\lambda },$РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Спектрально-люминесцентные свойства соединений 1–4
Спектры поглощения соединений 1 и 2 на основе дигидрохинолинов имеют три полосы поглощения в УФ-области спектра с максимумом длинноволновой полосы в области 340–370 нм (рис. 1 спектры 1 и 2, табл. 1). Спектры поглощения соединений 3 и 4 на основе тетрагидрохинолина сдвинуты в коротковолновую область и имеют длинноволновый максимум поглощения около 300 нм (рис. 1 спектры 3 и 4, табл. 1). Длинноволновые и коротковолновые максимумы поглощения этих соединений близки к соответствующим максимумам исходных гидрированных хинолинов. В случае соединения 4 длинноволновая полоса поглощения имеет вид плеча полосы поглощения в области 250–300 нм. Коротковолновый сдвиг этой полосы, по-видимому, обусловлен водородной связью между кислородом фуранового цикла и водородом группы NH. Максимумы поглощения в области 250–300 нм являются характерными для соединений с аннелированными фуро-группами [16, 18].
Рис. 1.
Нормированные по коротковолновой полосе спектры поглощения соединений: 1 – 1 (сплошная линия), 2 – 2 (пунктир), 3 – 3 (пунктир-точка), 4 – 4 (точки); растворитель: MeOH (1, 2), EtOH (3, 4).
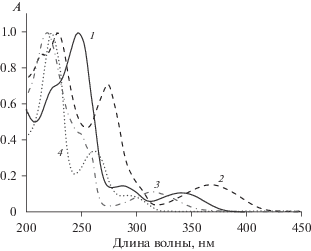
Таблица 1.
Спектрально-люминесцентные свойства соединений 1–4
| Вещество | Растворитель | λмакс, нм | ε, М–1 см–1 | λфл, нм | φfl | Стоксов сдвиг, см–1 (нм) |
|---|---|---|---|---|---|---|
| 1 | MeOH | 342 | 2440 | 417 | 0.18 | 5344 (76) |
| 288 | 3260 | |||||
| 247 | 22370 | |||||
| 341 | 3100 | |||||
| EtOH | 288 | 4250 | ||||
| 248 | 29200 | |||||
| 343 | 3550 | |||||
| MeCN | 289 | 4250 | ||||
| 249 | 27700 | |||||
| 368 | 2500 | 436 | 0.20 | 4240 (68) | ||
| 2 | МеOH | 275 | 11700 | |||
| 229 | 16500 | |||||
| 3 | EtOH | 315 | 2430 | 380 | 0.18 | 5430 (76) |
| 220 | 21760 | |||||
| 4 | 295 | 2180 | 411 | 0.12 | 9570 (116) | |
| EtOH | 262 | 8125 | ||||
| 225 | 26240 |
На рис. 2 представлены нормированные спектры поглощения и флуоресценции. Квантовые выходы флуоресценции для соединений 1–3 (табл. 1) близки к полученным ранее для других фуродигидрохинолинов [16, 18] и дигидрохинолинов [22]. Для соединения 4 квантовый выход флуоресценции несколько ниже, при этом наблюдается относительно большой Стоксов сдвиг, что, видимо, также обусловлено особенностями его строения.

Стационарный фотолиз
Фотохимические превращения соединения 1 существенным образом зависят от растворителя (рис. 3). При фотовозбуждении 1 в протонных растворителях в нерелаксированном возбужденном состоянии ${\text{S}}_{{\text{1}}}^{{{\text{FC}}}}$ происходит перенос протона через растворитель от группы NH к атому С3 с последующим образованием карбкатиона и продукта присоединения ROH (схема 3 , последовательность реакций (II)), как это происходит в случае дигидрохинолинов [11, 13]). Действительно, характер изменений спектров поглощения при стационарном фотолизе 1 в метаноле в присутствии кислорода показывает, что основным процессом является образование продукта присоединения метанола к двойной связи дигидрохинолинового фрагмента. В атмосфере аргона скорость фотолиза незначительно увеличивается (рис. 3б), что связано с процессом образования аминильных радикалов из триплетного состояния фуродигидрохинолинов (схема 3 , реакция (III)) с последующей их димеризацией. В ацетонитриле в атмосфере аргона фотолиз 1 происходит существенно медленнее, чем в метаноле (рис. 3в), и основным процессом является образование димерных продуктов из аминильных радикалов (схема 3 реакция (III)). Данные MALDI TOF МС анализа фотолизатов 1 подтверждают, что при его фотолизе в MeOH образуется продукт присоединения спирта (M/z = 302) и димерные продукты из аминильных радикалов в небольших количествах (M/z = 538).
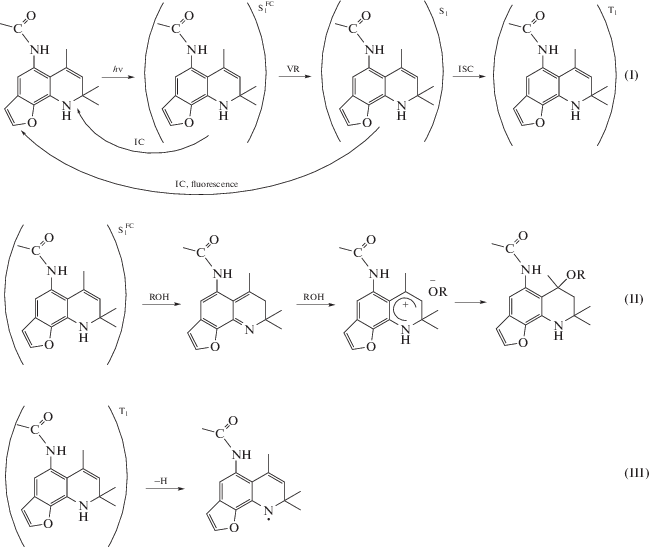
Рис. 3.
Изменения спектров поглощения при фотолизе 1 в МеОН в атмосфере воздуха (а), время (мин): 0 (1), 2 (2), 4 (3) 6 (4), 8 (5), 10 (6), и в MeCN в атмосфере аргона (б) время (мин): 0 (1), 1 (2), 4 (3), 10 (4), 20 (5), 35 (6), 55 (7), 80 (8), 110 (9).

Схема 3.
При стационарном фотолизе соединений 3 и 4 наблюдали небольшие изменения спектров поглощения (рис. 4), которые выражаются в сдвиге максимумов поглощения на 1–2 нм и небольшом падении оптической плотности, что свидетельствует об образовании димерного продукта, спектр которого не должен сильно отличаться от спектров исследуемых веществ, так как структура димера состоит из двух фрагментов исходного вещества. В случае тетрагидрохинолинов реакция присоединения протонного растворителя не имеет места, поэтому при фотовозбуждении генерируется только аминильный радикал из триплетного состояния аналогично реакциям (I) и (III) в схеме 3 для соединения 1.

Импульсный фотолиз фурогидрохинолинов 1 и 3
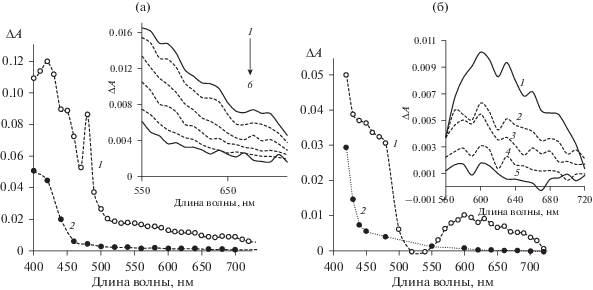
Соединения 1–4 являются потенциальными препаратами для фотодерматологии, поэтому образование триплетных состояний, способных реагировать с тиминовыми основаниями ДНК при фотовозбуждении светом в ближней УФ-области, является необходимым условием для проявления их лечебного действия. В данной работе проведено сопоставление интермедиатов, образующихся при импульсном фотолизе соединений 1 и 3 в отсутствие О2 и на воздухе в растворе в ацетонитриле, и исследованы их спектрально-кинетические характеристики. Дифференциальные спектры поглощения интермедиатов различаются в зависимости от наличия кислорода в растворе (рис. 5). Кроме появления поглощения в области 500–700 нм в отсутствие О2, которое может быть связано с образованием триплетов, в вакууме увеличивается поглощения в области около 400 нм, которое мы относим к поглощению аминильного радикала, и около 480 нм. Очевидно также различие в спектрах этих соединений в длинноволновой области в отсутствие О2: для 1 наблюдается постепенное уменьшение поглощения при λ > 500 нм (рис. 5а), вставка, спектр 1), а для 3 поглощение отсутствует в области 550 > λ > 500 нм, далее увеличивается, достигая максимума при λmax = 600 нм, после чего спадает до 0 при λ = = 720 нм (рис. 5б), вставка, спектр 1)).
Рис. 5.
Дифференциальные спектры поглощения после импульса света, (а) соединение 1 и (б) соединение 3, (1) – в отсутствие О2, (2) – на воздухе. Вставки. Изменения дифференциальных спектров поглощения во времени в диапазоне 550 < λ < 720 нм, (а) время регистрации, с: 0.00015 (1), 0.0002 (2), 0.0003 (3), 0.0005 (4), 0.001 (5), 0.002 (6). (б), с: 0.0008 (1), 0.0015 (2), 0.002 (3), 0.005 (4), 0.02 (5).
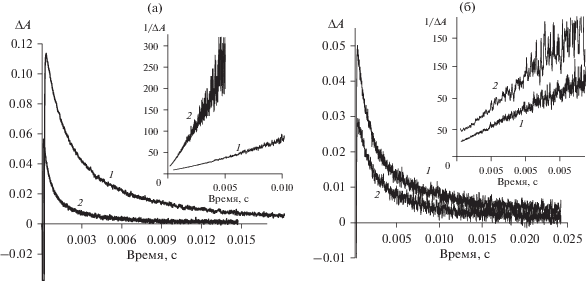
Кинетика гибели интермедиатов, поглощающих в области 400 нм, подчиняется уравнению 2‑го порядка (рис. 6), что является подтверждением их радикальной природы. Увеличение концентрации аминильных радикалов при фотолизе в отсутствие О2 может свидетельствовать об их образовании из триплетного состояния. Преимущественное образование аминильных радикалов из соответствующих триплетных состояний аминов давно обсуждается в литературе. Следует отметить, что в случае соединения 3 гибель радикальных интермедиатов происходит с близкими константами скорости как на воздухе, так и в вакуумированном растворе (рис. 6б), вставка), в то время как для соединения 1 константа скорости гибели интермедиата, образовавшегося в отсутствие О2, примерно в 6 раз ниже (рис. 6а), вставка). Это вероятно свидетельствует об изменении структуры радикала для 1, в котором ацетамидная группа может играть особую роль в реакциях триплетного состояния. Этот вопрос требует более детального исследования и выходит за рамки данной работы.
Рис. 6.
(а) Кинетика гибели дифференциального поглощения, образующегося при фотовозбуждении раствора соединения 1: (1) – в отсутствие О2, (2) – на воздухе, λрег = 400 нм; вставка: зависимость 1/ΔA от времени. (б) Кинетика гибели дифференциального поглощения, образующегося при фотовозбуждении раствора соединения 3: (1) – в отсутствие О2, (2) – на воздухе, λрег = 420 нм.
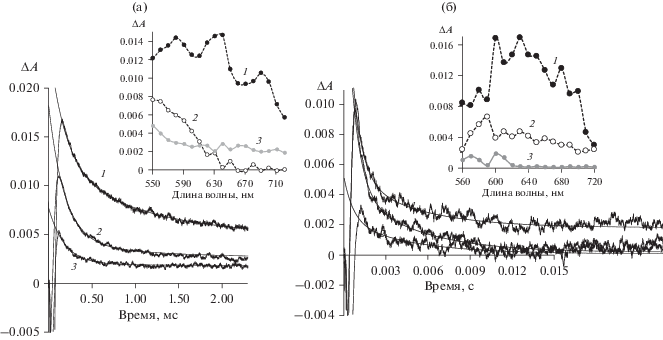
Изменения спектров дифференциального поглощения во времени в отсутствие О2 в длинноволновой области (рис. 5, вставки) показывает, что скорость этих изменений различна на разных длинах волн. Действительно, кинетические кривые гибели отчетливо демонстрируют наличие по крайней мере двух процессов, происходящих с разными константами скорости. Аппроксимация экспериментальных данных методом глобального анализа по уравнению (2) при i = 2 позволила определить константы скорости гибели и спектры поглощения для промежуточных частиц (рис. 7). Для соединения 1 k1 = (5.5 ± 0.5) × 103 с–1 (λmax = 630 нм) и k2 = (1.0 ± 0.1) × 103 с–1 (λmax ≤ 550 нм), а для соединения 3 k1 = (1.5 ± 0.2) × 103 с–1 (λmax = 630 нм) и k2 = (2.2 ± 0.2) × 102 с–1 (λmax = 590 нм). Экспериментальные результаты позволяют сделать предположение, что два короткоживущих интермедиата образуются независимо из синглетного возбужденного состояния фурогидрохинолина.
Рис. 7.
Экспериментальная кинетика и аппроксимирующие кривые гибели дифференциального поглощения в отсутствие О2, (а) соединение 1, λрег, нм: 550 (1), 620 (2) и 720 (3); (б) соединение 3, λрег, нм: 600 (1), 630 (2) и 720 (3). Вставки: рассчитанные спектры поглощения интермедиатов с константами гибели, с–1: (а) 5.5 × 103 (1) и 1.0 × 103 (2), (б) 1.5 × × 103 (1) и 2.2 × 102 (2), (3) спектр поглощения при t → ∞.
ЗАКЛЮЧЕНИЕ
Были синтезированы новые фуро- и тиенодигидрохинолины и фуротетрагидрохинолины и исследованы их спектрально-люминесцентные и спектрально-кинетические свойства. Показано, что состав продуктов стационарного и импульсного фотолиза зависит от наличия кислорода, что указывает на участие триплетного состояния исследуемых соединений в фотохимических процессах в отсутствие кислорода. Образование триплетных состояний исследуемых соединений с высоким выходом указывает на перспективность их дальнейшего исследования с целью применения в фотодерматологии.s
Список литературы
Llano J., Raber J., Eriksson L.A. // J. Photochem. Photobiol A: Chemistry. 2003. V. 154. P. 235.
Ando K., Akai Y., Kunimoto J., Yokomizo T., Nakajima H., Takeuchi T., Yamashita M., Ohta S., Ohishi T., Ohishi Y. // Org. Biomol. Chem. 2007. V. 5. P. 655.
Bethea D., Fullmer B., Syed S, Seltzer G., Tiano J., Ris-chko C., Gillespie L., Brown D., Gasparro F.P. // J. Dermatol. Sci. 1999.V. 19. P. 78.
Via L.D., Mammi S., Uriarte E., Santana L., Lampronti I., Gambari R., Gia O. // J. Med. Chem. 2006. V. 49, 4317.
Marzano C., Chilin A., Bordin F., Baccichetti F., Guiotto A. // Bioorg. Med. Chem. 2002. V. 10. P. 2835.
Muthukrishnan I., Sridharan V., Menéndez J. C. // Chem. Rev. 2019. V. 119. P. 5057–5191; https://doi.org/10.1021/acs.chemrev.8b00567
Некипелова Т.Д., Гагарина А.Б. // Нефтехимия. 1982. Т. 22. С. 278.
Некипелова Т.Д., Малкин Я.Н., Кузьмин В.А. // Изв. Акад. наук СССР, Сер. хим. 1980. № 1. С. 80.
Malkin Ya.N., Pirogov N.O., Kuzmin V.A. // J. Photochemistry. 1984. V. 26. P. 193.
Некипелова Т.Д., Кузьмин В.А., Шишков В.С. // Химия высоких энергий. 2002. Т. 36. С. 212.
Nekipelova T., Gostev F., Kuzmin V., Sarkisov O. // Photochem. Photobiol. Sci. 2006. V. 5. P. 815.
Лыго О.Н., Некипелова Т.Д., Ходот Е.Н. // Химия высоких энергий. 2010. Т. 44. С. 431.
Некипелова Т.Д., Кузьмин В.А. // Успехи химии. 2012. Т. 81. С. 983.
Кузьмин В.А., Мазалецкая Л.И., Некипелова Т.Д., Ходот Е.Н. // Изв. Акад. наук. Сер. хим. 2008. № 11. С. 2356.
Некипелова Т.Д., Лыго О.Н., Ходот Е.Н., Кузьмин В.А., Шахматов В.В., Варгин В.В., Белякова А.В., Зылькова М.В. // Химия высоких энергий. 2012. Т. 46. С. 211.
Лыго О.Н., Некипелова Т.Д., Ходот Е.Н., Кузьмин В.А., Шахматов В.В., Волнухин В.А., Варгин В.В., Шевелев А.Б., Шибаева А.В. // Химия высоких энергий. 2012. Т. 46. С. 216.
Лыго О.Н., Некипелова Т.Д., Ходот Е.Н., Шахматов В.В., Кононихин А.С., Николаев Е.Н., Кузьмин В.А. // Химия высоких энергий. 2012. Т. 46. С. 452.
Кузьмин В.А., Волнухин В.А., Егоров А.Е., Климович О.Н., Костюков А.А., Некипелова Т.Д., Ходот Е.Н., Шахматов В.В., Шевелев А.Б., Шибаева А.В., Штиль А.А. // Химическая физика. 2019. Т. 38. № 12. С. 3.
Пaтeнт (19)RU(11)2 686 692(13) C1
Zambias R.A., Hammond M.L. // Synthetic Communications. 1991. V. 21. № 7. P. 959.
Паркер С. // Фотолюминесценция растворов. М.: Мир. 1972. С. 247.
Некипелова Т.Д., Кузьмин В.А. // Химия высоких энергий. 2005. Т. 39. С. 449.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий