

Химия высоких энергий, 2022, T. 56, № 1, стр. 81-84
Фотофизические и структурные характеристики энантиомеров металлоценов дикарборанилов
Г. В. Лукова a, *, А. А. Милов b
a Федеральное государственное бюджетное учреждение науки Институт проблем химической физики Российской академии наук
142432 Московская обл., Черноголовка, проспект Академика Семенова, 1, Россия
b Южный научный центр Российской Академии наук
344006 Ростов-на-Дону, проспект Чехова, 41, Россия
* E-mail: gloukova@mail.ru
Поступила в редакцию 29.07.2021
После доработки 24.08.2021
Принята к публикации 28.08.2021
- EDN: ANCONO
- DOI: 10.31857/S0023119322010089
Металлоценовые комплексы подгруппы титана являются основными компонентами катализаторов многих органических реакций (как, например, полимеризации непредельных углеводородов [1]) и активации малых молекул, обладают самыми редкими и наименее изученными возбужденными состояниями с переносом заряда с лиганда на металл (ПЗЛМ) [2–10], в том числе уникальными фосфоресцентными 3ПЗЛМ [3–10].
В самое последнее время нами получен и систематически проанализирован [11] большой набор данных расчетов геометрии d0 металлоценового комплекса на примере титаноцена дикарборанила Ti(η5:η1-CpCMe2CB10H10C)2 (Cp – циклопентадиенил) и продемонстрировано хорошее соответствие большинства рассчитанных структурных данных с имеющимися кристаллографическими [12].
В настоящей работе впервые смоделированы фотофизические и структурные характеристики энантиомеров d0-металлоценов на примере триады структурно сложных комплексов подгруппы титана с карборанильными σ-лигандами, имеющих C(CH3)2< мостики между лигандами π- и σ‑типа: M(η5:η1-CpCMe2CB10H10C)2 (M = Ti, Zr и Hf (рис. 1а)). На примере Hf(η5:η1-CpC-Me2CB10H10C)2 впервые оценены изменения структуры и дипольный момент металлокомплекса при переходе из основного состояния в 1ПЗЛМ. Обнаружено, что одностадийная энантиомеризация между противоположными энантиомерами M(η5:η1-CpCMe2CB10H10C)2 не осуществляется в термических условиях.
Рис. 1.
Оптимизированная структура гафноцена (симметрия С1) в S0 и S1 состояниях (B3LYP/CEP-121G) (а) и спектр его электронного возбуждения ПЗЛМ в газовой фазе (TDDFT/TPSSh/SDD) (б).
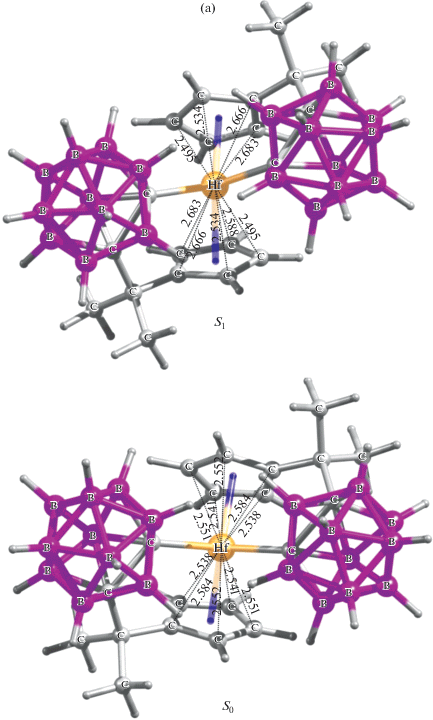
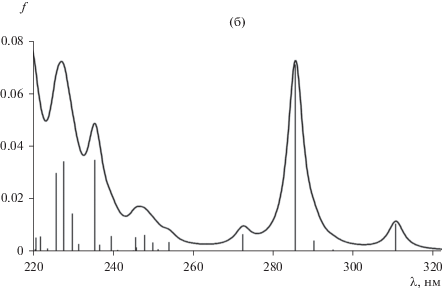
Кристаллографически описан только один из двух энантиомеров в случае каждого из комплексов M(η5:η1-CpCMe2CB10H10C)2 [12], при этом ошибочно указывалось, что их РСА характеристики соответствуют точечной группе симметрии С2; в действительности – С1. Существование зеркальных стереоизомеров для группы металлоцен дикарборанилов экспериментально не подтверждено. По данным наших DFT и HF расчетов (с использованием Gaussian 16 [13]), в случае изоструктурной триады парам энантиомеров M(η5:η1-CpCMe2CB10H10C)2 соответствуют достаточно глубокие локальные минимумы на поверхности потенциальной энергии (ППЭ). Кроме того, обнаружено, что конформационные переходы (малые структурные изменения с изменением точечной группы симметрии, например: С1 ↔ С2) в M(η5:η1-CpCMe2CB10H10C)2 протекают практически безбарьерно и приводят к значимым изменению электрического дипольного момента: Δμ = 0.1–0.2 Дебай и сдвигам полос ПЗЛМ в спектрах возбуждения (поглощения) на несколько нанометров (рассчитано набором TDDFT методов с хорошим совпадением с экспериментом, пример моделирования на рис. 1б), что, в общем случае, объясняет природу уширения полос в электронных спектрах клиносэндвичевых комплексов. О подобной чувствительности положения и формы полос электронных спектров к очень слабым конформационным изменениям металлоокомплексных хромофоров в литературе ранее не сообщалось. Отметим, что особенностью этого класса молекул являются очень малые сольватохромные сдвиги полос ПЗЛМ в спектрах [9, 10].
На примере гафноцена Hf(η5:η1-Cp-CMe2CB10H10C)2 впервые оценены (B3LYP/CEP-121G) изменения геометрии металлокомплекса при переходе из основного в 1ПЗЛМ состояние. Обнаружено, что в гафноцене в состоянии S1 по сравнению с S0 на ~7° сужаются двугранный угол Ср(центроид)–Hf–Ср´(центроид) и угол между σ-связями Hf–карборанил, сэндвичевый остов становится еще более скошенным, расстояния Hf–Ср(центроид) удлиняются на ~0.04 Å, но длины σ-связей Hf–карборанил не меняются (рис. 1). В состоянии S1 ослабляется координация устойчивых Cp-лигандов (“лигандов-зрителей” [1]), таким образом, π-связи активируются. Дипольный момент гафноцена в состоянии S1 возрастает (Δμ > > 0.5 Д), что подтверждает образование возбужденного состояния с переносом заряда.
Ранее на примере мостичного цирконоцена дихлорида [5–7] мы показали, что фотоизомеризация рац-форма <=> мезо-форма d0-металлоцена осуществляется из короткоживущего состояния 1ПЗЛМ (редкий случай установления мультиплетности фотохимически активного состояния) [6]. О реализации термически активированной энантиомеризации комплексов 4 группы, в общем случае d0-металлоценов – сообщений не было. Хотя лабильность обмена металл–лиганд часто вызывает энантиомеризацию, данные наших DFT-расчетов (B3LYP/CEP-121G, B3LYP/ LANL2DZ, M06/LANL2DZ) не подтверждают возможность протекания одностадийной энантиомеризации между противоположными энантиомерами структурно жестких комплексов M(η5:η1-CpCMe2CB10H10C)2 в термических условиях. С целью сравнения мы изучили возможность энантиомеризации родственных мостичных d0-металлоценов дихлоридов EtCp2MCl2 (M = Ti, Zr, Hf и Et = (CH2)2), близких по структуре к цирконоцену дихлориду из [5–7], (использованы функционалы: TPSSH, PBE0, B3LYP, CAM-B3LYP и M06-2X – и базисы: LANL2DZ, CEP-121G, SDD и 6-31G* (для Ti); все оптимизированные структуры соответствовали стационарным точкам на ППЭ). Обнаружено, что тепловая энантиомеризация EtCp2MCl2 протекает с низким энергетическим барьером в газовой фазе: Еа < 5 ккал/моль. Примечательно, что энантиомеризация EtCp2MCl2 осуществляется в термических условиях не за счет разрыва металлоценового остова, а благодаря скручивающему движению этиленового мостика и одновременным маятниковым движениям двух π-лигандов Cp и двух σ-лигандов Cl относительно d0-металла. Примечательно, что для перехода от точки минимума, соответствующей одному энантиомеру, к точке минимума, соответствующей другому энантиомеру, система должна преодолеть последовательно два несимметричных переходных состояния и связывающую их седловую точку второго порядка, как в [14].
Список литературы
Metallocene Complexes as Catalysts for Olefin Polymerization, Ed. Alt H.G. // Coord. Chem. Rev. 2006. V. 250. № 1–2.
Loukova G.V. // “Organometallic Compounds: Preparation, Structure and Properties”. Chapter 4 / Ed.: H.F. Chin. N.Y.: Nova Sci. Pub. 2010. P. 159.
Loukova G.V., Smirnov V.A. // Chem. Phys. Lett. 2000. V. 329. № 5–6. P. 437.
Loukova G.V. // Chem. Phys. Lett. 2002. V. 353. № 3–4. P. 244.
Лукова Г.В., Смирнов В.А., Зенчук Е.В. // Коорд. химия. 2005. С. 319.
Лукова Г.В., Смирнов В.А., Зенчук Е.В. // Доклады АН. 2006. Т. 410. С. 495.
Loukova G.V., Huhn W., Vasiliev V.P., Smirnov V.A. // J. Phys. Chem. A. 2007. V. 111. № 20. P. 4117.
Loukova G.V., Starodubova S.E., Smirnov V.A. // J. Phys. Chem. A. 2007. V. 111. № 43. P. 10928.
Loukova G.V., Vasiliev V.P., Milov A.A., Smirnov V.A., Minkin V.I. // J. Photochem. Photobiol. A: Chem. 2016. V. 327. P. 6.
Loukova G.V., Milov A.A., Vasiliev V.P., Minkin V.I. // Phys. Chem. Chem. Phys. 2016. V. 18. P. 17822.
Лукова Г.В., Милов А.А., Васильев В.П., Минкин В.И. // Изв. АН. Сер. хим. 2020. С. 218.
Shin C.H., Han Y., Lee M.H., Do Y. // J. Organomet. Chem. 2009. V. 694. P. 1623 (и цитир. ссылки).
Frisch M.J., Trucks G.W., Schlegel H.B. et al. // GAUSSIAN 16, Revision C.01, Gaussian Inc., Wallingford CT, 2016.
Миняев Р.М., Гетманский И.В., Квапп В. // Журн. физ. химии. 2004. Т. 78. С. 1700.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий