

Химия высоких энергий, 2022, T. 56, № 1, стр. 38-44
Моделирование реакции взаимодействия нитробензола с олефинами. Влияние донорных и акцепторных заместителей
С. Д. Плехович a, *, С. В. Зеленцов a, Ю. В. Минасян a, И. Т. Гримова a
a ФГАОУ ВО “Национальный исследовательский Нижегородский государственный университет
им. Н.И. Лобачевского”
603950 Нижний Новгород, пр. Гагарина, 23, Россия
* E-mail: senypl@mail.ru
Поступила в редакцию 25.06.2021
После доработки 23.08.2021
Принята к публикации 28.08.2021
- EDN: HPHRKZ
- DOI: 10.31857/S0023119322010107
Аннотация
Методом uB3LYP/6-311g++(d,p) в газовой фазе выполнено моделирование реакции нитробензола с этеном в T1 состоянии. Изучен механизм реакции и определены энергия активации для взаимодействия о,м,п – PhNO2-R, где R=CH3, CCl3, CF3, Cl. Установлено, что взаимодействие олефинов с нитрометаном в триплетном состоянии (Т1) протекает в три реакции. Выявлено увеличение энергии активации, связанное со стерическим фактором в реакциях присоединения этена к RNO2Ph, где R находится орто-положениях. Обнаружен факт уменьшения энергии активации в реакциях распада продукта первой стадии присоединения нитросоединений к олефинам – бирадикала, в котором R находится в орто-положении, связанный со стерическим фактором.
В 1956 г. Бучи и Айер [1] сообщили о том, что при облучении смеси нитробензола и 2-метил-2-бутена УФ светом образуется несколько продуктов с малыми выходами. Последующее установление их строения показало, что взаимодействие идет именно по этиленовой связи. Эти авторы предположили, что в качестве промежуточных веществ образуется 1,3,2-диоксазолидин.
Известно [2], что НС взаимодействует с олефином с образованием аддукта (1). В триплетном состоянии НС представляет собой активную частицу – бирадикал, который, как мы считаем, может взаимодействовать с непредельными углеводородами, в частности, с олефинами, по двойной связи с образованием бирадикального аддукта.
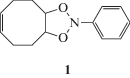
Доказательства корректности этого предположения приведены в работе [2], где показано, что облучение УФ светом смеси нитробензола и циклогексена при –78°С привело к появлению прозрачного раствора, который окрашивался при повышении температуры. В ИК спектре, зарегистрированном в хлороформе при –20°С отсутствовало поглощение карбонильной группы. Однако при комнатной температуре данное поглощение было найдено при 1720 см–1. Описанная в работе [2] совокупность спектральных данных согласуются со строением соединения 1 и с результатами работы [1].
Таким образом, в ходе реакций взаимодействия НС с олефинами, в зависимости от мультиплетности исходных реагентов, возможно получение соединения 1. Однако дальнейший путь превращения соединения 1 неизвестен. Неизвестно в каком состоянии в синглетном или триплетном протекает реакция. Кроме того, неясна структура переходных состояний изучаемых реакций, а также величины энергий активации всех стадий процессов.
Целью нашей работы явилось квантово-химическое моделирование механизма взаимодействия олефинов с ароматическими НС, с образованием окисей олефинов и определение влияния заместителей акцепторного и донорного типов на реакцию.
МЕТОДИКА ЭКСПЕРИМЕНТА
Для расчета использована программа Gaussian 03 [3]. Расчеты проводились в рамках метода uB3LYP/6-311g++(d,p) [4, 5]. Поиск геометрического строения переходных состояний проводили при помощи методов TS, QST2 и QST3 [6]. Энергии активации реакций рассчитывались как разности между полными энергиями переходных состояний и полными энергиями исходных соединений с учетом энергий нулевых колебаний.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Согласно работе [7] триплетном состоянии присоединение НС к олефинам включает следующие реакции: в первой реакции происходит возбуждение НС до состояния S1, а затем ИКК его в состояние T1 согласно схеме 1 . Разница энергий между S1 и T1 составляет 0.22 эВ [8 ] .
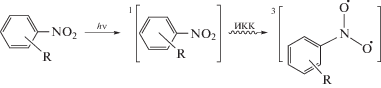
Схема 1 .
Следующей реакцией является присоединение триплетного НС по двойной связи олефина
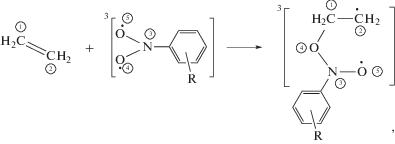
Схема 2 .
где R – CH3, CCl3, CF3, Cl.
В третьей реакции происходит распад получившегося аддукта в окись и нитрозометан, находящийся в триплетном состоянии (схема 3 ).
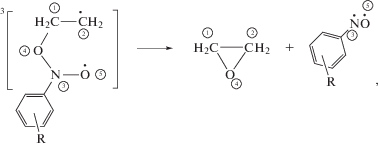
Схема 3 .
где, R – CH3, CCl3, CF3, Cl
Обсуждение механизма реакции взаимодействия этена с нитросоединениями
Для всех исследуемых реакций, находящихся в синглетном состоянии, третья реакция преимущественно протекает в сторону образования циклических соединений таких как: o-C8H10NO2(T), m-C8H10NO2(T), p-C8H10NO2(T). Это происходит из-за того, что более выгодным (низким по энергии) являются циклические соединения по сравнению с окисью этилена и нитрозосодержащими веществами. Для молекул в триплетном состоянии реакция протекает в сторону образования окисей этилена и нитрозосодержащих веществ.
Энергия активации изученных нами реакций представлена в табл. 1. Согласно табл. 1, в реакциях взаимодействия этилена с нитросоединениями прослеживается некоторая закономерность. Максимальное значение величины энергии активации определено для реакции взаимодействия этилена с o-ClNO2Ph = 3.4 ккал/моль (ΔrH = = –12.6 ккал/моль). Промежуточное положение занимает величина энергии активации для этилена с p-ClNO2Ph = 2.2 ккал/моль (ΔrH = = –15.5 ккал/моль). Минимальное для этилена с m‑ClNO2Ph = 1.9 ккал/моль (ΔrH = –15.7 ккал/моль). Снижение энергии активации в данном ряду, по всей видимости, связано с влиянием одного из факторов, а именно энтальпии реакции [7]. Согласно работе [7] для реакций присоединения и отрыва, одним из факторов, влияющих на увеличение энергии активации, является энтальпия реакции, при увеличении которой, происходит и увеличение энергии активации.
Таблица 1.
Энергии активации исследуемых реакций
| Реакция окисления | Cтадия 1 | Cтадия 2 | ΔHe (стадия 1) | ΔHe (стадия 2) |
|---|---|---|---|---|
| H2С=CH2 + o-ClNO2Ph | 3.4 | 12.4 | –12.6 | –26.6 |
| H2С=CH2 + m-ClNO2Ph | 1.9 | 14.9 | –15.7 | –20.2 |
| H2С=CH2 + p-ClNO2Ph | 2.2 | 14.9 | –15.5 | –19.9 |
Нитросоединение в триплетном состоянии, представляет собой бирадикал со спиновой плотностью локализованной, главным образом, на атомах кислорода. Нитросоединение испытывает 1,2-присоединение по двойной связи олефина с образованием фрагмента o-,m-,p-ClC8H10NO2.
В табл. 2 показано распределение спиновой плотности в исходных веществах, переходном состоянии и конечном продукте для первой реакции, с соединениями: o-PhNO2Cl, m-PhNO2Cl, p‑PhNO2Cl.
Таблица 2.
Результаты расчета распределения спиновой плотности на молекулах второй реакции
| № атома |
Исходные компоненты | Переходное состояние | Продукты реакции | |
|---|---|---|---|---|
| С2Н4(S) | para-PhNO2Cl(T) | para-C8H10NO2(T) | ||
| (1)С | – | –0.011 | –0.030 | |
| (2)С | – | 0.264 | 1.005 | |
| (3)N | – | 0.362 | 0.368 | 0.359 |
| (4)O | – | 0.720 | 0.645 | 0.062 |
| (5)O | – | 0.715 | 0.608 | 0.419 |
| С2Н4(S) | meta-PhNO2Cl(T) | meta-C8H10NO2(T) | ||
| (1)С | – | –0.007 | –0.033 | |
| (2)С | – | 0.210 | 1.007 | |
| (3)N | – | 0.356 | 0.367 | 0.358 |
| (4)O | – | 0.719 | 0.675 | 0.068 |
| (5)O | – | 0.721 | 0.637 | 0.423 |
| С2Н4(S) | orto-PhNO2Cl(T) | orto-C8H10NO2(T) | ||
| (1)С | – | –0.057 | –0.036 | |
| (2)С | – | 0.392 | 1.009 | |
| (3)N | – | 0.342 | 0.375 | 0.350 |
| (4)O | – | 0.624 | 0.565 | 0.057 |
| (5)O | – | 0.762 | 0.632 | 0.430 |
Исходя из данных, приведенных в табл. 2 видно, что соединения o-PhNO2Cl, m-PhNO2Cl, p‑PhNO2Cl представляют собой бирадикал со спиновой плотностью, сосредоточенной на атомах С(2) и O(5). В третьей реакции в результате которой происходит образование окиси этилена в синглетном состоянии и нитрозосоединений в триплетном состоянии при распаде соединений o-C8H10NO2, m-C8H10NO2, p-C8H10NO2. Максимальные значения энергии активации (табл. 2) прослеживаются для реакции распада продуктов присоединения этилена с m-PhNO2Cl и p-PhNO2Cl 14.9 ккал/моль (ΔrH = –20.2 и –19.9 ккал/моль соответственно). Меньшее значение для реакции с o-PhNO2Cl 12.4 ккал/моль (ΔrH = –26.6 ккал/моль). Увеличение энергии активации также согласуется с увеличением энтальпии реакции. Рассмотрим распределение спиновой плотности в третьей реакции (табл. 3). В табл. 3 показано распределение спиновой плотности в исходных веществах, переходном состоянии и конечном продукте для третьей реакции, с соединениями: o-PhNO2Cl, m-PhNO2Cl, p-PhNO2Cl. По данным таблицы можно сделать вывод, что в ходе реакции образуется бирадикал – нитрозосоединение с максимальной спиновой плотностью на атомах N(3) и O(5).
Таблица 3.
Результаты расчета распределения спиновой плотности на молекулах третьей реакции
| № атома |
Исходные компоненты | Переходное состояние | Продукты реакции | |
|---|---|---|---|---|
| p-C8H10NO2 (T) | Окись этилена (S) | p-PhNOCl(T) | ||
| (1)С | –0.030 | –0.048 | – | – |
| (2)С | 1.005 | 0.690 | – | – |
| (3)N | 0.359 | 0.705 | – | 0.877 |
| (4)O | 0.419 | –0.197 | – | – |
| (5)O | 0.062 | 0.626 | – | 0.834 |
| m-C8H10NO2 (T) | Окись этилена (S) | m-PhNOCl(T) | ||
| (1)С | –0.033 | –0.048 | – | – |
| (2)С | 1.007 | 0.690 | – | – |
| (3)N | 0.358 | 0.702 | – | 0.883 |
| (4)O | 0.068 | –0.196 | – | – |
| (5)O | 0.423 | 0.630 | – | 0.834 |
| o-C8H10NO2(T) | Окись этилена (S) | o-PhNOCl(T) | ||
| (1)С | –0.036 | –0.050 | – | – |
| (2)С | 1.009 | 0.715 | – | – |
| (3)N | 0.350 | 0.662 | – | 0.867 |
| (4)O | 0.057 | –0.206 | – | – |
| (5)O | 0.430 | 0.651 | – | 0.829 |
Влияние акцепторных и донорных заместителей на реакции взаимодействия ароматического нитросоединения с олефином
На энергию активации оказывает сильное влияние следующие факторы: энтальпия реакции, прочность образующейся связи, различная электроотрицательность атомов реакционного центра, стерический эффект и другие. Согласно данным табл. 4 для реакций со всеми заместителями во всех положениях наблюдается общая закономерность в реакциях присоединения: с уменьшением энтальпии реакции происходит уменьшение энергии активации. Так, например, для CH3 наименьшее значение энтальпии реакции –14.9 ккал/моль и энергия активации составляет 2.8 ккал/моль. Кроме того, хорошо видно влияние стерического эффекта, ярко выраженного для реакции присоединения, в которых радикалы находятся в орто-положении. Стерический эффект приводит к значительному увеличению энергии активации реакций присоединения. Кроме того, наблюдается и изменение угла атаки нитросоединения до максимального значения по сравнению с мета- и пара-положениями, а также уменьшение длины связи C–O – для соединений, в которых радикалы находятся в орто-положении, что способствует увеличению энергии активации образования такого соединения. Согласно табл. 4, максимальное значение величины энергии активации найдено для реакции этена с o-C8H10NO2 и равно 3.7 ккал/моль (∆rH = = –13.38 ккал/моль, где ∆rH – энтальпия реакции). Промежуточное значение занимают величины энергии активации для реакции взаимодействия этена с o-PhNO2ССl3 и o-PhNO2Cl, равные Еа (o-PhNO2Cl) = 3.39 ккал/моль (∆rH = = –12.57 ккал/моль), Еа (o-PhNO2ССl3) = = 2.95 ккал/моль (∆rH = –12.69 ккал/моль). Значительное снижение энергии активации наблюдается для реакций o-PhNO2CF3 с этеном Еа(o-PhNO2CF3) = = 1.8 ккал/моль (∆rH = –14.59 ккал/моль). Увеличение энергии согласуется с увеличением энтальпии реакции.
Таблица 4.
Геометрия переходного состояния, энергия активации и энтальпия второй реакции
| Нитробензол | 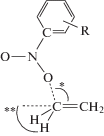 |
||
|---|---|---|---|
| r(C–O) = 2.27 φатак. = 97.9 φпирам. = 67.9 Ea = 2.7 ΔrH = –15.0 |
|||
| R | орто | мета | пара |
| CH3 | r(C–O) = 2.14 *φатак. = 100.8 **φпирам. = 67.9 Ea = 3.7 ΔrH = –13.4 |
r(C–O) = 2.26 *φатак. = 97.78 **φпирам. = 67.9 Ea = 2.8 ΔrH = –14.9 |
r(C–O) = 2.26 *φатак. = 97.85 **φпирам. = 67.9 Ea = 3.0 ΔrH = –14.8 |
| CCl3 | r(C–O) = 2.16 *φатак. = 100.42 **φпирам. = 67.8 Ea = 3.0 ΔrH = –12.7 |
r(C–O) = 2.43 *φатак. = 97.10 **φпирам. = 67.8 Ea = 1.7 ΔrH = –15.9 |
r(C–O) = 2.52 *φатак. = 98.6 **φпирам. = 67.8 Ea = 1.7 ΔrH = –15.9 |
| CF3 | r(C–O) = 2.23 *φатак. = 98.9 **φпирам. = 67.8 Ea = 1.8 ΔrH = –14.6 |
r(C–O) = 2.51 *φатак. = 97.9 **φпирам. = 67.8 Ea = 1.4 ΔrH = –16.1 |
r(C–O) = 2.56 *φатак. = 99.73 **φпирам. = 67.7 Ea = 1.3 ΔrH = –16.1 |
| Cl | r(C–O) = 2.14 *φатак. = 100.85 **φпирам. = 67.9 Ea = 3.4 ΔrH = –12.6 |
r(C–O) = 2.38 *φатак. = 97.03 **φпирам. = 67.81 Ea = 1.9 ΔrH = –15.7 |
r(C–O) = 2.33 *φатак. = 97.25 **φпирам. = 67.8 Ea = 2.2 ΔrH = –15.5 |
Для третьей реакции, согласно табл. 5, наблюдается обратная тенденция, заключающаяся в уменьшении энергии активации для соединений, в которых заместители находятся в орто положении в бензольном кольце. Это происходит за счет различной электроотрицательности атомов, которая вносит существенный вклад в энергию активации в реакциях распада: чем больше разница, тем меньше энергия активации, что и наблюдается для атомов азота и углерода в реакционном центре с заместителями в орто-положении о‑Δµ(N–C) = 1.204 по сравнению с заместителями в мета-положении Δµ(N–C) = 0.587 [9].
Таблица 5.
Геометрия переходного состояния, энергия активации и энтальпия третьей реакции
| Нитробензол | 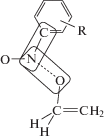 |
||
|---|---|---|---|
| r(N–O) = 2.14 Δµ(N–O) = 0.082 Δµ(N–C) = 1.204 Ea = 13.5 ΔrH = –24.0 |
|||
| R | орто | мета | пара |
| CH3 | r(N–O) = 2.14 Δµ(N–O) = 0.082 Δµ(N–C) = 1.204 Ea = 13.5 ΔrH = –24.0 |
r(N–O) = 1.87 Δµ(N–O) = 0.08 Δµ(N–C) = 0.587 Ea = 14.9 ΔrH = –20.2 |
r(N–O) = 1.88 Δµ(N–O) = 0.07 Δµ(N–C) = 0.307 Ea = 14.9 ΔrH = –19.3 |
| CCl3 | r(N–O) = 1.86 Δµ(N–O) = 0.299 Δµ(N–C) = 0.404 Ea = 12.4 ΔrH = –29.9 |
r(N–O) = 1.86 Δµ(N–O) = 0.302 Δµ(N–C) = 0.302 Ea = 14.9 ΔrH = –20.3 |
r(N–O) = 1.85 Δµ(N–O) = 0.203 Δµ(N–C) = 0.095 Ea = 14.9 ΔrH = –19.8 |
| CF3 | r(N–O) = 1.86 Δµ(N–O) = 0.195 Δµ(N–C) = 0.939 Ea = 12.9 ΔrH = –26.1 |
r(N–O) = 1.86 Δµ(N–O) = 0.113 Δµ(N–C) = 0.548 Ea = 14.9 ΔrH = –20.3 |
r(N–O) = 1.85 Δµ(N–O) = 0.141 Δµ(N–C) = 0.334 Ea = 14.9 ΔrH = –19.9 |
| Cl | r(N–O) = 1.87 Δµ(N–O) = 0.043 Δµ(N–C) = 1.5 Ea = 12.4 ΔrH = –26.2 |
r(N–O) = 1.86 Δµ(N–O) = 0.096 Δµ(N–C) = 0.575 Ea = 14.9 ΔrH = –20.2 |
r(N–O) = 1.87 Δµ(N–O) = 0.876 Δµ(N–C) = 0.510 Ea = 14.9 ΔrH = –19.9 |
Для третьей реакции, согласно табл. 5, минимальное значение энергии активации соответствует распаду бирадикала на окись этилена и нитрозо-соединение Еа (o-PhNOССl3) = 12.4 ккал/моль (∆rH = –29.9 ккал/моль), а также на окись этилена и нитрозосоединение Еа (o-PhNOCl) = = 12.4 ккал/моль (∆rH = –26.2 ккал/моль). Повышение энергии активации наблюдается для реакции распада на окись этилена и нитрозосоединение Еа (o-PhNO2CF3) = 12.9 ккал/моль (∆rH = = –26.1 ккал/моль). Максимальное значение энергии активации характерно для реакции распада бирадикала, в ходе которой образовалось нитрозо-соединение o-C8H10NO2 и равно 13.5 ккал/моль (∆rH = –13.38 ккал/моль, где ∆rH – энтальпия реакции).
ЗАКЛЮЧЕНИЕ
Установлено, что взаимодействие алкенов с ароматическими нитросоединениями в триплетном состоянии протекает в три реакции.
В первой реакции происходит возбуждение НС до состояния S1, а затем ИКК его в состояние T1.
Во второй реакции происходит взаимодействие ароматических нитросоединений в триплетном (Т1) состоянии с этеном с образованием промежуточного продукта-бирадикала.
В третьей реакции – распад продукта второй реакции до нитрозосоединений и окиси этилена.
Определены энергии активации реакции взаимодействия этена с o,m,p-ClNO2Ph; o,m,p-CCl3NO2Ph; o,m,p-CF3NO2Ph; o,m,p-CH3NO2Ph. Выявлено увеличение энергии активации, связанное со стерическим фактором в реакциях присоединения этена к RNO2Ph, где R находится орто-положениях. Обнаружен факт уменьшения энергии активации в реакциях распада продукта первой реакции – присоединения нитросоединений к олефинам – бирадикала, в котором R находится в орто-положении, связанный со стерическим фактором.
Список литературы
Buchi G., Ayer D. E. // J. Am. Chem. Soc. 1956. V. 78. Iss. 3. P. 689.
Charlton J.L., Liao C.C., de Mayo P. // J. Amer. Chem. Soc. 1971. V. 93. Iss. 10. P. 2463.
Gaussian R.A., Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A. et al. // Gaussian. Inc., Wallingford CT. 2003.
Mahadevan D., Periandy S., Ramalingam S. // Spectrochim. Acta. A. 2011. V. 64. № 1. P. 86.
Yi J. et al. // Sci. Rep. 2016. V. 6. P. 19364.
Peng C., Ayala P.Y., Schlegel H.B., Frisch M.J.J. // Comp. Chem. 1996. V. 17. Iss. 1. P. 49.
Плехович С.Д., Зеленцов С.В., Минасян Ю.В., Дегтяренко А.И. // Химия высоких энергий. 2018. Т. 52. № 6. С. 457.
Arenas J.F., Otero J.C., Pelaez, J.D., Soto J. // Chem. Phys. 2003. V. 119. Iss. 8. Р. 7814.
Денисов Е.Т. // Успехи химии. 2000. Т. 69. № 2. С. 166.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий