

Химия высоких энергий, 2022, T. 56, № 1, стр. 27-37
Флуоресцентные хемосенсорные материалы
В. А. Сажников a, b, *, Д. С. Ионов a, М. В. Алфимов a, b
a Центр фотохимии РАН, ФНИЦ “Кристаллография и фотоника” РАН
119421 Москва, ул. Новаторов, д. 7А, корп. 1, Россия
b Московский физико-технический институт (Государственный университет)
141701 Московская обл., Долгопрудный, Институтский пер., д. 9, Россия
* E-mail: sazhnikov@yandex.ru
Поступила в редакцию 17.07.2021
После доработки 29.08.2021
Принята к публикации 29.08.2021
- EDN: LUJPXX
- DOI: 10.31857/S0023119322010120
Аннотация
В обзоре изложены основные этапы разработки флуоресцентных хемосенсорных материалов, выполненных в ЦФ РАН за последние 15 лет. Описаны системы на основе оригинальных красителей акридинового ряда, синтезированных в ЦФ РАН, а также на основе известных флуоресцентных красителей и флуорофоров семейства дикетонатов дифторида бора. Полимерные пленки и микрочастицы, а также кремнеземные микро- и наночастицы были изучены в качестве твердотельных матриц для хемосенсоров.
ВВЕДЕНИЕ
Проблема обнаружения, идентификации и измерения малых концентраций летучих органических соединений (ЛОС) актуальна как для мониторинга состава воздуха в окружающей атмосфере или в замкнутых помещениях, так и для неинвазивной диагностики состояния здоровья человека, в том числе в стрессовых ситуациях.
Как известно, флуоресцентный анализ является одним из самых простых и в то же время самых чувствительных методик в аналитической химии. Непрерывный прогресс в создании новых миниатюрных источников света и фотодетекторов на базе нанотехнологий делают их все более перспективными с практической точки зрения. Однако в настоящее время промышленно выпускаемых флуоресцентных хемосенсоров практически нет, хотя разработки соответствующих материалов и устройств ведутся довольно широко. В данном обзоре представлены результаты исследований в области разработки трех типов флуоресцентных хемосенсорных материалов, выполненных в Центре фотохимии РАН (с 2016 г. Центр фотохимии ФНИЦ “Кристалография и фотоника” РАН) за последние 10–15 лет.
1. РАЗРАБОТКА СЕНСОРНЫХ МАТЕРИАЛОВ НА ОСНОВЕ 9-ДИАРИЛАМИНОАКРИДИНОВ
В ЦФ РАН было впервые установлено, что при УФ-облучении растворов дифениламина и тетра-бромметана в толуоле или гексане наряду с трифенилметановыми красителями образуется новый, неизвестный в литературе краситель 9-дифениламиноакридин (9-ДФАА) (рис. 1, соединение 1). Было показано также ([1, 3] и цитируемые там ссылки), что применение производных исходных дифениламина и тетрабромметана позволяет провести в растворах аналогичный фотоиндуцированный синтез 9-дитолиламиноакридина (9‑ДТАА) (соединение 2), а также более сложного производного (соединение 3).
Рис. 1.
Структурные формулы 9-дифениламиноакридина (1), 9-дидолиламиноакридина (2), полученный на основе ди-пара-толиламина (ДТА) и соединения 3, полученного из исходного (4,4'-бис(альфа,альфа-диметилбензил)дифениламина, ДMБДФА).
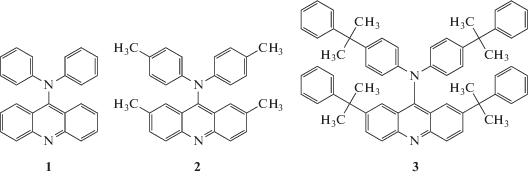
Следует отметить следующие структурные и электронные особенности этих соединений. На основе полученных рентгеноструктурных данных было установлено, что в соединениях 1–3 плоскость дифениламинного фрагмента развернута относительно плоскости акридинового фрагмента на угол от 45° до 80°. Наличие в соединениях 1–3 атома азота, обладающего неподеленной парой электронов, делает их достаточно сильными протонными акцепторами. Спектральные свойства нейтральных и заряженных форм этих молекул существенно отличаются. Проведенные квантово-химические расчеты (на основе DFT и TD-DFT методов с использованием оптимизированной геометрии основного и возбужденного состояний) показали, что длинноволновые полосы являются переходами с внутримолекулярным переносом электрона от дифениламинного фрагмента на акридиновый цикл. Это приводит к большому увеличению дипольного момента этих молекул при электронном возбуждении и к появлению ярко выраженных сольватофлуорохромных свойств. В частности, оценки на основе сдвигов максимумов спектров флуоресценции 9-ДТАА в различных полярных растворителях дали значения μg = 2.6 D для основного и μe = 12 D для первого синглетного состояния ([1, 2] и цитируемые там ссылки).
В качестве примера на рис. 2 приведен спектр поглощения соединения 2. Спектры поглощения соединений 1 и 3 в гексане практически совпадают со спектром 2, имеющим максимумы полос поглощения в районе 290, 360 и 450 нм.
Рис. 2.
Спектр поглощения 9-ДТАА в гексане (с = = 10–4 М). Цифры указывают положение макси-му мов полос.
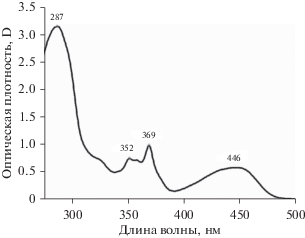
При переходе к полярным растворителям (ТГФ, ацетон, ДМСО) для всех трех соединений наблюдается небольшой батохромный сдвиг (от 300 до 500 см–1) длинноволновой полосы. Во всех растворителях коэффициенты экстинкции в максимуме длинноволновых полос 1 и 2 составляют в среднем около 11000 л моль–1 см–1, для 3 – около 15000 л моль–1 см–1.
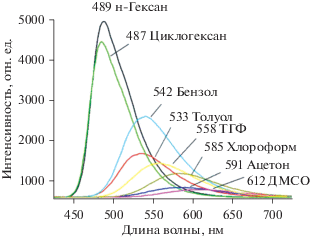
В отличие от спектров поглощения, максимумы полос флуоресценции нейтральных 9-дифениламиноакридинов 1–3 при переходе от неполярных к полярным растворителям испытывают значительные батохромные сдвиги (от 3000 до 5000 см–1) [2]. Общий вид зависимости основных параметров флуоресценции 9-ДТАА (интенсивности и положения полосы) от полярности растворителей, приведенный на рис. 3, указывает на возможность использования синтезированных диариламиноакридинов в качестве флуоресцентных молекулярных сенсоров, чувствительных к парам полярных растворителей.
Рис. 3.
Изменение интенсивности и положения полос флуоресценции 9-ДТАА в зависимости от полярности растворителей.
Протонированные формы соединений 1–3 также могут быть использованы для аналитических целей. Расчеты показывают, что при протонировании энергетический интервал между HOMO и LUMO уменьшается (все орбитали при этом резко сдвигаются вниз по энергиям). Это приводит к наблюдаемым значительным сдвигам длинноволновых полос поглощения и флуоресценции соединений 1–3 в красную область.
Ниже представлены некоторые результаты проведенных исследований. В качестве полимерных матриц для приготовления сенсорных слоев на основе нейтральных флуорофоров 1–3 использовали полистирол (ПС), полиметилметакрилат (ПММА) и этилцеллюлозу (ЭЦ). Для получения тонких полимерных пленок использовали методику спин-коатинга. Было найдено, что полимерные пленки, допированные соединениями 1–3, обладают достаточно яркой флуоресценцией с максимумом в районе 510–520 нм при возбуждении светодиодом с λ = 375 нм.
На рис. 4 представлены данные, относящиеся к ПС-пленкам, допированных 9-ДТАА. Видно, что выдерживание пленок в насыщенных парах толуола (рис. 4а) приводит к сдвигу максимума флуоресценции до 537 нм, что соответствует максимуму флуоресценции 2 в чистом толуоле (рис. 3). Наблюдаемые изменения отражают, по-видимому, процесс достраивания диффундирующими в пленку молекулами толуола сольватной оболочки вокруг молекул 2 до оболочки, характерной для чистого толуола. Процесс изменения флуоресценции полностью обратим: после удаления образца из атмосферы паров толуола спектр быстро (~20 с) и полностью восстанавливается.
Рис. 4.
Изменение со временем спектра флуоресценции ПС-пленок, допированных 9-ДТАА (2); (а) при выдерживании в насыщенных парах толуола; (б) – при выдерживании в насыщенных парах хлороформа; (в) тушение флуоресценции ПС-пленки с 9-ДТАА в насыщенных парах четыреххлористого углерода (кривые записаны с интервалами в 3 и 10 с соответственно).
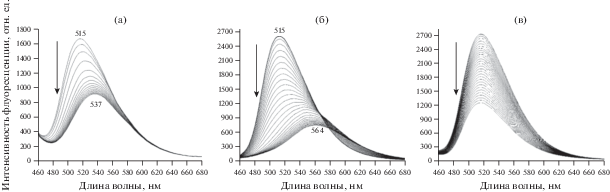
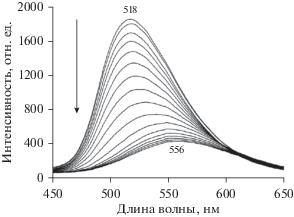
На рис. 4б представлено изменение спектра флуоресценции ПС-пленки после выдерживания ее в течение нескольких минут в атмосфере насыщенных паров хлороформа. Видно, что по мере диффузии молекул CHCl3 (ХФ) в пленку интенсивность флуоресценции уменьшается и одновременно наблюдается значительный сдвиг максимума полосы флуоресценции в сторону длинных волн (от 515 до 564 нм), хотя и меньший, чем в растворе хлороформа (сдвиг до 580 нм). Отметим, что сдвига до 580 нм не удается добиться даже при очень больших временах экспозиции. Кинетика падения интенсивности носит почти линейный характер.
Очевидно, что такое поведение может быть объяснено частичной сольватацией флуорофора 2 полярными молекулами ХФ. Наблюдаемые спектральные изменения в целом тоже носят обратимый характер. Однако при испарении молекул ХФ из пленки обратный сдвиг полосы в коротковолновую область происходит достаточно быстро (30 с) только для области от 560 до 530 нм, а процесс окончательного перехода от 530 нм до первоначального спектра с максимумом в районе 515 нм требует уже большего времени (до 100–300 с). Возможно, это следствие того, что энергия взаимодействия молекулы 9-ДТАА с одной или двумя оставшимися в сольватной оболочке молекулами ХФ имеет сравнительно большое значение.
Для дихлорметана процессы изменения спектров и кинетические кривые аналогичны случаю хлороформа. При насыщении парами дихлорметана (ДХМ) максимум полосы флуоресценции 9‑ДТАА сдвинут сильнее (до 574 нм), чем в случае ХФ, однако и в этом случае наблюдаемый сдвиг меньше, чем сдвиг для раствора ДХМ (586 нм). В отличие от случая полярных ХФ и ДХМ, выдерживания пленки ПС-ДТАА в парах неполярного тетрахлорметана (ТХМ) приводит только к равномерному падению интенсивности флуоресценции без изменения положения максимума полосы флуоресцении (рис. 4в). Сам по себе этот факт представляет интерес с точки зрения сенсорных свойств ПС-слоев, допированных 1–3, так как он означает, что по характеру изменения спектра флуоресценции можно отличить случаи наличия в воздухе паров ТХМ от случая присутствия паров ДХМ или ХФ.
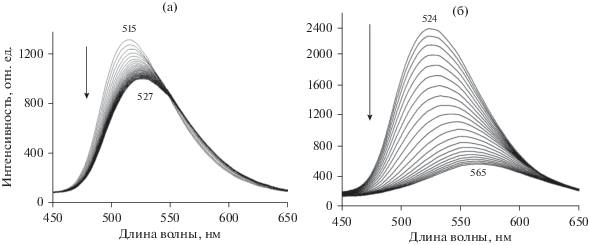
Аналогичные сдвиги наблюдаются для пленок на основе других полимеров. На рис. 5 показано изменение спектра флуоресценции полиметилметакрилатной пленки в парах ацетона.
Характер изменения спектров может сильно изменяться при варьировании природы полимера. В качестве иллюстрации на рис. 6а и 6б, приведены спектры пленок на основе полистирола и этилацетата. Видно, что как начальные, так и конечные спектры флуоресценции 9-ДТАА довольно сильно отличаются в этих двух пленках.
Рис. 6.
(а) – Изменение спектра флуоресценции ПС-пленки с 9-ДТАА в насыщенных парах этанола; (б) – Изменение спектра флуоресценции ЭЦ-пленки с 9-ДТАА в насыщенных парах этанола.
В описанном выше случае действия паров ТХМ на ПС-слои с нейтральным 9-ДТАА, одновременно с уменьшением интенсивности зеленой флуоресценции появляется новая, хотя и менее интенсивная полоса флуоресценции в красной области спектра с максимумом при 630 нм (рис. 7). При длительном экспонировании ПС-пленок в парах ТХМ (до 10 мин и более) наблюдается по-чти полный переход зеленой флуоресценции в красную.
Рис. 7.
Спектр флуоресценции ПС-пленки с 9-ДТАА после облучения светодиодом с λмах = 375 нм в течение нескольких минут в парах четыреххлористого углерода.
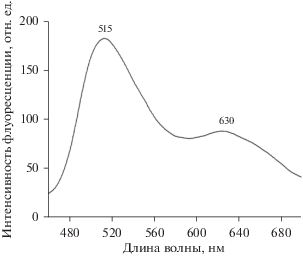
Свечение в красной области спектра связано с появлением протонированной формы 2Н+, которая легко отдает протон аммиаку или органическому основанию, обладающему большим чем у 2 сродством к протону. Протонированный 2Н+ может образоваться путем присоединения к 2 молекулы HCl, образующейся в ходе реакции фотопереноса электрона с молекулы 2 на молекулу CCl4 и последующей необратимой диссоциации ани-он-радикала CCl4 на ион Cl– и радикал CCl3.
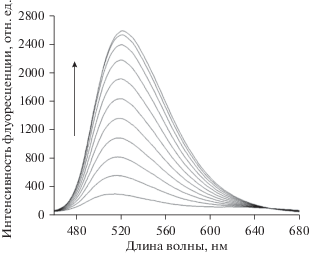
На рис. 8 показано, как начальная слабая красная флуоресценция пленки преобразуется в яркую зеленую флуоресценцию под действием насыщенных паров пиридина. Аналогичные изменения наблюдаются в случае паров аммиака.
2. РАЗРАБОТКА СЕНСОРНЫХ МАТЕРИАЛОВ НА ОСНОВЕ КРАСИТЕЛЕЙ РАЗЛИЧНЫХ КЛАССОВ
Поскольку сольватофлуорохромные свойства присущи большому количеству широко известных красителей, некоторые из них наряду с 9‑диариламиноакридинами были изучены в ЦФ РАН в качестве перспективных молекулярных сенсоров. Ниже приведены структуры некоторых соединений, относящихся ко второй группе индикаторов-флуорофоров.
Прежде всего, был изучен краситель Нильский красный (НК) (рис. 9), спектральные свойства которого близки к свойствам 9-диариламиноакридинов.
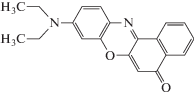
Сдвиги максимумов полос флуоресценции НК и изменения их интенсивности аналогичны изменениям, которые были описаны выше для 9‑диариламиноакридинов. Отметим, что НК, как и 9-ДТАА, является нейтральным красителем, однако в отличие от 9-ДТАА он имеет несколько гетероатомов, способных образовывать межмолекулярные водородные связи (прежде всего это атомы кислорода и азота центрального кольца, которые имеют неподеленные пары электронов).
Для других красителей – акрифлавина, флуоресцеина, эозина, родаминов Б и 6Ж, характерно присутствие боковых функциональных групп, способных образовывать различного типа водородные связи, а также возможность образования в некоторых случаях заряженных или лактонных форм.
Перечисленные выше красители вводили в полимерные слои и изучали флуоресцентные отклики на присутствие в воздухе паров аналитов. Однако скорость диффузии аналитов внутрь полимерных пленок сравнительно невелика. Кроме того, в пленки не всегда удается ввести достаточно большую концентрацию красителей. Поэтому, чтобы увеличить интенсивность флуоресцентного сигнала и уменьшить время отклика образцов, сенсорные слои изготавливались, как правило, на основе силикагельных микрочастиц (размером 6–10 микрон) с иммобилизованными на их поверхности или в объеме красителями. Использовали микрочастицы немодифицированного силикагеля С0 (Диасфер-SIL, Диасфер-С0) диаметром 10 мкм и порами порядка 100 Å или аналогичные модифицированные микрочастицы С1, С8, С18 (Диасфер-С1, -С8, С-18), на поверхности которых часть ОН-групп замещена на алкильные группы (табл. 1).
Таблица 1.
Немодифицированные и модифицированные силикагельные микрочастицы
| Матрица | |
|---|---|
| название | функциональная группа |
| Диасфер-С0, -SIL | 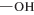 |
| Диасфер-С1 | 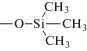 |
| Диасфер-С8 | 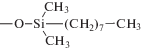 |
| Диасфер-С18 | 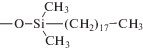 |
Иммобилизация достигалась путем помещения микрочастиц в растворы красителей и выдерживании их в течение определенного времени до достижения нужной концентрации. На рис. 10 приведены типичные изображения окрашенных полимерных микрочастиц, полученные с помощью конфокального флуоресцентного микроскопа.
Рис. 10.
Изображения флуоресцирующих микрочастиц, полученные с помощью конфокального флуоресцентного микроскопа.
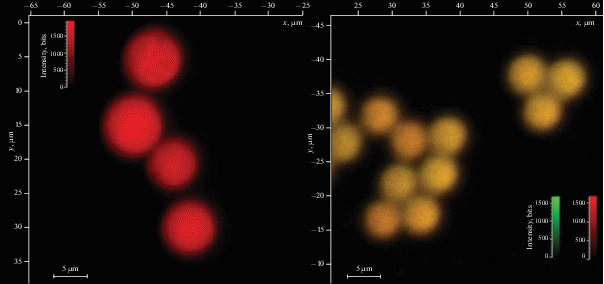
Флуоресценция адсорбированных красителей зависит от взаимодействия красителя с присутствующими на поверхности микрочастиц функциональными группами. Например, ввзаимодействие НК с ОН-группами С0 (образование водородных связей) приводит к сильному гипсохромному сдвигу его спектра флуоресценции. С другой стороны, взаимодействие НК с нейтральными метильными группами С1 и С18 почти никак не сказывается на его спектре флуоресценции.
Очевидно, что молекулы аналитов также взаимодействуют с поверхностными функциональными группами матриц (например, этанол или ацетон образуют водородные связи с силанольными группами). В общем случае схема взаимодействий включает все возможные варианты взаимодействий между компонентами систем матрица/индикатор/аналит.
Возможность образования димеров красителей проиллюстрирована на рис. 11. Из зависимости спектра возбуждения флуоресценции Родамина 6Ж от его концентрации на поверхности кремнеземных частиц С0 (флуоресценция наблюдалась на 18 000 см–1) видно, что по мере увеличения концентрации спектр возбуждения сдвигается в длинноволновую область, что связано с образованием димеров.
Рис. 11.
Зависимость спектра возбуждения флуоресценции Родамина 6Ж от его концентрации на поверхности кремнеземных частиц С0 (флуоресценция наблюдалась на 18 000 см–1).
Таким образом, в общем случае из адсорбированной (или ковалентно-связанной) молекулы флуорофора-индикатора и некоторого количества молекул функциональных групп за счет межмолекулярных взаимодействий на поверхности микрочастиц формируется наноразмерный супрамолекулярный рецепторный центр. Поверхностные функциональные группы (например, силанольные группы Si–OH) обеспечивают затем адсорбцию молекул аналитов и их диффузию к рецепторам.
Результаты модельных расчетов строения и энергий образования комплексов показывают, в частности, что для акридина в качестве индикатора полистирольные матрицы в целом предпочтительнее с точки зрения сенсорных свойств, чем кремнеземные. Для детектирования метанола, этанола и хлороформа подходят обе матрицы, а для ацетона и ацетонитрила обе матрицы оказываются не самым лучшим вариантом и т.п.
При выдерживании сенсорных элементов в насыщенных или ненасыщенных парах аналитов наблюдается изменение интенсивности флуоресценции. Вследствие того, что в разных матрицах на поверхности расположены разные функциональные группы, взаимодействие одного и того же флуорофора-индикатора с такими группами может привести как к тушению, так и к разгоранию флуоресценции. В табл. 2 просуммированы данные, полученные для трех матриц (полистирол, С0 и С1) и семи красителей (флуоресцеин, акрифлавин, 9-ДТАА, Нильский красный, эозин, родамин Б и родамин 6Ж).
Таблица 2.
Комбинации матрица/краситель, флуоресценция которых изменяется при экспонировании в парах аналита
| Матрица | Флуорофор | Изменение интенсив-ности |
|---|---|---|
| Аммиак | ||
| Полистирол | Акрифлавин | Увеличение |
| Полистирол | 9-ДТАА | Увеличение |
| Полистирол | Родамин Б | Уменьшение |
| Силикагель-С0 | Родамин 6Ж | Уменьшение |
| Силикагель-С0 | Родамин Б | Уменьшение |
| Силикагель-С0 | Эозин | Уменьшение |
| Силикагель-С0 | 9-ДТАА | Увеличение |
| Силикагель-С1 | Флуоресцеин | Увеличение |
| Ацетон | ||
| Полистирол | Акрифлавин | Увеличение |
| Полистирол | 9-ДТАА | Уменьшение |
| Силикагель-С0 | Родамин 6Ж | Уменьшение |
| Силикагель-С0 | Родамин Б | Уменьшение |
| Силикагель-С0 | Эозин | Увеличение |
| Силикагель-С0 | Акрифлавин | Уменьшение |
| Силикагель-С1 | Флуоресцеин | Уменьшение |
| Этанол | ||
| Полистирол | Акрифлавин | Увеличение |
| Полистирол | 9-ДТАА | Уменьшение |
| Полистирол | Нильский красный | Увеличение |
| Силикагель-С0 | Акрифлавин | Уменьшение |
| Силикагель-С1 | 9-ДТАА | Увеличение |
| Силикагель-С1 | Нильский красный | Уменьшение |
| Силикагель-С1 | Флуоресцеин | Увеличение |
Очевидно, что данные такого рода могут быть очень полезными для формирования матриц из сенсорных элементов. Например, на основе силикатных микрочастиц были сформированы матрицы из четырех сенсорных элементов, каждый из которых содержал один из 4 красителей (флуоресцеин, эозин, акрифлавин и 9-ДТАА). Затем эти матрицы подвергались действию паров аммиака и других аналитов. Анализ суммарных флуоресцентных откликов позволял идентифицировать каждый из аналитов. Очевидно, что такой подход может быть использован в дальнейшем для создания высокоселективных флуоресцентных сенсорных материалов. Для систематических исследования материалов данного класса был разработан специальный контрольно-измерительный стенд (КИС), способный осуществлять их высокопроизводительный скрининг [4, 5].
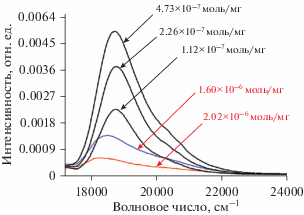
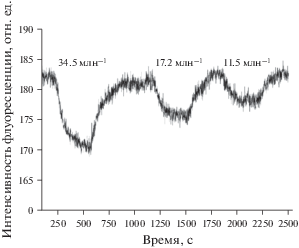
С помощью контрольно-измерительного стенда была изучена возможность регистрации флуоресцентных откликов сенсорных материалов на низкие концентрации ЛОС. В качестве примера на рис. 12 показан флуоресцентный отклик (λвозб = = 500 нм, λнабл = 550 нм) на пары ацетона сенсорного элемента на основе силикатных частиц, модифицированных красителем Родамин 6Ж (аналогичные результаты были получены с помощью элементов на основе НК). Было показано, что с помощью КИС легко могут быть созданы и измерены концентрации аналитов в диапазоне от 100 до 1 млн–1, а в некоторых случаях до долей млн–1.
3. ДИБЕНЗОИЛМЕТАНАТ ДИФТОРИДА БОРА И ЕГО ПРОИЗВОДНЫЕ В КАЧЕСТВЕ МОЛЕКУЛЯРНЫХ ФЛУОРЕСЦЕНТНЫХ ХЕМОСЕНСОРОВ
Еще одним вариантом формирования супрамолекулярного рецепторного центра для аналитических целей является образование флуоресцентных комплексов между флуорофорами и молекулами аналитов. Такой вариант был предложен в ЦФ РАН при решении задачи количественного анализа смесей бензола, толуола и ксилолов. Согласно литературным данным, дибензоилметанат дифторида бора (DBMBF2, хелатный комплекс BF2 и дибензоилметана) в возбужденном состоянии образует в растворах с бензолом и его метилпроизводными флуоресцентные комплексы (эксиплексы). Проведенные в ЦФ РАН исследования впервые показали, что DBMBF2, адсорбированный на силикагельных микрочастицах или ковалентно-привитой на поверхности микро- или наночастиц, также образует флуоресцентные эксиплексы с метилпрозводными бензола при их сорбции из газовой фазы ([6, 7] и цитируемые там ссылки). При появлении в воздухе паров бензола или метилбензолов наблюдается одновременное тушение флуоресценции адсорбированного DBMBF2 и разгорание флуоресценции соответствующих эксиплексов, в то время как при экспонировании образца в парах ацетона или этанола наблюдается просто тушение флуоресценции. На рис. 13а приведена упрощенная схема реакции образования эксиплекса на примере DBMBF2 и толуола. На рис. 13б представлены спектры флуоресценции DBMBF2, его эксиплекса с толуолом и суммарный спектр, наблюдаемый в эксперименте при экспонировании материала в парах толуола. Отсутствие образования эксиплексов с молекулами других классов, таких как алканы, спирты и др., и, следовательно, неизменность формы спектра флуоресценции образцов в парах этих соединений делает материалы на основе флуорофоров данного класса селективными к ароматическим веществам [8–12].
Рис. 13.
(а) – схема реакции образования эксиплекса на примере DBMBF2 и толуола; (б) – спектры флуоресценции флуорофора DBMBF2, его эксиплекса с толуолом и суммарный спектр, наблюдаемый в эксперименте; (в) – спектры флуоресценции флуорофора DBMBF2 и его эксиплексов с бензолом, толуолом и п-ксилолом.
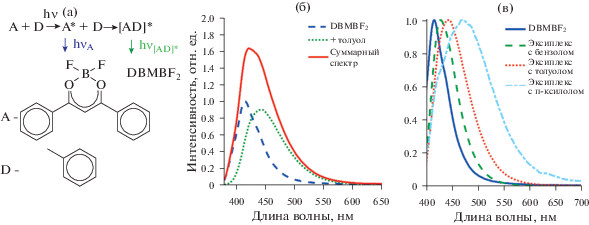
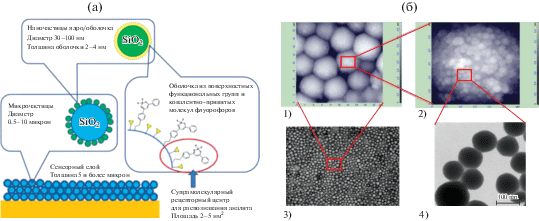
Спектр излучения эксиплекса и эффективность его образования зависит от типа молекул, между которыми происходит комплексообразование, а также от параметров микроокружения. Уменьшение потенциала ионизации в ряду бензол–толуол–п-ксилол (9.24, 8.83, 8.44 эВ соответственно) сдвигает максимум флуоресценции эксиплекса в длинноволновую область. Эта зависимость может быть использована для достижения селективности между структурно близкими ароматическими молекулами. Для применения предлагаемого подхода к анализу бензола и его производных в воздухе необходимо создать материал, в котором молекулы-флуорофоры располагаются в заданном окружении и доступны для взаимодействия с ароматическими молекулами, поступающими из газовой фазы. Для достижения максимальной чувствительности в материале необходимо наличие как можно большего количества флуорофоров, однако они должны находиться достаточно далеко друг от друга, чтобы не взаимодействовать между собой, так как в этом случае происходит агрегация, и возможность детектировать ароматические молекулы теряется. Совместить максимальную доступность молекул и максимизировать их количеств возможно, если использовать материалы с развитой поверхностью. Такие материалы были получены путем самосборки из наночастиц, на поверхности которых располагались молекулы-флуорофоры. Как показано на рис. 14а, материал состоит из следующих структурных элементов, имеющих различный масштаб: микрочастиц диаметром 1–10 мкм, наночастиц диаметром 30–100 нм и наноразмерных поверхностных супрамолекулярных рецепторных центров площадью 2–4 нм2, включающих молекулу-флуорофор и ее окружение. Данная структура позволяет получить проницаемый для молекул газа материал с высокой чувствительностью к ароматическим соединениями и коротким временем отклика с использованием технологий струйной печати из заранее синтезированных чернил на основе микро- и наночастиц [13, 14].
Рис. 14.
(а) – Схематическое изображение строения сенсорного материала; (б) – экспериментальные данные о строении разработанного сенсорного материала: оптическая микрофотография сенсорного слоя на поверхности стекла ((б1) – размер поля 220 × 160 мкм), АСМ-изображения группы микрочастиц (б2) – размер поля 20 × 20 мкм, одной микрочастицы, покрытой наночастицами (б3) – размер поля 3 × 3 мкм и ТЕМ-изображение группы наночастиц типа ядро-оболочка (б4) – размер поля 0.4 × 0.4 мкм [13, 14].
Была разработана физико-химическая модель, позволяющая описать зависимость спектров флуоресценции хемосенсорных материалов от концентрации анализируемых веществ в газовой смеси [15]. Согласно полученной модели, спектр флуоресценции материала в присутствии ароматических веществ представляет собой линейную комбинацию спектров флуоресценции флуорофора и его эксиплексов с данными соединениями. Коэффициенты перед спектрами зависят от концентрации ароматических молекул. Зная формы этих спектров, с помощью решения обратной задачи можно найти коэффициенты линейной комбинации, а из них получить концентрации. Формы спектров и другие параметры могут быть найдены путем калибровки измерительной системы по отдельным соединениям, калибровка по смесям веществ не требуется. Разработанный способ позволяет непрерывно и в реальном времени измерять концентрации ароматических веществ в двух- и трехкомпонентных смесях в воздухе, что было продемонстрировано экспериментально [16, 17].
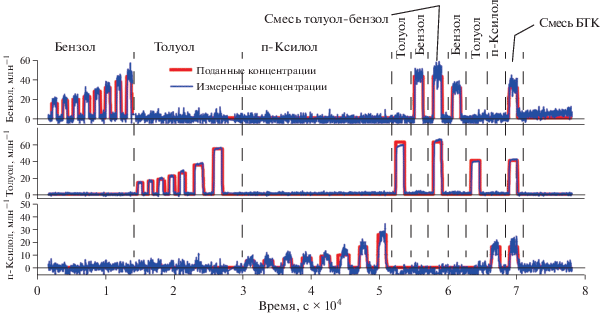
На рис. 15 приведено сравнение значений концентраций бензола, толуола и п-ксилола в воздухе, который поступал в экспериментальную ячейку, и значений, измеренных с использованием разработанного хемосенсорного материала. Измерение проводилось путем поиска коэффициентов разложения спектра флуоресценции материала на сумму четырех спектров и последующего вычисления по найденным коэффициентам концентраций. Спектры, на которые производилось разложение, получали заранее в процессе калибровки, используя процедуру многомерной нелинейной регрессии в соответствии с разработанной моделью. На верхнем графике представлено сравнение значений концентрации бензола, на среднем – толуола, на нижнем – п-ксилола. Вначале в ячейку подаются наборы концентраций бензола, потом толуола, затем п-ксилола. После этого подается одна концентрация бензола и одна концентрация толуола и двухкомпонентная смесь данных веществ. Затем подается последовательно по одной концентрации бензола, толуола и п‑ксилола и их трехкомпонентная смесь. Из графиков видно, что как в случае отдельных веществ, так и в случае смесей, измеренные концентрации хорошо совпадают с поданными в ячейку. Как следует из представленных данных, разработанный способ позволяет измерять концентрации ароматических соединений в двух- и трехкомпонентных смесях в воздухе.
Рис. 15.
Сравнение концентраций бензола, толуола и ксилола, поданных в экспериментальную ячейку и измеренных с помощью разработанных материалов, в ходе эксперимента. На рисунке красным показано изменение концентрации в ходе эксперимента, синим показаны показания датчика. Сверху обозначены моменты подачи ароматических веществ [16].
На основе исследований и разработок, описанных выше, были созданы и успешно испытаны образцы датчиков и газоанализаторов, в которых внедрен разработанный метод [18–20] (см. рис. 16). Разработанные газоанализаторы способны определять бензол, толуол и п-ксилол в смесях с пределом детектирования на уровне 0.5 ПДК для воздуха рабочей зоны. Время реакции для бензола не более 30 с.

ЗАКЛЮЧЕНИЕ
Фундаментальные и прикладные исследования, проведенные в ЦФ РАН, позволили разработать рациональный подход к конструированию флуоресцентных хемосенсорных материалов для детектирования летучих органических соединений. В основе предлагаемого метода лежит концепция рецепторного центра, включающего молекулу-индикатор с изменяющимися при взаимодействии с ЛОС спектрально-люминесцентными характеристиками и окружающие группы матрицы носителя. Проведенные исследования позволили установить взаимосвязь между составом и строением рецепторного центра (химическим составом и строением матрицы, структурой и спектрально-люминесцентными характеристиками флуорофоров) и функциональными характеристиками хемосенсорных материлов (чувствительностью и селективностью к ароматическим молекулам). Рассматриваемый подход был успешно применен при разработке новых типов газоанализаторов, способных селективно измерять ароматические вещества в многокомпонентных газовых смесях.
Список литературы
Сажников В.А. Аристархов В.М., Сазонов С.К., Ведерников А.И. и др. // Химия высоких энергий. 2013. Т. 47. № 6. С. 490.
Ионов Д.С., Коваленко В.А., Хлебунов А.А. и др. // Химия высоких энергий. 2011. Т. 45. № 4. С. 342.
Sazhnikov V.A., Khlebunov A.A., Sazonov S.K. et al. // J. Mol. Struct. 2013. V. 1053. P. 79.
Хлебунов А.А., Ионов Д.С., Комаров П.В. и др. // Приборы и техника эксперимента. 2009. № 1. С. 145.
Юрасик Г.А., Ионов Д.С. // Приборы и техника эксперимента. 2020. № 1. С. 121.
Сажников В.А., Аристархов В.М., Мирочник А.Г. и др. // Доклады Академии наук. 2011. Т. 437. № 2. С. 201.
Сажников В.А., Музафаров А., Копысов В.Н. и др. // Российские нанотехнологии. 2012. Т. 7. № 1–2. С. 24.
Arias Espinoza J.D., Sazhnikov V., Smits E.C.P. et al. // J. Fluoresc. 2014. V. 24. № 6. P. 1735.
Ионов Д.С., Сажников В.А., Юрасик Г.А. и др. // Химия высоких энергий. 2018. Т. 52. № 6. С. 473.
Kononevich Y.N., Sazhnikov V.A., Belova A.S. et al. // New J. Chem. 2019. V. 43. № 35. P. 13725.
Kononevich Y.N., Belova A.S., Sazhnikov V.A. et al. // Tetrahedron Lett. 2020. V. 61. № 31. P. 152176.
Kononevich Y.N., Temnikov M.N., Korlyukov A.A. et al. // Chempluschem. 2020. V. 85. № 6. P. 1111.
Ионов Д.С., Юрасик Г.А., Кононевич Ю.Н. и др. // Российские нанотехнологии. 2017. Т. 12, № 7–8. С. 13.
Ионов Д.С., Юрасик Г.А., Молчанов С.П. и др. // Российские нанотехнологии. 2016. Т. 11. № 7–8. С. 55.
Ионов Д.С., Юрасик Г.А., Антонов А.В. и др. // Химия высоких энергий. 2015. Т. 49. № 3. С. 210.
Ionov D., Yurasik G., Kononevich Y. et al. // Procedia Eng. 2016. V. 168. P. 341.
Katanov I., Kupriyanov A., Ionov D. et al. // CEUR Workshop Proceedings. 2020. V. 2667.
Ионов Д.С., Сажников В.А., Алфимов М.В. // пат. 2649038 Рос. Федерация, опубл. 27.08.2011. Бюл. № 24. 8 с.
Ионов Д.С., Алфимов М.В., Сажников В.А. и др. // пат. 2534729 Рос. Федерация, опубл. 29.03.2018. Бюл. № 10. 10 с.
Espinoza J.D.A., Sazhnikov V., Sabik S. et al. // Procedia Engineering. 2012. V. 47.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий