

Химия высоких энергий, 2022, T. 56, № 3, стр. 215-222
Защитные эффекты при радиолизе подкисленных углеводородных растворов трибутилфосфата
Ю. В. Серенко a, Е. В. Белова a, А. В. Пономарев a, *
a ФГБУН Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, корп. 4, Россия
* E-mail: ponomarev@ipc.rssi.ru
Поступила в редакцию 27.12.2021
После доработки 27.12.2021
Принята к публикации 03.01.2022
- EDN: QSSVPM
- DOI: 10.31857/S0023119322030020
Аннотация
Исследовано влияние радиолиза и пост-радиационного кислотного гидролиза на состав продуктов деградации трибутилфосфата (ТБФ, 30 мас. %) в подкисленном Изопар-М. Показано, что одновременное присутствие азотной кислоты и изо-парафинов в облучаемом растворе оказывает защитное действие по отношению к ТБФ. Основные радиолитические продукты обусловлены нитрованием, нитроксилированием и алкилированием ТБФ. При пост-радиационном термолизе, нитропроизводные и нитраты распадаются с частичной регенерацией ТБФ. Нагрев выше 110°C также провоцирует деалкилирование радиолитических продуктов с образованием легколетучих соединений, что увеличивает пожаро- и взрывоопасность использования облученных экстракционных смесей.
ВВЕДЕНИЕ
Радиационно-химические исследования занимают важное место в выборе экстракционных систем для промышленного разделения радиоизотопов, содержащихся в отработавшем ядерном топливе [1, 2]. Важна не только радиационная стойкость экстрагента и разбавителя, но также термическая, химическая и радиолитическая стабильность накапливающихся продуктов [3]. Основным экстрагентом в PUREX, UREX и CO-EX процессах служит три-н-бутилфосфат (ТБФ). Его деградация обусловлена совокупностью процессов радиолиза, кислотного гидролиза и термолиза [4]. Поэтому для определения эффективности экстракции, факторов разделения радиоизотопов и долговечности рециркуляции ТБФ в составе экстракционных систем, нужен широкий набор данных о радиационно-химических и пост-радиационных превращениях ТБФ в зависимости от последовательности и условий контакта с азотной кислотой. Особую важность представляют исследования, связанные с повышением пожаровзрывобезопасности процесса экстракции [5], а также улучшением гидродинамических и экстракционных показателей растворов ТБФ [6].
Одним из перспективных углеводородных растворителей может служить Изопар-М – смесь изопарафинов с диапазоном кипения 208–257°С. Это коммерчески доступный разбавитель, обладающий повышенной растворяющей способностью в отношении сольватов четырехвалентных актинидов с ТБФ [3]. В настоящей работе исследовано влияние поглощенной дозы ионизирующего излучения и термо-окислительных условий пост-радиационного хранения на механизм превращений и состав экстракционного раствора 30 об. % ТБФ в Изопар-М, насыщенном 3.4–4.0 М HNO3.
МАТЕРИАЛЫ И ЭКСПЕРИМЕНТАЛЬНЫЕ МЕТОДЫ
Реактивы и облучение
Три-н-бутилфосфат (99%, от “ACROS”) очищали от окисляющихся продуктов синтеза путем промывки водным раствором 10 г/дм3 перманганата калия и 15 г/дм3 гидроксида натрия (время контакта 30 мин, соотношение объемов органической и водной фаз 2 : 1). Образующийся осадок MnO2, удаляли промывкой с водным раствором 15 г/дм3 Н2С2О4 и 0.3 М HNO3 (время промывки 1 ч, соотношение объемов органической и водной фаз 2 : 1). Далее продукт дважды (по 30 мин) промывали равным объемом водного раствора 15 г/дм3 NaOH, а затем дистиллированной водой до нейтральной реакции. Очищенный ТБФ растворяли в Изопар-М (от “ExxonMobil”) в объемном соотношении 3 : 7. Далее образцы насыщали азотной кислотой (водный раствор 3.4 или 4.0 М HNO3). Проводили три повторных насыщения по 20 мин при равных объeмах органической и водной фаз. Облучение проводили при 20 ± 3°С в стеклянном цилиндрическом сосуде с гидрозатвором, заполненным водным раствором 4 М HNO3.
Излучателями служили линейный ускоритель УЭЛВ-10-10-С-70 (энергия электронов 8 МэВ, длительность импульса 6 мкс, частота повторения импульсов 300 Гц, средний ток пучка 700 мкА) или γ-установка РХМ-гамма-20 (НПО РАДОН). Электронный пучок сканировался вдоль вертикальной оси сосуда с частотой 1 Гц. Электронно-лучевое воздействие осуществлялось в прерывистом режиме: интервал облучения до дозы 4.7 ± 0.1 кГр (средняя мощность дозы 0.22 кГр/с) чередовался с интервалом остывания образца в течение 10 мин. Мощность дозы на γ-установке составляла 9.4 ± 0.4 кГр/ч. Для дозиметрии использовали сополимер СО ПД(Ф)Р-5/50 (ГСО 7865-2000).
Гидролиз
Облученные образцы либо подвергались высокотемпературному кислотному гидролизу (при 90–170°С, в контакте с 8 или 12 М HNO3), либо хранились 3 года без доступа света при температуре +6°С в пробирках с притертыми пробками. Термолиз проводился в течение 6 ч в 300 мл автоклаве с термостатированием. Выпадения осадка, помутнения или расслоения образцов в процессе пост-радиационного хранения не наблюдалось. Для контроля рН состаренных растворов использовался анализатор HANNA HI 98127 PHEP 4.
Анализ
Анализ группового состава продуктов проводили на ИК-спектрометре IR Prestige-21 (Shimadzu) в однолучевом режиме. Спектры регистрировались с использованием стекол CaF2 и кюветы со свинцовой прокладкой. Измерения проводились с использованием калибровочных кривых: –NO2 – 1556 см–1 (2-нитрооктан), –ОNO2 – 1639 см–1 (1‑о-ктилнитрат), –СООН – 1730 см–1 (миристиновая кислота), –СО – 1721 см–1 (4-метил 2 пентанон), –СООR – 1740 см–1 (гексиловый эфир масляной кислоты).
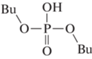
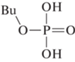
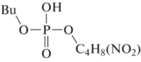
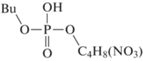
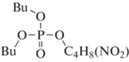
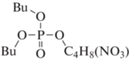
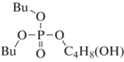
Компонентный состав образцов анализировали с помощью газожидкостного хроматографа ThermoScientific Trace 1310 с моноквадроупольным анализатором ISQ 8000 (ионизация электронным ударом, 70 эВ) и колонкой Thermo (TG-5MS, 15 м · 0.25 мм) с соотношением полидифинилсилоксан : полидиметилсилоксан = 5 : 95. Температура испарителя составляла 275°С, нагрев термостата – от 50 до 320°С, объем пробы 0.5 мкл. Пробы предварительно нейтрализовывались гидрокарбонатом натрия. Идентификацию продуктов проводили по масс-спектрам и индексам удерживания с использованием программ NIST MS Search 2.3 (2017) и ThermoXcalibur 2.2. Обозначения и строение зарегистрированных фосфорсодержащих продуктов приведены в табл. 1, где Bu, R1, R2 и R3 соответствует бутильной и более тяжелым алкильным группам.
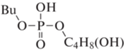
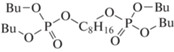
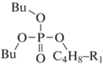
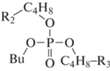
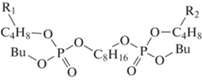
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Радиолиз свежего экстракционного раствора приводит к изменению компонентного состава (рис. 1) и небольшому повышению плотности (от 0.849 до 0.862 кг дм–3 при 2 МГр). Как при электронно-лучевой обработке (рис. 1а), так и при γ‑радиолизе (рис. 1б), доминирующими продуктами являются органические нитропроизводные. Причем их концентрация мало меняется в пост-радиационных процессах (рис. 1в). При дозах до 0.5 МГр радиационно-химические выходы накопления нитропроизводных (–NO2) и нитратов (–NO3) максимальны (до 0.3 мкмоль/Дж). Более высокие дозы снижают эти выходы (до 0.06–0.09 и 0.01 мкмоль/Дж для продуктов нитрования и нитроксилирования соответственно). Также возникают карбоновые кислоты и карбонильные соединения. Прирост их содержания наиболее существенен при дозах выше 0.5 МГр (до 0.03–0.05 мкмоль/Дж). В следовых количествах появляются сложные эфиры. Содержание карбоксильных и карбонильных соединений продолжает увеличиваться и во вторичных, пост-радиационных, процессах.
Рис. 1.
Зависимость группового состава продуктов радиолиза от дозы для свежих образцов при облучении электронным пучком (а) и γ-излучением ((б), 1 МГр), а также состаренных (в) экстракционных растворов после облучения электронным пучком. Для γ-излучения дано относительное содержание фракций.
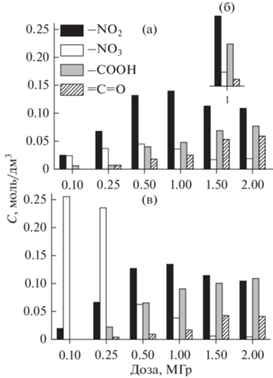
Длительное хранение также влияет на состав образцов, прежде всего, на содержание нитратов (рис. 1в). Их пост-радиационному накоплению способствует высокая остаточная концентрация азотной кислоты и, соответственно, низкое значение рН (рис. 2). Нитраты возникают преимущественно в облученных растворах и малозаметны в растворах, избежавших облучения. Низкое содержание нитратов наблюдается также в образцах, облученных при 1 МГр и подвергнутых последующему термолизу при >110°С (табл. 2). В этих же образцах, наблюдается пониженное содержание нитропроизводных. Ключевой причиной служит низкая термическая стабильность органических нитратов и нитропроизводных, формируемых в процессе радиолиза, хотя при комнатной и более низкой температуре эти соединения сравнительно стабильны.
Таблица 2.
Содержание продуктов в растворе 30% ТБФ в Изопар-М при дозе γ-облучения 1 МГр в зависимости от условий обработки, где [HNO3]0 – концентрация азотной кислоты при предварительном насыщении, t и [HNO3]T – температура и концентрация азотной кислоты при пост-радиационном гидролизе
| [HNO3]0, М | – | 4 | – | ||||
| t, °С | – | – | 110 | 150 | 90 | 90 | 110 |
| [HNO3]T, М | – | – | 12 | 12 | 8 | 12 | 12 |
| Продукт | С, мас. % | ||||||
| Р1 | 2.2 | 5.1 | 4.0 | 4.6 | 4.4 | 3.2 | 3.7 |
| Р2 | 0.05 | 0.23 | 0.17 | 0.22 | 0.14 | 0.03 | 0.15 |
| Р3 | 0 | 3.5 | 0.28 | 0.35 | 0.14 | 0.11 | 0.19 |
| Р6 | 0 | 7.8 | 1.54 | 1.14 | 0.75 | 0.5 | 0.71 |
| Р7 | 0 | 4.0 | 0.49 | 0.29 | 0.24 | 0.11 | 0.25 |
| Р8 | 0.12 | 4.1 | 0.35 | 0.1 | 0.1 | 0.17 | 0.12 |
| Р9 | 0.05 | 0.08 | 0.8 | 0.76 | 1.1 | 0.86 | 0.8 |
| Р10−Р12 | 14.2 | 3.55 | 11.7 | 13.02 | 21.0 | 19.9 | 18.2 |
Эффективное расходование HNO3 обусловлено, в частности, быстрыми, диффузионно-контролируемыми реакциями со свободными электронами, возникающими при ионизации всех компонентов раствора [7]. Причем электроны захватываются как ионами Н+, так и нитратом. В реакции электрона с Н+ образуется менее реакционноспособный радикал ⋅Н. В свою очередь, реакция электрона с нитратом дает ⋅NO2
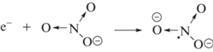
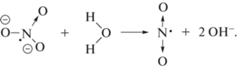
Расходование Н+ в реакции с электроном и ОН– приводит к росту рН и, соответственно, к ослаблению пост-радиационного кислотного гидролиза. Электроны являются наиболее сильными восстановителями. Эффективный захват электронов в первичных радиолитических процессах с кислотой обусловливает снижение вероятности их дальнейшего участия в деградации ТБФ. Однако нитрат также расходуется в реакциях с катион-радикалами (⋅R+) ТБФ и Изопар [7]

Естественно, реакции (1)–(3) ослабляются с ростом поглощенной дозы, т.е. по мере убыли концентрации нитрата-иона.
По сравнению с электроном, радикал ⋅Н восстанавливает нитрат гораздо медленнее [8]. Однако, он также вносит заметный вклад в конверсию нитрата до ⋅NO2 [9], тем самым способствуя последующему доминированию нитрования над нитроксилированием. Но большинство радикалов ⋅Н гибнет в реакциях отрыва водорода от алкильных групп ТБФ и компонентов Изопар. Следствием Н-отрыва является образование широкого ассортимента более громоздких свободных радикалов алкильного типа. Мобильность ⋅NO2 и ⋅О–NO2 обеспечивает им активное участие в рекомбинации с радикалами алкильного типа. В случае локализации радикальных центров на бутильных группах ТБФ, продуктами рекомбинации являются Р3, Р4, Р6 и Р7. Соответственно, легкие радикалы ⋅NO2 и ⋅О–NO2 снижают вероятность рекомбинации алкильных радикалов между собой. В частности, табл. 2 демонстрирует, что по сравнению с неподкисленным образцом, радиолиз предварительно подкисленного раствора дает больше Р1, Р2 и Р8, однако выход алкилированных продуктов Р10−Р12 уменьшается до 4 раз.
Благодаря накоплению радиолитических продуктов, содержание ТБФ и Изопар в неподкисленном растворе при дозе 1 МГр снижается с 36 до 22 и с 64 до 56 мас. % соответственно. В свою очередь, остаточное содержание ТБФ и Изопар при радиолизе подкисленного раствора составляет около 29 и 54 мас. % соответственно. Следовательно, в присутствии HNO3 радиолитическая деградация ТБФ ослабляется, а разложение Изопар несколько усиливается. Это означает, что реакции ⋅NO2 и ⋅О–NO2 с алкильными радикалами, возникающими из Изопар, более конкурентоспособны, чем с радикалами из ТБФ. Таким образом, в присутствии HNO3, компоненты Изопар осуществляют защиту ТБФ от радиолитической деградации.
При старении и термостимулируемом гидролизе, содержание ТБФ изменяется сравнительно мало. Даже после 3 лет хранения, наблюдается почти пропорциональная зависимость концентрации ТБФ от дозы (рис. 2). Это указывает, что главным фактором деградации ТБФ является радиолиз. Благодаря высокой исходной концентрации ТБФ, образование его возбужденных молекул и катион-радикалов может происходить как по механизму прямого действия излучения, так и в результате переноса избыточной энергии и заряда от компонентов углеводородного разбавителя. В катион-радикале TBФ положительный заряд и неспаренный электрон равномерно распределяются по трем атомам кислорода, соединяющим атомы фосфора и углерода, с небольшим участием связей C–C и атомов углерода в α-положении [7]. Это делает вероятным разрыв связи Р–О с элиминированием бутокси-радикала
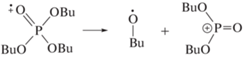
Нейтрализация остаточного катиона ионом ОН– приводит к образованию стабильного Р1. Распад возбужденных молекул ТБФ может приводить как к отщеплению бутил-, так и бутокси-радикалов. Образование анион-радикала происходит в результате захвата электрона молекулой ТБФ, однако роль этого процесса мала из-за преимущественного захвата электронов азотной кислотой. В анион-радикале отрицательный заряд локализован на кислороде, а радикальный центр на атоме фосфора. Происходит значительное ослабление связей P–O и C–O, что облегчает удаление бутокси- или бутил-радикала, однако образование бутокси-радикала более вероятно [7]
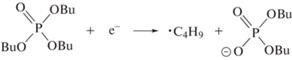
Нейтрализация катион-радикалов и анион-радикалов ТБФ может служить важным источником Р1:
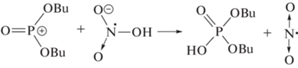
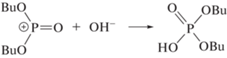
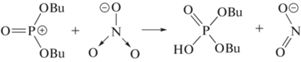
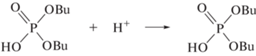
Другой путь образования Р1 реализуется при взаимодействии алкокси-радикалов, получаемых при радиолитическом дебутилировании ТБФ, с углеводородными молекулами (R–H)
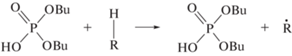
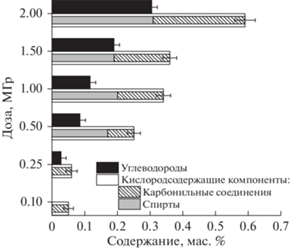
Бутокси радикал, образуемый, например, в реакции (4), подобно другим алкокси радикалам [10, 11], обладает способностью отрывать Н атом от алкильных групп. Соответственно, бутокси радикал служит главным предшественником 1-бутанола, доминирующего среди легких продуктов радиолиза. Доля 1-бутанола среди совокупности легких спиртов составляет не менее 90 мас. %, а средний наблюдаемый выход, измеренный в состарившемся растворе – 0.015 ± 0.005 мкмоль/Дж. Хотя фракции карбонильных соединений, спиртов и углеводородов сопоставимы между собой (рис. 3), доля 1-бутанола от 3 до 16 раз выше, чем любого другого легкого продукта. Среди сравнительно легких алканов в облученных образцах доминирует н-октан. Очевидно, он представляет собой продукт димеризации бутильных радикалов, возникающих, в частности, в реакции (2). Н-октан и бутанол образуются только на стадии радиолиза, т.е. в радикальных процессах. В свою очередь, пост-радиационный гидролиз образует ксилолы и этилбензол, количество которых монотонно увеличивается с повышением температуры в диапазоне 110–170°С. Соответственно, н-бутанол и н-октан могут служить индикаторами вклада радиолиза, а ксилолы и этилбензол – вклада термолиза, в суммарное разложение исследованных растворов.
Рис. 3.
Влияние дозы на содержание легких органических продуктов (легче октана) в состарившихся образцах.
Наряду с радикалами ⋅Н, в образовании алкильных радикалов могут участвовать другие легкие радикалы, в частности, ⋅СН3 и ⋅ОН, а также алкокси радикалы. Образование ⋅Н и ⋅СН3 неизбежно при радиолизе алканов, входящих в состав Изопар-М [12]. В свою очередь, радикалы ⋅ОН могут возникать при прямом и косвенном действии излучения на растворенную воду или спирты. Н-отрыв возможен в различных положениях молекулы алкана или бутильной группы ТБФ [12, 13]. Например, при распаде возбужденных молекул ТБФ, отщепление вероятнее всего происходит в γ-положении относительно атома кислорода [14]. Разнообразие строения алкильных радикалов, соответственно, приводит к разнообразию продуктов их рекомбинации (табл. 1, рис. 5). Очевидно, радикальные продукты Н-отрыва от бутильных групп молекул дибутилфосфата, являются предшественниками Р3–Р5, а радикал (⋅TBP), сформированный Н-отрывом от ТБФ, – предшественником продуктов Р6–Р12. Выход радикалов ⋅NO2 существенно выше, чем ⋅NO3 и, поэтому, доля нитропроизводных выше, чем доля нитратов (рис. 2а). В свою очередь, из-за преобладания ТВФ над Р1, доля Р6 и Р7 в продуктах нитрования от 4 до 7 раз выше, чем доля Р3 и Р4. Фракция продуктов Р3–Р5 в состарившихся образцах относительно невелика – 0.2–0.4 мас. % для Р3 + Р4 и 0.2% для Р5.
Рис. 4.
Влияние поглощенной дозы на содержание фосфор-содержащих продуктов Р1, Р6–Р9 в состарившихся образцах.
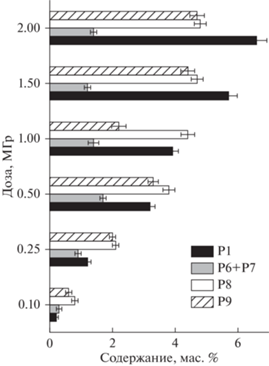
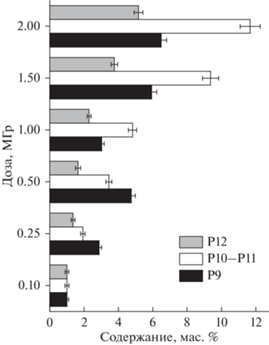
Продукт Р9 (рис. 4 и 5), очевидно, образуется в результате димеризации ⋅TBP. Другие высокомолекулярные продукты формируются несколькими путями. В частности, радиолиз Изопар-М служит источником непредельных углеводородов, которые участвуют в образовании Р10–Р12 (рис. 5). Присоединение короткого алкильного радикала к алкену дает более длинный алкильный радикал [8, 12], например
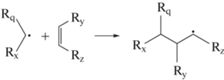
где длина радикалов (q, x, y и z) может быть различной, прежде всего, из-за разнообразия углеводородных компонентов в Изопар-М. Удлинение углеводородного скелета может также происходить в результате ион-молекулярного взаимодействия – присоединения первичного карбокатиона к молекуле алкена с последующей нейтрализацией удлиненного карбокатиона. Процесс образования Р10–Р12 может быть длительным из-за сравнительно низкой скорости укрупнения радикалов и затрудненной комбинации громоздких радикалов друг с другом. Во фракции алкил-бутилфосфатов Р10 и Р11 (m/z 379–564) доминируют соединения, где добавленная алкильная группа содержит 14‒15 атомов С. Среди димеров Р12 (m/z 600–1000) преобладают соединения с мольной массой 740–780. Независимо от поглощенной дозы, общая доля тяжелых фосфорсодержащих продуктов Р9–Р12 в состаренных образцах составляет 31 ± 4% от суммы всех фосфорсодержащих продуктов.
Естественно, радиолитическая деградация азотной кислоты оказывает ключевое влияние на эффективность пост-радиационных гидролитических процессов. В частности, гидролиз вносит основной вклад в образование Р5 и Р8. При дозе 1 МГр и выше, когда рН раствора во много раз выше, чем в необлученном образце, гидролиз мало меняет соотношение между продуктами, достигнутое на стадии радиолиза. Однако при более низких дозах (рис. 1), а также при повышении температуры и добавлении кислоты на пост-радиационной стадии (табл. 2), гидролиз эффективен. Причем, высокотемпературный кислотный гидролиз при >110°C приводит к эффективному разложению радиолитических нитропроизводных и нитратов, в том числе, к частичной регенерации ТБФ. В облученных образцах без стадии последующего термолиза обнаруживается около 1–1.5 мас. % легколетучих продуктов, включая н‑бутан, н-октан и н-бутанол. В свою очередь, при температурах выше 110°C, пост-радиационный термолиз приводит к росту выхода легколетучих продуктов – вплоть до 16–17 мас. % при 170°C. Важную роль в этом эффекте играют процессы деалкилирования компонентов облученного раствора.
ЗАКЛЮЧЕНИЕ
Радиолитическая деградация ТБФ в его подкисленных смесях с алканами складывается из множества процессов, в которых активное участие принимают все компоненты раствора. Определяющее значение играют реакции азотной кислоты первичными продуктами ионизации – электронами и катион-радикалами. Тем самым, снижается вероятность реакций этих первичных интермедиатов с ТБФ. Кроме того, радикалы ⋅NO2 и ⋅О–NO2 рекомбинируют с алкильными радикалами, образуемыми из Изопар-М, что также ослабляет воздействие на ТБФ. При этом реакции радикалов ⋅NO2 и ⋅О–NO2 с радикалами ТБФ приводят к образованию нестабильных нитро- и нитрокси- производных ТБФ, которые склонны к распаду при нагреве с частичной регенерацией ТБФ. В отсутствие кислоты, разбавитель более эффективно участвует в образовании продуктов радиолитической деградации ТБФ и, тем самым, не проявляет функций радиационной защиты. Однако совместное присутствие кислоты и разбавителя значительно уменьшает радиолитическую деградацию ТБФ, причем основной вклад в защитный эффект связан с присутствием кислоты.
Дебутилирование ТБФ с образованием кислых фосфатов Р1 и Р2 происходит как в результате радиолиза, так и в результате гидролиза. Причем по мере роста дозы, пост-радиационные гидролитические процессы приобретают малое значение по сравнению с радиолитическими процессами. Пост-радиационный нагрев образцов способствует гидролитическому образованию бутилфосфатов P1 и P2, в то время как образование N-содержащих продуктов и оксигенатов в основном определяется концентрацией кислоты. Более того, продукты радиолитической конверсии ТБФ более чувствительны к нагреву, чем исходный ТБФ. Образование дибутилового эфира, ксилолов и этилбензола может служить специфическим индикатором вклада термолиза в разложение раствора. Индикаторами относительного вклада радиолиза как в подкисленных, так и в нейтральных растворах ТБФ, могут служить н-бутан, н-бутанол, н-октан и фосфорсодержащие продукты P9–P12.
Список литературы
Pearson J., Nilsson M. // Solvent Extraction and Ion Exchange. 2014. T. 32. C. 584. https://doi.org/10.1080/07366299.2014.924305
Aneheim E., Ekberg C., Fermvik A., Foreman M., Grűner B., Hájková Z., Kvičalová M. // Solvent Extraction and Ion Exchange. 2011. T. 29. C. 157. https://doi.org/10.1080/07366299.2011.539462
Dzhivanova Z., Kadyko M., Smirnov A., Belova E. // J. Radioanalytical and Nuclear Chemistry. 2019. T. 321. C. 439. https://doi.org/10.1007/s10967-019-06593-8
Peterman D., Mincher B., Riddle C., Tillotson R. // Office of Scientific and Technical Information (OSTI) (10.2172). 2010. https://doi.org/10.2172/993164
Belova E., Egorov G., Nazin E. // Radiochemistry. 2000. V. 42. P. 238.
Mincher B., Modolo G., Mezyk S. // Solvent Extraction and Ion Exchange. 2009. V. 27. P. 1. https://doi.org/10.1080/07366290802544767
Serenko Yu.V., Yudin N.V., Gritcenko R.T., Rodin A.V., Belova E.V., Ponomarev A.V. // Radiat. Phys. Chem. 2021. V. 185. P. 109495. https://doi.org/10.1016/j.radphyschem.2021.109495
Woods R., Pikaev A. // Applied Radiation Chemistry. Radiation Processing. Wiley, N.Y. 1994.
Ponomarev A.V., Bludenko A.V., Makarov I.E. // Mendeleev Communications. 2002. V. 12. P. 92. https://doi.org/10.1070/MC2002v012n03ABEH001583
Ponomarev A.V., Vlasov S.I., Kholodkova E.M. // High Energy Chem. 2019. V. 53. P. 314. https://doi.org/10.1134/S0018143919040106
Ponomarev A.V., Vlasov S.I., Kholodkova E.M., Chul-kov V.N., Bludenko A.V. // Radiat. Phys. Chem. 2019. V. 165. P. 108405. https://doi.org/10.1016/j.radphyschem.2019.108405
Metreveli A.K., Ponomarev A.V. // High Energy Chem. 2016. V. 50. P. 254. https://doi.org/10.1134/S0018143916040135
Lesage D., Virelizier H., Jankowski C.K. // Spectroscopy. 1997. V. 13. P. 275. https://doi.org/10.1155/1997/565194
Wang F., Horne G.P., Pernot P., Archirel P., Mostafavi M. // J. Phys. Chem. B. 2018. V. 122. P. 7134. https://doi.org/10.1021/acs.jpcb.8b03715
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий