

Химия высоких энергий, 2023, T. 57, № 1, стр. 3-8
Спектрально-люминесцентное исследование кислотно-основного равновесия 5- и 6-аминоурацила в водных растворах
С. С. Остахов a, Р. Р. Каюмова a, А. А. Ахияров a, С. П. Иванов a, С. Л. Хурсан a, *
a Уфимский институт химии УФИЦ РАН
450054 Уфа, просп. Октября, 71, Россия
* E-mail: khursansl@anrb.ru
Поступила в редакцию 14.07.2022
После доработки 02.09.2022
Принята к публикации 04.09.2022
- EDN: DCKNTA
- DOI: 10.31857/S0023119323010102
Аннотация
Исследованы спектрально-флюоресцентные характеристики 5‑аминоурацила (5AU) и 6‑аминоурацила (6AU) в нейтральных и щелочных водных растворах. С помощью теории функционала плотности показано, что урацилы преимущественно диссоциируют по N(1)–H связи. Флуоресцентным (ФЛ) методом определены константы кислотно-основного равновесия: рКа1(5AU) = 9.4, рКа1(6AU) = 8.95. Сделан вывод, что ультракороткое время жизни возбужденных синглетов 5AU и 6AU препятствует измерению константы кислотно-основного равновесия урацилов в электронно-возбужденном состоянии.
ВВЕДЕНИЕ
С середины XX века известно, что природные урацилы, являющиеся компонентами нуклеиновых кислот [1], принимают участие в их репликации для передачи генетической информации. Однако воздействие различных факторов (УФ-радиация, химические вещества, вирусные инфекции и др.) может приводить к нарушению в репликации ДНК, что зачастую обусловлено образованием редких таутомерных форм нуклеиновых кислот [1], а также их анионов [2].
В щелочных водных растворах производные урацила выступают как слабые двухосновные кислоты [3]. Диссоциация может происходить как по N(1)–H, так и по N(3)–H связи урацилов и зависит от природы заместителей в 5- и 6-положениях пиримидинового кольца [4]. Диссоциацию по первой ступени с образованием анионов AN1 и AN3 можно представить следующими равновесиями:
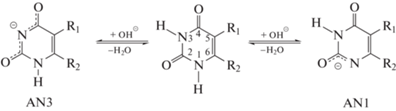
Общеизвестно, что константа диссоциации pKa1 является термодинамической характеристикой этих равновесий. Для 5- и 6-замещенных производных урацила величины pKa1 в водных растворах при 298 К изменяются в широком интервале от pKa1 = 5.3 у 5‑нитроурацила [5] до pKa1 = = 9.8 у тимина [6].
На сегодняшний день, спектрофотометрическое (СФ) и потенциометрическое титрование (ПТ) являются наиболее используемыми методами экспериментального определения pKa [7–9], наряду с квантово-химическими методами расчета констант диссоциации [9, 10]. Однако применение спектрофотометрии при определении pKa некоторых производных урацила ограничивается их низкой растворимостью в водных растворах и относительно малыми величинами коэффициентов молярной экстинкции. В частности, исследуемые в настоящей работе 5‑аминоурацил (5AU) и 6‑аминоурацил (6AU) довольно плохо растворимы в воде: согласно [11] предельная растворимость 5AU и 6AU при 25°C составляет 0.0047 моль/л, в работе [12] для 6AU приведено значение 0.006 моль/л. В этой связи возникает необходимость поиска альтернативных методов исследования кислотно-основных равновесий в урацилах, требующих меньших концентраций исследуемого вещества в растворе.
Ранее, основываясь на значительном увеличении квантового выхода φ флюоресценции (ФЛ) при переходе от молекулярной к анионной форме 5‑фторурацила [13], нами был предложен высокочувствительный спектрально-люминесцентный метод определения pKa1, успешно апробированный на ряде водных растворов пиримидиновых оснований [14].
Целью настоящей работы является сравнительное спектрально-люминесцентное исследование кислотно-основных равновесий 5- и 6‑аминоурацилов в водных растворах.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
6-Аминоурацил (873-83-6, “Реахим”) марки “ч”, 5-аминоурацил (932-52-5, “Реахим”) марки “хч”, КОН (1310-58-3, “Реахим”) марки “хч” и соляную кислоту 0.1 моль/л использовали без предварительной очистки. Спектрально-люминесцентные измерения водных растворов 5AU и 6AU проводили в интервалах рН 5.2–11.9 и 5.8–11.8 соответственно (298 K). Для определения рН использовался рН-метр рН-150 МИ с комбинированным стеклянным электродом ЭСК-10307. Калибровка электродов проводилась с помощью стандартных буферных растворов. Растворы исследуемых соединений получены в бидистилированной воде.
Электронные спектры поглощения записывали на спектрофотометре Shimadzu UV-1800. Скорректированные спектры ФЛ регистрировали на спектрофлюориметре СМ-2203. Квантовые выходы φ флюоресценции 5AU и 6AU определяли по известной методике [15] с использованием внешнего стандарта триптофана (Trp) по уравнению:
(1)
${{{{\varphi }}}_{i}} = {{{{{{\varphi }}}_{{{\text{Trp}}}}}({{S}_{i}}{{A}_{{{\text{Trp}}}}})} \mathord{\left/ {\vphantom {{{{{{\varphi }}}_{{{\text{Trp}}}}}({{S}_{i}}{{A}_{{{\text{Trp}}}}})} {({{S}_{{{\text{Trp}}}}}{{A}_{i}})}}} \right. \kern-0em} {({{S}_{{{\text{Trp}}}}}{{A}_{i}})}},$где φi – квантовый выход ФЛ, φTrp – квантовый выход ФЛ Trp (φTrp = 0.14, Н2О, pH 7.0, 298 K) [16], Si и Ai – светосумма под полосой ФЛ и оптическая плотность поглощения исследуемых растворов на длине волны возбуждающего света λex, соответственно, STrp и ATrp – аналогично для триптофана.
Квантово-химические расчеты выполнены с использованием программного пакета Gaussian 09, Rev.C.01 [17]. Визуализацию и обработку молекул осуществляли с использованием программы ChemCraft [18]. Полная оптимизация геометрических параметров, расчет частот, учет специфической сольватации и расчет энергии Гиббса выполнены с применением теории функционала плотности в приближении обменного TPSS τ-зависимого градиентно-скорректированного функционала [19]. Использовали базисный набор тройного валентного расщепления, дополненный поляризационными функциями d- и p-типа, а также набором диффузных функций 6-311+G(d,p) [20]. Неспецифическую сольватацию в воде оценивали с использованием поляризационно-континуальной модели Томази (РСМ) [21].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
На рис. 1 представлены спектры ФЛ 6AU при рН 6.5 и 11.8, приведенные к оптической плотности на длине волны возбуждающего света.
Рис. 1.
Спектры ФЛ 6-аминоурацила при рН 6.5 (1) и 11.8 (2). Условия: λex = 260 нм, с [6AU] = = 5 × 10–5 моль/л, l = 1 см, 298 К.
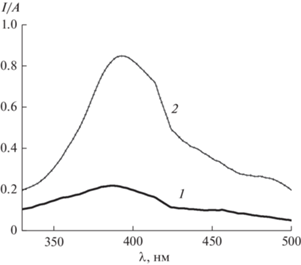
При увеличении рН растворов 6AU в интервале 5.8–11.8 спектральный состав и смещение максимума ФЛ (λem = 392 нм) не регистрируется (рис. 1, табл. 1). Квантовый выход ФЛ при рН 6.5 составляет φ = 0.3 × 10–4 и соответствует данным [22]. В то же время, при изменении рН наблюдается рост квантового выхода ФЛ 0.3 × 10–4 (рН 6.5) до 1.1 × 10–4 (рН 11.8) (табл. 1). Ранее в работе [13] квантово-химическим методом ядерно-независимых химических сдвигов [23, 24] нами было показано, что увеличение квантового выхода ФЛ анионов 5‑фтор-урацила по сравнению с его молекулярной формой связано с ростом величины константы магнитного экранирования, соответствующей увеличению сопряжения в пиримидиновом цикле аниона.
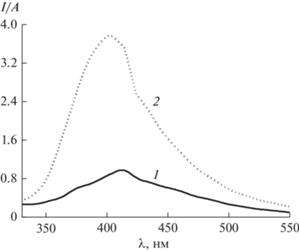
Спектры ФЛ 5AU при рН 6.7 и 11.9 представлены на рис. 2. Максимум ФЛ 5AU в нейтральных водных растворах смещен в длинноволновую область спектра (414 нм) по сравнению с 6AU и соответствует спектральному диапазону, приведенному в работах [22, 25]. В отличие от 6AU, для 5AU при увеличении рН среды наряду с ростом φ ФЛ регистрируется сдвиг максимума ФЛ до 402 нм. По-видимому, спектрально-люминесцентные характеристики 5AU и 6AU как в нейтральных [22, 25], так и в щелочных водных растворах определяются значительными различиями в их временах жизни (τ) люминесценции [22]. На основании TD-DFT расчетов сделан вывод [22], что эти различия связаны с различной пространственной конфигурацией их молекул в электронно-возбужденном состоянии, разница в величинах времен жизни ФЛ довольно существенна: 6AU (τ < 10–13 с), 5AU (τ ≈ 10–12 с) [22].
Рис. 2.
Спектры ФЛ 5-аминоурацила при рН 6.7 (1) и 11.9 (2). Условия: λex = 280 нм, с [5AU] = = 5 × 10–5 моль/л, l = 1 см, 298 К.
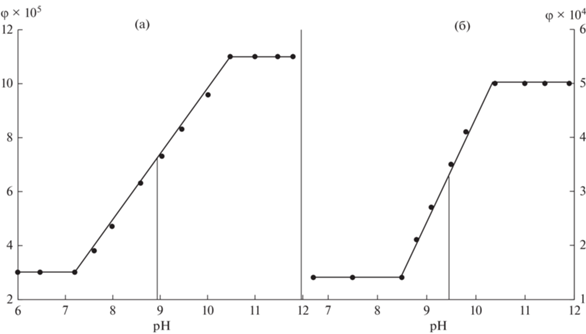
Как было показано выше, смещение кислотно-основного равновесия в растворе как 6AU, так и 5AU при изменении рН среды влияет на φ ФЛ (табл. 1) и, таким образом, позволяет определить их pKa1 в водных растворах.
Для 6AU константа кислотно-основного равновесия, определенная графическим методом [26] из зависимостей φ ФЛ от рН (рис. 3а) составляет 8.95. Значения pKa1 5AU определяли аналогично, из зависимости квантового выхода φ ФЛ от рН среды (рис. 3в). Найдено, pKa1 = 9.4. Полученные величины в пределах погрешности измерений совпадают с данными, полученными СФ и ПТ методами, а также с теоретическими оценками pKa высокоуровневыми квантово-химическими методами (табл. 2).
Рис. 3.
Зависимость φ ФЛ от рН водных растворов 6‑аминоурацила (а) и 5‑аминоурацила (б). Условия: 5 × 10–5 моль/л, 298 К; λex (6АU) = 260 нм, λex (5АU) = 280 нм.
Диссоциация какой связи, N(1)–H или N(3)‒H ответственна за наблюдаемые величины pKa1? Ответ на данный вопрос дает относительная стабильность соответствующего аниона. В работе [10] найдено, что AN1 5AU термодинамически стабильнее AN3 всего на 2.4 кДж/моль, что соответствует содержанию анионов в водном растворе при комнатной температуре в соотношении 0.72 : 0.28. Сведения об относительной стабильности анионов 6AU отсутствуют. Мы провели DFT расчеты сольватных кластеров AN1 ⋅ 9H2O и AN3 ⋅ 9H2O (рис. 4) и установили, что AN1 анион 6AU существенно стабильнее AN3, причем разница свободных энергий Гиббса анионов составляет 20.0 кДж/моль, т.е. в водном растворе 6AU присутствует исключительно анион AN1.
Рис. 4.
Строение девятиводных комплексов анионов 6‑аминоурацила. Расчет в приближении TPSSTPSS/6-311 + + G(d,p) + IEFPCM(SMD).
Физическая природа наблюдаемых спектрально-люминесцентных закономерностей достаточно очевидна: при установлении кислотно-основного равновесия, положение которого определяется pH среды, регистрируется интенсивность ФЛ, зависящая от содержания в растворе урацила и его ионизированной формы. Однако, наряду с “обычным” кислотно-основным равновесием, в электронно-возбужденном состоянии может протекать депротонизация без потери энергии возбуждения за время жизни ФЛ исследуемых соединений, характеризуемая величиной $pK_{{{\text{a1}}}}^{*}$ [15]. Для ряда люминофоров – органических X–H кислот (X = C или гетероатом) – определенные из спектрально-люминесцентных измерений значения $pK_{{{\text{a1}}}}^{*}$ оказались существенно ниже, чем pKa1 в основном состоянии [32]. Разность между величинами pKa1 и $pK_{{{\text{a1}}}}^{*}$ с хорошей степенью точности пропорциональна разнице максимумов ФЛ кислой и щелочной форм [15]. Для 6AU и 5AU эта разница близка к нулю (рис. 1 и 2), что свидетельствует о близости показателей кислотности урацилов в основном и возбужденном состоянии и ставит вопрос о природе исследуемого нами равновесия. Для ответа на этот вопрос следует принять во внимание, что, в отличие от большинства органических люминофоров, времена жизни ФЛ которых составляют τ = 10—8—10–9 с [15], для пиримидиновых оснований характерно ультракороткое время жизни возбужденных состояний, τ = 10–12–10–13 с [22, 33]. Таким образом, наиболее вероятно, что ФЛ урацилов определяется сверхбыстрыми процессами S1 → S0 конверсии, превалирующими над скоростью фотодепротонизации с сохранением возбуждения на анионе.
На основании полученных результатов, ФЛ метод может быть предложен в качестве альтернативного СФ и ПТ титрованию при определении констант рКа1 кислотно-основного равновесия органических люминофоров (в частности, урацилов) с ультракороткими временами жизни в электронно-возбужденном состоянии (τ ≈ 10–12—10–13 с) в водных растворах.
Список литературы
Watson J.D., Crick F.H.C. // Nature. 1953. V. 171. № 4361. P. 964.
Sowers L.C., Shaw B.R, Veigl M.L., Sedwick W.D. // Mutat. Res. 1987. V. 177. P. 201.
Abdrakhimova G.S., Ovchinnikov M.Yu., Lobov A.N., Spirikhin L.V., Ivanov S.P., Khursan S.L. // J. Phys. Org. Chem. 2014. V. 27. P. 876.
Abdrakhimova G.S., Ovchinnikov M.Yu., Lobov A.N., Spirikhin L.V., Khursan S.L., Ivanov S.P. // J. Mol. Struct. 2018. V. 1158. P. 51.
Privat E.S., Sowers L.C. // Mutat. Res. 1996. V. 354. P. 151.
Wittenberg E. // Chem. Ber. 1966. V. 99. P. 2391.
Berens K., Shugar D. // Acta Biochim. Polonica. 1963. V. 10. № 1. P. 25.
Wempen I., Fox J.J. // J. Am. Chem. Soc. 1964. V. 86. № 12. P. 2474.
Ilyina M.G., Khamitov E.M., Ivanov S.P., Mustafin A.G., Khursan S.L. // J. Phys. Chem. A. 2017. V. 122. P. 341.
Ilyina M.G., Khamitov E.M., Ivanov S.P., Khursan S.L. // Comput. Theor. Chem. 2016. V. 1078. P. 81.
Zielenkiewicz W., Poznanski J., Zielenkiewicz A. // J. Solution Chem. 2000. V. 29. P. 757.
Ахияров А.А., Иванов С.П. // Вестник Башкирского университета. 2021. Т. 26. С. 631.
Ostakhov S.S., Sultanbaev M.V., Ovchinnikov M.Yu., Kayumova R.R., Khursan S.L. // High Energy Chem. 2017. V. 51. P. 108.
Ivanov S.P., Ostakhov S.S., Abdrakhimova G.S., Akhiyarov A.A., Khursan S.S. // Biophys. Chem. 2020. V. 266. 106432.
Паркер С. Фотолюминесценция растворов / Под ред. Васильева Р.Ф. М.: Мир, 1972. 510 с.
Tatischeff I., Klein K. // Photochem. Photobiol. 1975. V. 22. P. 221.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery Jr. J. A., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. // Gaussian 09, Revision D.1, Gaussian, Inc., Wallingford CT. 2009.
Andrienko G.A. // Chemcraft. Version 1.8 (build 489). URL: http://www.chemcraftprog.com.
Tao J., Perdew J.P., Staroverov V.N., Scuseria G.E. // Phys. Rev. Lett. 2003. V. 91. 146401.
Krishnan R., Binkley J.S., Seeger R., Pople J.A. // J. Chem. Phys. 1980. V. 72. P. 650.
Tomasi J., Mennucci B., Cammi R. // Chem. Rev. 2005. V. 105. P. 2999.
Bányász Á., Karpati S., Mercier Ya., Reguero M., Gustavsson T., Markovitsi D., Improta R. // J. Phys. Chem. B. 2010. V. 114. P. 12708.
Jimenez-Halla J.O.C., Matito E., Robles J., Sola M. // J. Organomet. Chem. 2006. V. 691. № 21. P. 4359.
Stanger A. // J. Org. Chem. 2005. V. 71. № 3. P. 883.
Gustavsson T., Improta R., Bányász Á., Vaya I., Markovitsi D. // J. Photochem. Photobiol. A. 2012. V. 234. P. 37.
Васильев В.П. Аналитическая химия. Ч. 1. Гравиметрический и титриметрические методы анализа. М.: Высшая школа, 1989. 320 с.
Langen P., Etzold G., Barwolff D., Preussel B. // Biochem. Pharm. 1967. V. 16. P. 1833.
Stankevich E.I., Popelis Yu.Yu., Grinshtein E.E., Ozola A.Ya., Dubur G.Ya. // Khim. Geterot. Soed. 1970. V. 6. P. 122.
Barlin G.B. // J. Chem. Soc. B. 1971. P. 1425.
Hatada T., Mamori M., Nakashima K., Yoshimura M. // Yakugaku Zasshi. 1978. V. 98. № 5. P. 668.
Mori M., Teshima S., Yoshimoto H., Fujita S., Taniguchi R., Hatta H., Nishimoto S. // J. Phys. Chem. B. 2001. V. 105. № 10. P. 2070.
Барлтроп Дж., Койл Дж. Возбужденные состояния в органической химии / Под ред. Кузьмина М.Г. М.: Мир, 1978. 446 с.
Gustavsson T., Bányász Á., Lazzarotto E., Markovitsi D., Scalmani G., Frisch M.J., Barone V., Improta R. // J. Am. Chem. Soc. 2006. V. 128. № 2. P. 607.
Дополнительные материалы отсутствуют.
Инструменты
Химия высоких энергий