

Физикохимия поверхности и защита материалов, 2021, T. 57, № 6, стр. 609-617
Формирование металл-оксидных гетероструктур при низкотемпературном окислении сплавов на основе системы Fe–18Cr
В. А. Котенев *
Федеральное государственное бюджетное учреждение науки Институт физической химии и электрохимии
им. А.Н. Фрумкина РАН
119071 Москва, Ленинский пр-т, 31, корп. 4, Россия
* E-mail: m-protect@mail.ru
Поступила в редакцию 12.05.2021
После доработки 14.06.2021
Принята к публикации 14.07.2021
Аннотация
Методами спектральной эллипсометрии, Раман-спектроскопии, Оже-электронной спектроскопии с послойным травлением исследованы кинетика окисления и распределение химического состава по глубине металл-оксидного слоя при низкотемпературном (300°С) газовом окислении сплава Fe–18Cr при разных степенях вакуумирования системы (0.0001–1 Торр). Зарегистрировано сильное активирующее действие давления кислорода (0.02 Торр), соответствующего активно-пассивному переходу с экстремумом скорости оксидирования. Оно сопровождается перераспределением химического состава по глубине поверхностного оксида, связанном как с прямым окисление железа и хрома на границе оксид–газ, так и с обменными реакциями (типа реакции замещения) оксидов железа и хрома в объеме оксидного слоя. Первоначально на поверхности в результате прямого окисления быстро вырастает структурно-чувствительный слой окислов железа и хрома в количестве, недостаточном для пассивации, а далее при диффузии хрома из металла и реакции замещения хрома с окислами железа в объеме слоя окислов железа формируются оксиды хрома типа полуторной окиси хрома и хромистой шпинели. При пассивации сплава, падение скорости роста оксида при дальнейшем повышении давления кислорода связано с локализацией формирования защитных окислов хрома в тонком неоднородном слое при перемещении фронта реакции магнетита с хромом от границы раздела оксид–газ к границе металл–оксид.
ВВЕДЕНИЕ
Контроль и управление твердофазными реакциями синтеза новых металл-оксидных наноматериалов становится в настоящее время одной из насущных проблем современных нанотехнологий. Реакции этого типа лежат в основе многочисленных процессов химической промышленности, металлургии, микроэлектроники и др. [1–7], связанных с формированием и трансформацией сложных металл-оксидных наноструктур.
Состав и структура многослойных металл-оксидных слоев, как правило, зависят от состава внешней среды, состава оксидируемого металла (сплава), температуры окисления [8]. Существующий опыт по исследованиям процессов оксидирования сплавов на основе системы Fe–Cr позволяет заключить, что в состав оксидного слоя при изотермическом оксидировании сплавов на основе железа и хрома входит железо-хромистая шпинель с формулой Fe(Fe2 –xCrx)O4 или твердый раствор Fe2 –xCr2O3 (0 < х < 2), а также окислы железа [9, 10]; последовательность слоев в окалине, образованной на воздухе, следующая: Fe–Cr/шпинель/Fe3O4/Fе2O3. Согласно данным [11], ниже 500–600°С на сталях аналогичного состава образуется оксидный слой, основную часть которого составляет твердый раствор Fe2 –xCr2O3 (0 < х < 2), т.е. полупроводник переменного состава, где x меняется в зависимости от температуры и времени окисления. В зависимости от величины x ромбоэдрический Fe–Cr оксид способен проявлять как n-, так и p-тип проводимости [12]. Внешний тонкий слой оксида составляет α-Fe2O3 – оксид n-типа с незначительным отклонением от стехиометрии, доминирующими дефектами которого являются анионные вакансии [13]. Очевидно, многокомпонентный металл-оксидный слой, в котором тип проводимости меняется по его толщине, представляет несомненный интерес в микро- и наноэлектронике при создании новых гетероструктур [6].
Однако вышеописанные примеры относятся в основном к случаям окисления при средних и высоких температурах (500°С и выше), чему посвящено значительное количество коррозионных работ по толстослойному термооксидированию нержавеющих сталей с получением, как правило, весьма дефектной окалины11, и что не позволяет однозначно предсказывать свойства гораздо более совершенных оксидов, формируемых при гораздо более низких температурах. В этой связи исследование процессов оксидирования при пониженных температурах (до 300–500°С) и различных давлениях окислителя может предоставить большие возможности для мягкого (низкотемпературного) роста металл-оксидных гетероструктур на многокомпонентных сплавах. Особый интерес в последнее время представляют гетероструктуры с изменяющимся по глубине составом тонкого поверхностного металл-оксидного слоя (электроника, катализ, “фотонно-кристаллическая” оптика и др.).
В отличие от высокотемпературных “керамических” реакций, рассматриваемых в рамках термодинамических моделей оксидирования типа Вагнеровской [8], низкотемпературные топохимические реакции позволяют сформировать метастабильные фазы, осуществляя кинетический, а не термодинамический контроль, используя различие скоростей диффузии в твердом состоянии разных частиц, поскольку они соответствуют различиям в реакционной способности [2–4]. Как правило, во многих сложных оксидах один класс ионов, будь то катион или анион, часто значительно более подвижен при данной температуре. Как следствие, эти подвижные частицы (катионы или анионы) могут быть встроены или удалены из материнской фазы при низкой температуре в условиях, в которых окружающая решетка является фактически неизменной, с образованием метастабильных фаз, которые обычно недоступны при высокотемпературном синтезе. Твердофазные реакции этого типа (например, реакции замещения) можно сопоставить с “топохимическими”, которые, в отличие от большинства высокотемпературных реакций с трансформацией фаз, сохраняют основную структурную топологию материнской фазы.
Очевидно, наноматериалы и наноструктуры, где формирование градиента состава осуществляется за счет топохимических реакций с образованием метастабильных фаз, могут обладать выраженными функциональными свойствами (полупроводниковыми, каталитическими, адсорбционными, фотонно-кристаллическими и др.), существенно превосходящими свойства простых оксидных слоев. Особый интерес представляют электронные свойства металл-оксидных гетероструктур. В частности, в работе [6] был описан новый 3-мерный металл-оксидный мемристор на основе магнетит-гематитовых гетероструктур.
Ранее при исследовании окисления железа и бинарных сплавов железо-хром был зарегистрирован эффект низкотемпературноой пассивности (или активно-пассивного перехода) в условиях вакуумирования при росте в системе парциального давления окислителя, скорость окисления металла сначала растет, а по достижении критической степени вакуумирования начинает падать [15]. В отличие от явления высокотемпературной пассивности [16], которое проявляется для хромистых сталей при температурах свыше 1000°С и объясняется исходя из термодинамической модели Вагнера [17], явление низкотемпературной пассивности сплавов железо-хром проявляется при достаточно умеренных температурах (200–300°С и выше) и может быть рассмотрено исходя из кинетической модели формирования системы окисных фаз в объеме растущего оксидного слоя за счет топохимических реакций обменного типа. Очевидно, развиваемые в последнее время представления о метастабильности фаз и структур в ходе низкотемпературных топохимических реакций могут быть полезными при разработке новых металл-оксидных гетероструктур, формируемых при мягком оксидировании многокомпонентных сплавов.
В данной работе с использованием комплекса физических и структурных методов (методов спектральной эллипсометрии, Раман-спектроскопии, Оже-электронной спектроскопии) исследовано перераспределение химического состава по глубине оксидного слоя при низкотемпературном (300°С) газовом окислении сплава Fe–18Cr при различных степенях вакуумирования в области активно-пассивного перехода, соответствующего экстремальной скорости оксидирования.
ЭКСПЕРИМЕНТ
Зеркально отполированный образец сплава Fe–18Cr промывали дистиллированной водой и спиртом, высушивали и вносили в вакуумную камеру, снабженную печкой. В качестве вакуумной системы использовалась препаративная камера электронного спектрометра LAS-300 фирмы RIBER, в которой проводилось окисление. Заданная степень вакуумирования достигалась штатными безмасляными средствами откачки. Предельный вакуум 10–10 Торр. Предусматривалось вымораживание водяных паров системой азотных ловушек. Состав остаточных газов контролировался магнитным масс-спектрометром.
Окисление при низких степенях вакуумирования (10–4–10–1 Торр, 570 K) проводили по методике с предварительной вакуумно-термическкой обработкой образца. Для этого предварительно осуществлялся высоковакуумный отжиг при вакууме 10–8 Торр и температуре 670 K при непрерывном эллипсометрическом контроле. Отжиг прекращался, когда эллипсометрические параметры прекращали ощутимо изменяться (примерно 2 ч).
Эллипсометрические измерения проводили на спектральном эллипсометре, описанном в [18], совмещенном на базе обратно-проходной схемы с препаративной камерой электронного спектрометра RIBER. В качестве спектрального источника излучения эллипсометра использовали монохроматор МДР-4, снабженный ксеноновой лампой с телескопической системой для фокусировки излучения на поверхность образца.
Распределение химического состава по глубине оксида и толщину поверхностного оксида определяли методом эллисометрической спектральной томографии [19, 20], априорно допуская его состав: полуторный оксид хрома Cr2O3, гематит Fe2O3, магнетит Fe3O4, хромит FeCr2O4, термодинамически обусловленные и экспериментально регистрируемые (по результатам спектроскопии комбинационного рассеяния) для выбранных условий оксидирования. Спектры оптических параметров данных оксидов брали из [21–23], сплава (подложки) – определялись в контрольном эксперименте на исходном отожженном образце в приближении эффективной подложки [24].
Фазовый состав металл-оксидной пленки на поверхности образцов контролировался также с использованием зондовой Раман-спектроскопии комбинационного рассеяния (КР) света на модульном спектрометре Ava-Raman-532, снабженном волоконно-оптическим зондом AvaRaman-PRB-FC-532 с возможностью фокусировки возбуждающего излучения. Для этого пластину с закрепленными образцами размещали на микрометрический столик с возможностью фокусирования возбуждающего лазерного излучения Раман-спектрометра на выбранных участках образцов. Спектры регистрировали на длине волны возбуждения λвозб = 532 нм и мощности лазера 10 мВт в области 200–1600 см–1. Идентификация фазового состава пленок спектров велась сравнением полученных КР-спектров с известными и контрольными спектрами КР индивидуальных оксидов и КР-спектрами оксидных пленок.
Для контроля атомного состава поверхностных оксидных слоев и распределения элементов по глубине оксидных слоев, образовавшихся после окисления, был использован метод Оже-электронной спектроскопии [25–27] в комбинации с распылением поверхности пучком ионов аргона [25]. В расчетах использовали пик хрома 526 эВ, пик железа 650 эВ и пик кислорода 510 эВ [26].
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Для контроля фазового состава оксидных слоев первоначально были исследованы спектры комбинационного рассеяния (КР). На рис. 1 приведены спектры КР от образца, окисленного при 2 × 10–2 Торр.
Рис. 1.
Спектры комбинационного рассеяния от оксидированного образца, полученного при оксидировании сплава Fe–18Cr, при температуре 300°C при степени вакуумирования 0.01 Торр.
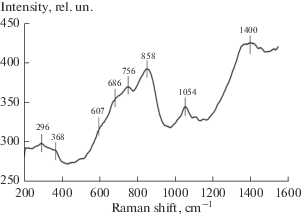
Наблюдаемые в спектрах КР полосы в основном соответствуют оксидам железа, хрома и шпинели [28–30]. Слабые полосы 296, 368 cм–1 в низкочастотной области спектра можно отнести к гематитовой фазе α-Fe2O3. Полосу 607 см–1 можно отнести к гематитовой фазе, а полосу 686 см–1 – как к магнетитовой фазе, так и фазе хромита. Полоса 756 см–1 характерна для фаз γ-Fe2O3 или Fe3O4, а широкая полоса 858 см–1 ранее сопоставлялась с окисными фазами хрома в валентном состоянии Cr(III)/Cr(VI). Полосы 1054 и 1140 см–1 относятся к фазам полуторной окиси хрома (Cr2O3), а широкая полоса 1400 см–1 относится к фазе хромита FeCr2O4. Таким образом, спектры КР образца соответствуют наличию в оксиде фаз окислов железа (Fe2O3 и Fe3O4), а также Cr2O3 и хромита FeCr2O4. Полосы окислов железа выражены слабее окислов хрома.
Спектры эллипсометрических параметров, полученные при различных степенях вакуумирования на различных стадиях процесса окисления приведены на рис. 2. Томографическая обработка спектров проводилась исходя из термодинамических данных и полученных экспериментальных данных КР, по которым при окислении сплавов железо-хром в изотермических условиях печного нагрева при температуре 570 К термодинамически возможно образование окислов железа (магнетит и гематит) и окислов хрома (Cr2O3 и хромита FeCr2O4). Использовалась расчетная методика, описанная в [19, 20]. Для каждого момента времени определялось по отдельности полное по всей глубине оксидного слоя содержание окислов железа и хрома в неоднородном поверхностном оксиде (рис. 3, 4).
Рис. 2.
Спектры эллипсометрических параметров ψ и Δ на различных стадиях окисления сплава Fe–18Cr при 570 К и степенях вакуумирования 0.0009 (а), 0.008 (б), 0.02 (в), 0.5 Торр (г).
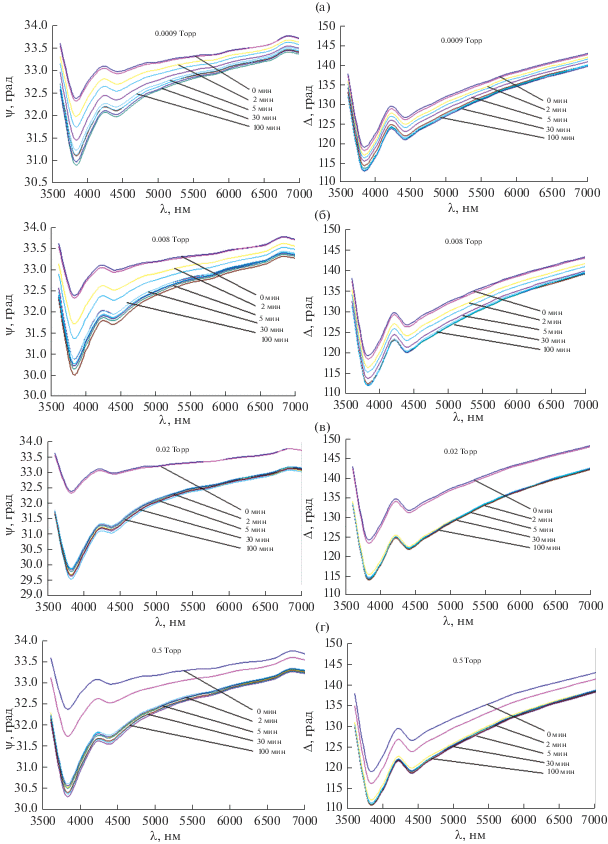
Рис. 3.
Кинетика роста окислов железа при окислении сплава Fe–18Cr при 570 К и различных степенях вакуумирования.
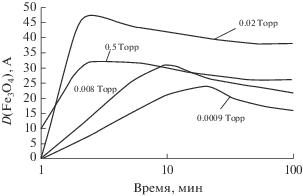
Рис. 4.
Кинетика роста окислов хрома при окислении сплава Fe–18Cr при 570 К и различных степенях вакуумирования.
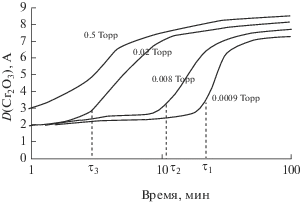
Из полученных кинетических кривых содержания индивидуальных оксидов (рис. 3, 4) видно, что на самой начальной стадии окисления быстро вырастает слой окислов хрома с толщиной ~2 А, практически независимой от степени вакуумирования и, судя по толщине – практически незащитный. Далее процесс окисления сплава по существу – двухстадиен.
На первой стадии в течение 3–20 мин (в зависимости от степени вакуумирования) преимущественно растут окислы железа и в небольшом количестве окислы хрома (рис. 3, 4), диффундирующие через оксидный слой из металла к границе раздела металл–газ. Сравнение спектров отожженного сплава со спектром оксида после начальной стадии окисления (2 мин) показали, что оптически параметры окислов железа более всего соответствовали фазам магнетита и гематита.
Характерной особенностью кривых рис. 3 является все более раннее отступление от логарифмического закона роста на начальных стадиях окисления по мере роста давления кислорода. Период, в течение которого окислы железа образуются с высокой скоростью, с ростом парциального давления кислорода сокращается (рис. 5), что при неизменной экспозиции выражается как максимум прироста толщины оксидного слоя при определенной степени вакуумирования. На рис. 6 приведена зависимость прироста толщины оксида за 1 ч, от степени вакуумирования, рассчитанная по экстремальным точкам кинетических кривых (рис. 3), откуда следует, что в области 2 × 10–2 Торр реализуется активно-пассивный переход.
Рис. 5.
Период ускоренного роста окислов железа при окислении сплава Fe–18Cr при 570 К в функции степени вакуумирования Р.
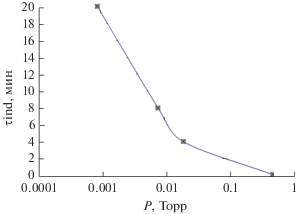
Рис. 6.
Прирост общей толщины оксида при окислении стали Х18 при 570 К за 1 ч при различных степенях вакуумирования Р.
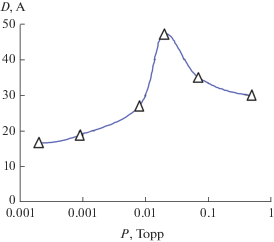
В целом ряде работ, где исследовались начальные стадии изобарного окисления железа и сплавов на его основе предполагалось, что торможение окисления железа в основном обусловлено образованием и развитием во времени оксида α-Fe2O3 , который растет на поверхности Fe3O4 и блокирует рост последнего вследствие своего высокого диффузионного сопротивления [15]. Исходя из этого, активно-пассивный переход железа можно связать с тем, что при повышении парциального давления кислорода оксид α-Fe2O3 образуется из Fe3O4 все быстрее (рис. 5) и, соответственно, раньше блокирует рост слоев Fe3O4. Средняя скорость окисления во времени при этом существенно падает. Очевидно, активно-пассивный переход (переход с 1 стадии на 2 стадию) сплавов Fe–Cr при данных условиях окисления также связан с участием в образовании на внешней границе оксид-газ блокирующего слоя α-Fe2O3 c включением Cr2O3.
На второй стадии наблюдается процесс падения содержания окислов железа, сопровождаемый медленным ростом окислов хрома (рис. 3, 4). Оба процесса примерно совпадают по началу. Отсюда следует, что данная картина соответствует началу протекания реакции замещения, когда рост оксидов хрома протекает за счет медленного восстановления железа из оксидов железа [8, 31]:
(1)
${\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}} + 2{\text{Cr}} = > {\text{FeC}}{{{\text{r}}}_{2}}{{{\text{O}}}_{4}} + 2{\text{Fe}},~$(2)
 $3{\text{FeC}}{{{\text{r}}}_{2}}{{{\text{O}}}_{{4~}}} + 2{\text{Cr}} = > 4{\text{C}}{{{\text{r}}}_{2}}{{{\text{O}}}_{3}} + 3{\text{Fe}}.~~$
$3{\text{FeC}}{{{\text{r}}}_{2}}{{{\text{O}}}_{{4~}}} + 2{\text{Cr}} = > 4{\text{C}}{{{\text{r}}}_{2}}{{{\text{O}}}_{3}} + 3{\text{Fe}}.~~$Результатом этой реакции замещения может быть накопление шпинели FeCr2O4 во время частичного восстановления или оксида Cr2O3 во время полного восстановления FeCr2O4 в оксидной пленке. Большее сродство к кислороду Cr по сравнению с Fe, является движущей силой данной реакции замещения. Кинетику накопления шпинели можно аппроксимировать логарифмической функцией времени.
Количество Cr, переносимого через границу раздела сплав–оксид во время окисления, ограничено диффузией в сплаве и условиями на этой границе раздела и на начальных стадиях не подвержено диффузионным ограничениям. Однако при больших экспозициях может происходить истощение хрома в металлической матрице и результирующее торможение реакции замещения.
Таким образом, в процессе оксидирования происходит трансформация оксидного слоя как результат процессов роста оксидов железа и их взаимной трансформации (магнетита в гематит) на 1 стадии, и далее реакции замещения окислов железа с хромом с образованием слоя окислов хрома по реакциям (1), (2).
Восстановление Fe3O4 может быть представлено как двухэтапный процесс: перенос Cr в реакционную зону и реакция восстановления Fe3O4 и формирования FeCr2O4 (илиCr2O3). Причиной ускорения реакции замещения (1) с ростом давления кислорода может быть интенсификация катодной реакции ионизации кислорода на границе оксид–газ и соответствующее ускорение пассивации сплава сплошным слоем гематита. Действительно, после формирования в конце 1 стадии сплошного пассивирующего слоя α‑Fe2O3, активность кислорода в оксидном слое резко падает, поэтому доля катионов хрома, ранее потреблявшихся реакцией прямого окисления на границе оксид–газ, существенно уменьшается, что, очевидно, увеличивает долю катионов хрома в реакции замещения (1), приводя к ее ускорению. При этом, очевидно, происходит смещение профиля шпинели FeCr2O4 в оксидной пленке от внешней границы оксид–газ к внутренней границе металл–оксид.
Вышеприведенные доводы о причинах пассивации сплава можно существенно дополнить данными спектротомографии в пространстве химических составов неоднородного поверхностного оксида, когда в результате предварительного высоковакуумного отжига на поверхности уже создан слой оксида, максимально обогащенный хромом [14].
Неоднородность оксидного слоя выражается в изменяющемся по глубине объемном соотношении окислов железа и хрома, оптические параметры которых сильно различаются. На полуколичественном уровне это означает, что объемная доля $\theta = {{V({\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}})} \mathord{\left/ {\vphantom {{V({\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}})} {V({\text{C}}{{{\text{r}}}_{2}}{{{\text{O}}}_{3}} + {\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}})}}} \right. \kern-0em} {V({\text{C}}{{{\text{r}}}_{2}}{{{\text{O}}}_{3}} + {\text{F}}{{{\text{e}}}_{3}}{{{\text{O}}}_{4}})}}$ окислов железа – функция координаты x по глубине слоя ($\theta = \theta (x)$). Ограничение двумя простыми окислами представляется оправданным, так как оптические параметры окисла хрома и хромита близки, и у магнетита с гематитом они гораздо ближе, чем, например, у магнетита с окислами хрома.
На рис. 7 приведены спектры эллипсометрических параметров: (а) после 1 ч отжига сплава при вакууме 10–8 Торр и температуре 670 К и (б) после часового окисления отожженного образца при 2 × 10–2 Торр и температуре 570 К.
Рис. 7.
Спектры эллипсометрических параметров $\Delta $ (а) и $\psi $ (б), полученные на отожженной поверхности (1 ч, 10–8 Торр, 670 К) и после 1 ч окисления (0.02 Торр) при 570 К; $\lambda $ – длина волны света, нм.
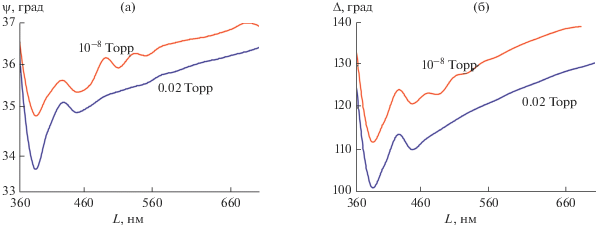
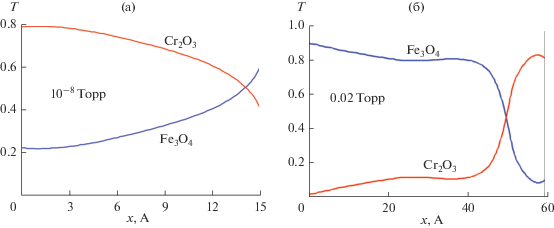
Дискретизацию полученных спектров осуществляли с шагом 20 нм. При этом поиск $d = ({{d}_{1}},...,{{d}_{N}})$ соответствовал поиску толщин N-подслоев неоднородного слоя, разбитого мысленно на 6-подслоев, в каждом из которых $\theta $ (объемная доля окислов железа) имела заданное значение от 0 до 1 с шагом 0.2 ($\theta $ = 0 : 0.2 : 1). Рассчитанные по полученным спектрам сглаженные профили содержания оксидов железа и хрома по глубине приведены на рис. 8.
Рис. 8.
Рассчитанные профили объемного содержания Т окислов хрома и окислов железа в неоднородном поверхностном оксид, полученные на отожженной поверхности (1 ч, 10–8 Торр, 670 К) и после 1 ч окисления (0.02 Торр) при 570 К; x – координата “в глубину” оксида.
Для контроля данных спектротомографии элементный состав полученных оксидных пленок параллельно контролировали методом Оже-спектроскопии в сочетании с бомбардировкой поверхности пучком ионов аргона. В расчетах использовали пик хрома 526 эВ, пик железа 650 эВ и пик кислорода 510 эВ [25–27].
Полученные Оже-профили распределения элементов по глубине оксида как на отожженном, так и на окисленном образце приведены на рис. 9. Видно, что на полуколичественном уровне профили по данным эллипсометрии соответствуют данным Оже-профилирования. Что касается несовпадения Оже-профилей при “приближении” к подложке (5 и 20 мин), то при этом на Оже-спектр окисла начинает накладываться спектр подложки. При таких тонких общих толщинах анализируемых слоев (порядка десятков А), когда глубина съема Оже-сигнала – порядка общей толщины слоя, говорить о локальности Оже-спектрального метода достаточно проблематично. Использованный в данной работе томографический метод лишен этого недостатка и позволяет четко отделить и выявить окисленные формы сплава.
Рис. 9.
Оже-профили содержания А (%) элементов (кислорода, хрома, железа) в неоднородном поверхностном оксиде, полученные на отожженной поверхности (1 ч, 10–8 Торр, 670 К) и за 1 ч окисления (0.01 Торр) при 570 К; t – время травления аргоновым пучком, мин.
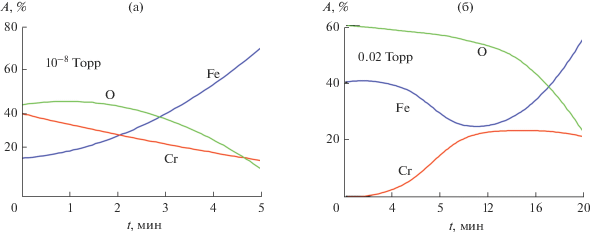
Как и следовало ожидать [14], после вакуумного отжига поверхностный слой в основном обогащен окислами хрома и обеднен окислами железа. Первые, по данным электронной спектроскопии, локализованы у границы раздела оксид–газ. С дальнейшим окислением образца в вакууме 2 × 10–2 Торр при 570 К объемное содержание окислов железа растет. Область формирования окисленного хрома смещается к границе металл-оксид. Общая толщина поверхностного оксида существенно увеличивается. Поскольку окислялся образец, поверхность которого уже была обогащена окислами хрома, то можно заключить, что при 2 × 10–2 Торр на начальных стадиях преимущественно растут окислы железа, а судя по полученным профилям, его катионы в ходе окисления диффундируют через полученный в результате высоковакуумного отжига тонкий слой окислов хрома, не обладающий высокими защитными свойствами.
Из рис. 7, 8 также видно, что для двух различных условий оксидирования были получены два типа профилей распределения состава оксидного слоя. При отжиге (670 К, 10–8 Торр) поверхностный оксид состоит главным образом из Сr2О3 и частично окислов железа. При этом окислы хрома концентрируются около границы раздела оксид–газ. При понижении температуры до 570 К и повышении давления в камере до 0.02 Торр происходит образование оксидного слоя, преимущественно обогащенного окислами железа с наличием окислов хрома, концентрирующихся около границы раздела металл–оксид.
Очевидно, априорно предположенная в томографических расчетах смесь Fe3O4 + Cr2O3 может формально рассматриваться как шпинель переменного (по глубине) состава Fe(Fe2 –xCrx)O4, по существу представляющая собой твердый раствор Cr2O3 в Fe3O4. Когда локальное содержание хрома в такой шпинели превышает критическое значение, в данной точке толщины оксида реализуются условия формирования оксидной фазы Cr2O3. При этом Cr2O3 может выделяться в виде индивидуальной фазы по реакции (2). Место локализации данной реакции зависит от температуры и парциального давления кислорода в камере. Поэтому можно подобрать условия, при которых избирательность оксидирования хромистых сплавов определяется не просто соотношением общего объемного содержания окислов железа и хрома в неоднородном поверхностном оксиде, а пространственной локализацией субслоя окислов хрома в объеме оксида.
Из предложенной модели вытекают основные причины пассивации и механизм формирования гетероструктуры оксидного слоя на сплаве Fe–Cr при его оксидировании в области давлений активно-пассивного перехода. Это рост блокирующих слоев гематита на внешней границе оксид–газ и формирование защитных окислов хрома в очень тонком неоднородном слое при смещении фронта реакции магнетита с хромом от границы раздела оксид–газ к границе металл–оксид. При росте парциального давления кислорода в вакуумной камере фронт формирования слоя защитных окислов хрома Cr2O3 смещается от границы раздела оксид–газ все ближе к границе раздела металл–оксид, а слой получается все менее проницаемым.
ВЫВОДЫ
Таким образом, описать явление низкотемпературного окисления и пассивации сплавов на основе системы Fe18Cr с формированием металл-оксидной гетероструктуры в области активно-пассивного перехода можно следующим образом. Первоначально на поверхности сплава происходит прямое окисление компонентов сплава (железа и хрома) с преимущественным формированием оксидов железа (магнетит). По достижении критической толщины слой магнетита покрывается сплошным пассивирующим слоем гематита, блокирующим поверхность и резко понижающим активность кислорода в нижележащем оксидном слое магнетита. В результате доля катионов хрома, диффундирующих из сплава к границе оксид–газ, существенно уменьшается, что увеличивает долю катионов хрома в реакции замещения (1) в объеме оксидного слоя. В результате происходит смещение области формирования шпинели в оксидной пленке от внешней границы оксид-газ к внутренней границе металл–оксид. Реакция (1) не зависит от активности кислорода в оксиде. Большее сродство к кислороду Cr по сравнению с Fe является движущей силой данной реакции замещения. Поэтому в результате последующей обменной реакции хрома с окислами железа (реакции замещения) в объеме слоя магнетита формируются защитные оксиды хрома типа полуторной окиси хрома (2) и хромистой шпинели переменного состава (1). При пассивации сплава, падение скорости роста оксида при повышении парциального давления кислорода в камере, как видно из результатов томографического анализа, связано с локализацией формирования защитных окислов хрома в очень тонком неоднородном слое при смещении фронта реакции замещения магнетита с хромом от границы раздела оксид–газ к границе металл–оксид.
Список литературы
Розовский А.Я. Кинетика топохимических реакций. М: “Химия”, 1974. 224 с.
Gopalakrishnan J. // Chem. Mater. 1995. V. 7. № 7. P. 1265–1275.
Hayward M.A. // Semicond. Sci. Technol. 2014. V. 29. P. 064010.
Testa-Anta M., Rodríguez-González B., Salgueiriño V. // Part. Part. Syst. Charact. 2019. P. 1900283.
Продан Е.А., Павлюченко М.М., Продан С.Л. Закономерности топохимических реакций. Минск: “Наука и техника”, 1976. 264 с.
Kotenev V.A., Vysotskii V.V. // Prot. Met. Phys. Chem. Surf. 2019. V. 55. P. 880–887.
Revina A.A., Suvorova O.V., Pavlov Y.S. et al. // Prot. Met. Phys. Chem. Surf. 2019. V. 55. P. 888–894.
Окисление металлов / Под ред. Бенара Ж. М.: Металлургия, 1968. Т. 2. 448 с. [Oxydation des Metaux / Ed. Benard J. Paris: Gauthier-Villars, 1962. V. 2.]
Rahmel A., Schenk W. Korrosion und Korrosionsschutz von Stahlen. Weinhein: Chemie, 1977. S. 214.
Shibagaki S., Koga A., Shirakawa Y. et al. // Thin Solid Films. 1997. V. 303. P. 101–106.
Haiduga M., Kucera J. // Korore Mater. 1986. V. 24. № 6. P. 666.
Whittle P.D., Wood G.C. // J. Electrochem. Soc. 1968. V. 115. № 2. P. 133.
Ettabirov M., Dupre B., Gleitzer C. // Reactivity of Solids. 1986. V. l. P. 329.
Greyling C.J., Roux J.P. // Corr. Sci. 1984. V. 24. № 8. P. 675.
Котенев В.А. // Защита металлов. 2003. Т . 39. № 4. С. 341–351.
Фокин М.Н., Опара Б.К., Медведева Н.И., Серегин Е.П. // ДАН СССР. 1972. T. 206. № 6. С. 1405–1408.
Hoar T.R., Price L.E. // Trans. Faraday Soc. 1958. V. 34. № 8. P. 867–874.
Котенев В.А. // Защита металлов. 2000. Т. 36. № 4. С. 405–413.
Kotenev V.A. // Proc. SPIE. 1992. V. 1843. P. 259.
Котенев В.А. // Защита металлов. 1997. Т. 33. С. 270.
Tanaka T. // Jap. J. Appl. Phys. 1979. V. 18. P. 1043.
Idczak E., Oleszkiewicz E. // Thin Solid Films. 1981. V. 77. P. 301.
Винчелл А.Н., Винчелл Г.В. Оптические свойства искуственных минералов. М.: Мир, 1967. С. 98.
Azzam R., Bashara N. Ellipsometry and Polarized Light. M.: Mir, 1981.
Методы анализа поверхностей / Под ред. Зандерны А.Н. М.: Мир, 1979. С. 31.
Jensen C.P., Mitchell D.F., Graham M.J. // Corros. Sci. 1982. V. 22. № 12. P. 1125.
Allen G.C., Dyke J.M., Harris S.J., Morrist A. // Oxidation of Metals. 1988. V. 29. № 5–6. P. 391–408.
Cvelbar U., Chen Z., Sunkara M.K., Mozetic M. // Small. 2008. V. 4. P. 1610.
McCarty K.F., Boehme D.R. // J. Solid State Chemistry. 1989. V. 79. P. 19.
Ningshen S., Kamachi Mudali U., Ramya S., Baldev R. // Corrosion Sci. 2011. V. 53. P. 64.
Акимов А.Г. // Поверхность. Физика, химия, механика. 1984. № 10. С. 17.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов