

Физикохимия поверхности и защита материалов, 2022, T. 58, № 1, стр. 86-112
Биокоррозия, биообрастание и современные методы борьбы с ними
Т. А. Кочина 1, *, Ю. А. Кондратенко 1, **, О. А. Шилова 1, Д. Ю. Власов 1, 2
1 Институт химии силикатов им. И.В. Гребенщикова РАН
199034 Санкт-Петербург, наб. Макарова, 2, Россия
2 Санкт-Петербургский государственный университет
199034 Санкт-Петербург, Университетская набережная, 7–9, Россия
* E-mail: t-kochina@mail.ru
** E-mail: kondratencko.iulia@yandex.ru
Поступила в редакцию 11.06.2021
После доработки 20.09.2021
Принята к публикации 29.09.2021
- EDN: VRBHTF
- DOI: 10.31857/S0044185622010120
Аннотация
Микробиологически обусловленная коррозия (МБК) и биообрастание в морской среде являются двумя основными механизмами морской коррозии. В настоящем обзоре обобщены результаты последних исследований и показано, что и МБК, и морское биообрастание тесно связаны с биопленками на поверхности материалов, образованными морскими микроорганизмами и их метаболитами. В результате, чтобы предотвратить возникновение МБК и биообрастание, важно контролировать микроорганизмы в биопленках или предотвращать адгезию и образование биопленок. В этом обзоре представлены исследовательские подходы, включающие применение новых материалов и инновационных технологий в сочетании с традиционными химическими веществами для достижения более длительных эффектов при наименьшем загрязнении окружающей среды за счет возникающего синергетического эффекта.
ВВЕДЕНИЕ
Коррозия ‒ это глобальная проблема, которая затрагивает широкий спектр отраслей промышленности и муниципальных служб, таких как судоходство, нефтеперерабатывающие предприятия, строительство, канализация и системы питьевого водоснабжения, а также содержание исторических зданий и скульптурных памятников. Одной из главных причин разрушения различных материалов является биокоррозия и биодеструкция материалов, возникающие вследствие жизнедеятельности микроорганизмов. Идея об участии микроорганизмов в разрушении материалов была выдвинута еще в 1910 г. [1]. Различные термины были использованы для описания коррозии, вызванной микроорганизмами, такие как биокоррозия, микробная коррозия, микробиологически обусловленная коррозия [2] и др. По смыслу все эти понятия очень близки. Биокоррозия и микробная коррозия обычно указывают на то, что микробы являются основной причиной коррозии. Микробиологически обусловленная коррозия (МБК) (в англоязычной литературе используется термин “microbiologically influenced corrosion, MIC”) предполагает влияние на коррозию не только самих микробов, но и их метаболитов. Формулируя более точно понятие “биокоррозия”, можно сказать, что биокоррозия ‒ это результат электрохимических реакций, на которые влияют или стимулируют микроорганизмы, часто присутствующие на материалах в виде биопленок. Таким образом, понятия МБК и биокоррозия имеют очень близкое значение. Теоретические исследования по биокоррозии металлических и композиционных материалов наиболее полно приведены в работах [3–6].
Важными объектами, которые наиболее остро страдают от биоповреждений, являются морские суда, портовые сооружения, водоводы и трубопроводы, теплообменники, нефтяные и газовые платформы на шельфе, навигационное и подводное оборудование, гидротехнические сооружения и др. Наиболее агрессивными биодеструкторами материалов являются микроорганизмы – бактерии и грибы. На их долю приходится более 40% всех биоповреждений. Ущерб, вызываемый микроорганизмами, исчисляется десятками миллиардов долларов ежегодно [4, 7–9]. Однако в ряде случаев микробы могут ингибировать или защищать от коррозии [10].
Биокоррозию можно рассматривать как самостоятельный вид разрушения, но чаще всего процессы биологической коррозии протекают параллельно с другими, например, морской, почвенной, атмосферной, коррозией в электролитах, водных растворах. Наиболее распространенными видами коррозии, наносящими существенный ущерб народному хозяйству, являются атмосферная и почвенная коррозия. В связи с этим изучение законов их развития и совершенствование методов защиты не теряют своей актуальности [11–14]. Морская коррозия, наряду с почвенной, является наиболее разрушительной [15, 16]. Биологическая коррозия в зависимости от вида микроорганизмов делится на бактериальную и микологическую, а также может быть смешанной [17–21]. Чаще всего протекает бактериальная коррозия [22–24], она же и наиболее разрушительна. Опасность бактериальной коррозии состоит в том, что бактерии быстро размножаются и легко приспосабливаются к изменениям физических, химических и биологических условий среды [25].
Исследования показали, что микробиологическая коррозия и морское биологическое обрастание являются двумя основными составляющими морской коррозии, вызванной сложной морской средой и морскими организмами.
Морская среда является чрезвычайно агрессивной средой для металлов и других материалов, используемых в морской промышленности [26]. Во-первых, сама морская вода является электролитом с высокой коррозионной активностью. Во-вторых, в окружающей среде океана морские организмы формируют на твердых объектах сложные сообщества и воздействуют на них одновременно, вызывая коррозию материалов [27].
По оценкам зарубежных специалистов, коррозия наносит ущерб экономике развитых стран, составляющий 3–3.5% от стоимости валового национального продукта. А потери металла достигают 20% [28]. Общий мировой ущерб от биоповреждения и морского обрастания по неполным оценкам составляет около 50 млрд долларов США в год. Морское биообрастание происходит из-за нежелательного заселения и накопления морских микроорганизмов, растений и животных на погруженных в водную среду поверхностях материалов и оказывает огромное неблагоприятное воздействие на инфраструктуру и оборудование, обслуживаемое в морских отраслях промышленности [29–32]. Морское обрастание увеличивает вес и шероховатость корпусов судов, что повышает сопротивление трения и тем самым, вызывает дополнительный расход топлива.
В обзоре будут рассмотрены механизмы микробиологической коррозии и биообрастания, а также способы предотвращения и борьбы с ними.
МИКРОБИОЛОГИЧЕСКАЯ КОРРОЗИЯ
В настоящее время большое внимание уделяется микробиологически обусловленной коррозии или для простоты микробиологической коррозии (МБК). При этом основной интерес исследователей вызывают конкретные микробы как основная причина коррозии, а не другие факторы коррозии, такие как сопутствующие химические превращения (образование CO2, сероводорода и др.). MБК ‒ это коррозия материалов, которая вызывается непосредственно жизнедеятельностью микроорганизмов и/или их метаболитами [33, 34]. Большая часть экономических потерь в морской промышленности вызвана MБК. Согласно статистике на ее долю приходится около 20% общих экономических потерь [35].
Бактерии, жизнедеятельность которых протекает в кислородсодержащих средах, относят к аэробным, а в бескислородных средах — к анаэробным. В природных средах аэробные и анаэробные микроорганизмы существуют совместно. При этом условия для существования анаэробных бактерий часто могут быть созданы деятельностью аэробных бактерий. МБК часто производится смесью анаэробных сульфатвосстанавливающих бактерий (СВБ) или нитратвосстанавливающих бактерий (НВБ) и аэробных металлоокисляющих бактерий [36–40]. Процесс коррозии обусловлен использованием СВБ и НВБ элементного железа Fe0 в качестве источника энергии [41, 42]. Эксперименты показали, что Fe0 может служить единственным источником энергии для некоторых СВБ и НВБ, которые используют металлы в качестве доноров электронов. Аэробные металлоокисляющие бактерии можно разделить на две основные группы: железоокисляющие бактерии (ЖОБ) и марганецокисляющие бактерии (MOБ) [43, 44]. Аэробные ЖOБ легко обнаружить в водах нефтяных месторождений, и они обычно считаются коррозионно-активными микроорганизмами, способствующими MБК.
Сульфатвосстанавливающие бактерии (СВБ) являются наиболее изученными бактериями в условиях МБК. СВБ относятся к хемолитотрофным и хемоорганотрофным бактериям, насчитывающим 220 видов и использующим сульфат в качестве акцептора электронов [45–47]. СВБ анаэробны, что означает, что они не нуждаются в кислороде для роста и активности [40]. Это позволяет СВБ выживать в экстремальных условиях. Эти бактерии восстанавливают сульфат до сульфида. Электрохимическая реакция протекает внутри биопленки, в которой железо взаимодействует с водой и СВБ. В результате образуются гидроксильные группы OH‒, которые способствуют протеканию реакции. СВБ активны в диапазоне рН от 4 до 9.5 и могут переносить давление до 500 атм [45, 47]. На рис. 1 представлен процесс МБК, вызванный СВБ на поверхности железа.

Как уже отмечалось, СВБ обычно используют сульфат в качестве акцептора электронов. Однако некоторые штаммы СВБ также могут использовать серу, тиосульфат или даже CO2 в качестве окислителей.
Согласно литературным данным механизм коррозии, вызванной СВБ, можно описать с помощью теории катодной деполяризации. Однако в настоящее время известны и другие теории, описывающие данный процесс. Согласно теории катодной деполяризации электрохимическая реакция протекает на двух участках: анодном и катодном. На анодном участке железо переходит в ионную форму (Fe2+). Ионы Fe2+ отделяются от поверхности, теряя при этом электроны, которые перемещается к катодному участку. Из-за отрыва ионов железа на аноде образуются вогнутые участки (“ямы”). Далее ионы железа вступают в реакцию с сульфидом (S2–) от СВБ с образованием сульфида железа (FeS), являющегося побочным продуктом. На катодном участке электроны перемещаются к поверхности и вступают в реакцию с ионами водорода (H+) с образованием газообразного водорода (H2). Благодаря природе биопленки молекулы воды ионизируются до гидроксида (OH–) и ионов водорода. Ионы водорода понижают уровень рН в пределах биопленки до кислотного. Гидроксид реагирует с ионами железа, образуя гидроксид железа (Fe(OH)2) или ржавчину. На схеме 1 представлены электрохимические реакции, протекающие внутри биопленки, формируемой СВБ, на поверхности железа [46, 49].
Анод: 4Fe → 4Fe2+ + 8e;
Диссоциация/ионизация воды: 8H2O → 8H+ + 8OH–;
Катод: 8H+ + 8e → 8H;
Катодная деполяризация, вызванная СВБ: ${\text{SO}}_{4}^{{2 - }}$ + 8H → S2– + 4H2O;
Продукты коррозии: Fe2+ + S2– → FeS; 3Fe2+ + 6OH– → 3Fe(OH)2;
Общая реакция: 4Fe + 4H2O + ${\text{SO}}_{4}^{{2 - }}$ → 3Fe(OH)2 + FeS + 2OH–.
Схема 1.
Следует отметить, что окисление нерастворимого элементного железа происходит вне СВБ, тогда как восстановление сульфата происходит внутри СВБ [45]. Таким образом, электроны, высвобождаемые при окислении железа, необходимо переносить через клеточную стенку в цитоплазму СВБ для участия в восстановлении сульфата. Для объяснения того, как электроны пересекают клеточную стенку СВБ, вводится термин внеклеточный перенос электронов (ВПЭ) [50]. Существует два основных типа ВПЭ, включая прямой перенос электронов и опосредованный [51, 52]. Когда “сидячие” клетки прикрепляются к металлу напрямую, для переноса электронов используется цитохром С. А в случае, когда “сидячие” клетки находятся близко к поверхности железа, секретируются токопроводящие нанопроволоки (пили) для связывания микроорганизмов с поверхностью железа.
Следует отметить, что в отличие от коррозии НВБ, коррозия СВБ протекает более сложно из-за выделения H2S. Можно сказать, что их основным метаболитом становится сероводород — сильный стимулятор коррозии стали [53]. Существует несколько гипотез о механизме анаэробной коррозии стали, железа, алюминия и их сплавов под влиянием СВБ. Одна из гипотез состоит в том, что при высоком содержании сульфида железа в среде он образует гальваническую пару с железом, в которой сульфид является катодом, а железо, являясь анодом, подвергается коррозии [6].
Другая гипотеза предполагает, что высокая концентрация H2S, приводящая к образованию сульфида железа, фактически пассивирует поверхность железа против дальнейшей коррозии [2]. Является ли пленка FeS причиной коррозии, вызываемой СВБ [54] или напротив пассивирует поверхность железа ‒ это вопрос дискуссионный. Прежде всего, S2– это не акцептор электронов, а состояние восстановления для элемента серы. Это означает, что пленка FeS в лучшем случае служит проводящей минеральной пленкой между железом и биопленкой, что может повлиять на прилипание биопленки к поверхности железа. Кроме того, предположение участия пленки FeS в качестве причины коррозии СВБ не может объяснить, почему НВБ являются коррозийными, если в случае этих бактерий вообще не образуется сульфид.
Нитратвосстанавливающие бактерии
Азот является одним из основных элементов всех форм жизни и необходим для производства амино- и нуклеиновых кислот [55]. Бактерии используют нитрат в качестве альтернативного акцептора электронов вместо кислорода и восстанавливают его до N2 в анаэробных окислительно-восстановительных процессах [55, 56]. НВБ в основном гетеротрофны и часто анаэробны, обладая способностью переключаться между кислородным и нитратным дыханием в зависимости от условий окружающей среды [57].
Коррозия металла в присутствии НВБ протекает следующим образом: нитрат и железо образуют редокс-пару. Металлическое железо (Fe0) окисляется до двухвалентного железа (Fe2+) или двухвалентное железо окисляется до трехвалентного (Fe3+) с сопутствующим восстановлением нитрата до азотистых соединений с более низкой степенью окисления (например, ${\text{NH}}_{4}^{ + }$, N2) [58]. В ряде работ сообщалось, что химическое восстановление нитратов может происходить в присутствии металлического (Fe0) или двухвалентного железа и что медь катализирует эту реакцию [59–62]. Установлено, что химическое восстановление нитрата железом Fe2+ происходит самопроизвольно в диапазоне рН 7.0–8.8 [62, 63].
В работе [64] исследован процесс коррозии, вызванной НВБ Prolixibacter sp. MIC1-1. Было установлено, что основными продуктами коррозии, образующимися в анаэробных условиях культуры Prolixibacter sp. MIC1-1, были FePO4 и FeCO3. Это указывает на то, что Fe0 окислялся как до ионов Fe2+, так и до Fe3+. Окисление Fe0 до ионов Fe3+ в анаэробных условиях является необычным поскольку соединения двухвалентного железа (Fe2+), как правило, обнаруживали в качестве продуктов коррозии в случае СВБ и метаногенов [41, 65]. Авторы предложили следующий механизм протекания коррозии, вызванной Prolixibacter sp. MIC1-1. Данный штамм окисляет Fe0 преимущественно до ионов Fe2+, как и в случае других агрессивных микроорганизмов. Далее ионы Fe2+ могут дополнительно окисляться до ионов Fe3+. Реакции окисления железа сопровождаются восстановлением нитрата либо до нитрита, либо до ионов ${\text{NH}}_{4}^{ + }$. Предполагаемая модель коррозии Fe0, вызванной НВБ Prolixibacter sp. MIC1-1, представлена на рис. 2.
Рис. 2.
Предполагаемая модель коррозии Fe0, вызванной НВБ Prolixibacter sp. MIC1-1. Сплошные линии ‒ биотические процессы; пунктирные линии ‒ абиотические химические превращения [64].

ЖЕЛЕЗООКИСЛЯЮЩИЕ БАКТЕРИИ (ЖОБ)
Железо (Fe) уже давно признано потенциальным источником энергии для бактерий, и упоминания о бактериях, как об организмах, которые питаются железом, можно датировать серединой 1800-х годов [66, 67]. Однако долгое время понимание процесса окисления железа оставалось неясным. И только начиная с 1990-х годов стали появляться первые обзорные работы, посвященные ЖОБ [68].
ЖОБ − это бактерии, которые окисляют железо в результате своего метаболизма, и многие из них могут использовать электроны, захваченные в результате этого процесса, в качестве своего единственного источника энергии для роста. Из всех потенциальных источников энергии окисление Fe(II) дает наименьшую свободную энергию Гиббса (G◦) для клеточного метаболизма. Количество энергии, которое бактерия может извлечь из реакции Fe2+ + 0.25O2 + H+ → Fe3+ + 0.5H2O, составляет 29 кДж моль−1 [69]. Однако, если Fe(III) осаждается в виде гидроксида железа (ржавчина), как это происходит при нейтральном рН [Fe2+ + 0.25O2 + 2.5H2O → Fe(OH)3 +2H+], то энергетический выход удваивается. Кроме того, при окислении Fe(II) при низких парциальных давлениях кислорода энергетический выход, по оценкам, увеличивается до 90 кДж моль−1 [68, 70].
Бактерии могут быть литотрофными или гетеротрофными в зависимости от того, откуда они получают энергию и углерод.
Литотрофные микроорганизмы получают энергию от окисления двухвалентного железа до трехвалентного железа и используют эту энергию для усвоения углекислого газа (CO2) в качестве основного источника углерода в клетке. Гетеротрофное Fe-окисление относится к микробам, которые активно катализируют окисление Fe(II), но не получают энергию от этого процесса и не усваивают CO2, вместо этого используют органический субстрат в качестве источника углерода и энергии. Примерами этого процесса являются такие организмы, как Leptothrix discophora и Sphaerotilus natans, которые производят белки или ферментные системы, активно катализирующие окисление Fe, но не получающие от этого никакой энергетической выгоды [66, 71].
Основная роль ЖОБ в МБК заключается в развитии экологических ассоциаций внутри более крупного микробного сообщества или микробиома, ответственного за МБК. Эти ассоциации могут быть более значимыми для процесса, чем прямые физиологические взаимодействия между ЖОБ и поверхностью металла. Таким образом, на основании ряда исследований можно постулировать, что наиболее важная роль ЖОБ заключается в ранней колонизации стальных поверхностей в сочетании с производством трехмерной биопленки [71].
В работе [72] предложена модель, составленная на основе детальных исследований ранних фаз колонизации стали чистыми культурами Zetaproteobacteria (рис. 3). Основываясь на этой модели, на ранней стадии колонизации стальных поверхностей, подвергшихся воздействию O2, Zetaproteobacteria прикрепляются и растут с поверхности, используя преимущества высвобождения Fe(II) из стали. Бактерии Epsilonbacteraeota также были обнаружены на ранних стадиях формирования биопленки, однако их общая роль в этом процессе не ясна. По мере того как биопленка на поверхности стали растет и стареет, развиваются аноксические области, вероятно, из-за потребления О2 как литотрофными, так и гетеротрофными бактериями, причем последняя группа растет на органическом субстрате внутри биопленки. Эти области открывают возможности для анаэробов, таких как СВБ и метаногенов, которые в свою очередь растут и развивают более зрелый микробиом, ускоряющий процесс коррозии. В этой модели на начальном этапе количество ЖОБ может уменьшаться, однако поверхности из оксидов железа, которые они оставляют после себя, облегчают процесс колонизации другими микробами.
Рис. 3.
Схематическая модель обрастания стали в морской среде в присутствии Zetaproteobacteria [72].
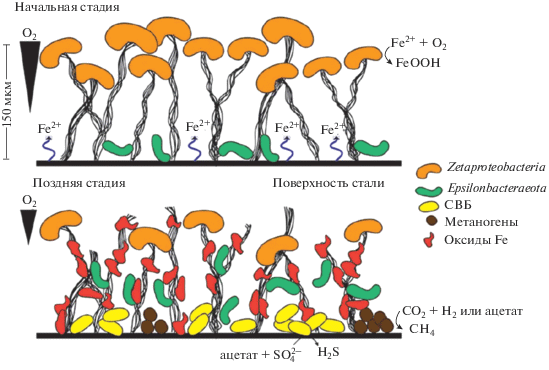
Таким образом, учитывая общую сложность процесса МБК с точки зрения как микробных, так и физико-химических факторов, влияние ЖОБ носит скорее ситуативный, нежели однозначно отрицательный или положительный характер.
МАРГАНЕЦОКИСЛЯЮЩИЕ БАКТЕРИИ (МОБ)
Марганецокисляющие бактерии широко распространены в природе, а также их легко обнаружить в системах питьевого водоснабжения [73, 74]. МОБ характеризуются способностью катализировать окисление Mn(II) до Mn(IV) с последующим осаждением диоксида марганца (MnO2) [44, 75]. Биогенные оксиды марганца часто взаимодействуют с различными металлами. Смешанные оксиды Fe и Mn являются одними из наиболее распространенных в природе. Оксиды марганца считаются сильными окислителями [76], способными окислять различные органические и неорганические соединения [77]. Окисление Mn(II) до Mn(IV) термодинамически благоприятно в аэробных условиях с отрицательной свободной энергией примерно 16 ккал/моль [78–80]. Однако большая энергия активации окисления Mn(II) делает его очень стабильным в большинстве водных сред [78, 79]. Энергетический барьер активации может быть преодолен путем повышения рН (рис. 4) или добавлением Mn-связывающих компонентов, включая сами оксиды Mn, которые являются превосходными хелаторами Mn(II) [79]. Катализ окисления Mn(II) оксидами Mn (автоокисление) затрудняет различие между химически и микробиологически катализируемым окислением Mn, особенно в естественных средах, где органические хелаторы и частицы оксида Mn находятся в изобилии [78].

ФОРМИРОВАНИЕ БИОПЛЕНОК
Во время процесса коррозии СВБ и ЖОБ взаимодействуют друг с другом, образуя биопленки на металлических поверхностях. В естественных условиях микроорганизмы могут существовать либо в виде планктонных (свободно плавающих) клеток, либо в виде биопленок. По современным представлениям 95–99% микроорганизмов в природных местах обитания существуют в виде биопленок. Биопленка играет очень важную роль в МБК [35, 54], и в процессе своего развития она проходит ряд этапов [37]. На первом этапе происходит адгезия или сорбция микроорганизмов к поверхности субстрата из окружающей среды, создается адсорбированная пленка. Эта стадия обратима, так как сорбированные клетки могут вернуться к планктонной форме существования. На второй стадии планктонные микроорганизмы мигрируют на поверхность материала, притягиваемые адсорбированной пленкой. Третий этап заключается в окончательном прикреплении клеток к поверхности и называется фиксацией. На этой стадии микробы выделяют внеклеточные полимеры, обеспечивающие прочную адгезию, планктонные микроорганизмы прикрепляются к активным участкам на поверхности материала и превращаются в “сидячие” микроорганизмы. На четвертом этапе “сидячие” микроорганизмы растут и производят метаболиты с образованием биопленок. На пятой стадии микробы выделяют внеклеточные полимеры, обеспечивающие прочную адгезию. При этом образуются микроколонии ‒ отдельные скопления сорбированных клеток. На этой стадии клетки активно делятся, а выделяемый матрикс удерживает вместе всю колонию. И, наконец, микроколонии сливаются, и образуется зрелая биопленка, которая начинает вызывать коррозию. С течением времени стабильность биопленок уменьшается, а затем часть из них выпадает, создавая гетерогенные биопленки. Многие исследования показали, что состав биопленок влияет на коррозию материалов. Неоднородные биопленки, вызванные выпадением нестабильных биопленок, создают локальную коррозию материалов и ускоряют ее [38, 81–83]. Причину, по которой гетерогенные биопленки вызывают локальную коррозию, можно объяснить с помощью теории концентрации кислорода [84]. Когда на поверхности материала возникают гетерогенные биопленки, участки с плотными биопленками препятствуют распространению кислорода к ним, а аэробные бактерии в биопленках также способствуют уничтожению кислорода под биопленками. Оба эти процесса приводят к созданию мест с низкой концентрацией кислорода. Следовательно, эти места служат анодными площадками для коррозии материала. В то же время участки с менее плотными биопленками или без биопленки и с более высокими концентрациями кислорода служат катодными участками для реакции восстановления кислорода и потребления электронов.
ДРУГИЕ БАКТЕРИИ, ВЫЗЫВАЮЩИЕ КОРРОЗИЮ
Кроме СВБ и НВБ существуют и другие микроорганизмы, вызывающие коррозию, например, сероокисляющие бактерии (СОБ). Нейтрофильные (предпочитающие нейтральную среду) СОБ, такие как Thiobacillus spp. и Thiomonas spp. окисляют сульфиды и другие соединения серы (${{{\text{S}}}_{2}}{\text{O}}_{3}^{ - }$ и S0) до серной кислоты и политионовых кислот, тем самым снижая рН примерно до 3.5–5.0 [85–87]. Скорость бактериального окисления сульфидов может быть в миллионы раз больше, чем скорость обычного химического окисления. При рН 5.0 и ниже ацидофильные СОБ, такие как Acidithiobacillus thiooxidans, продолжают окисление серы, производя большое количество серной кислоты, которая снижает рН до 1.0–2.0 [88, 89]. H2SO4 реагирует с цементной матрицей, приводя к образованию гипса (CaSO4 · 2H2O) и эттрингита (3CaO · Al2O3 · 3CaSO4 · 32H2O) [90]. Эти сульфатсодержащие соли приводят к внутренним трещинам в бетоне и, в конечном счете, к разрушению конструкции [91].
Кислотообразующие бактерии способны метаболизировать органические соединения (например, этанол, лактат, ароматические углеводороды и даже CO2) и производить органические и неорганические кислоты. Эти продуцируемые кислоты могут увеличить скорость коррозии, вызвать растрескивание трубопроводов или служить питательными веществами для некоторых других агрессивных микробов, таких как СВБ [92, 93]. Некоторые кислотообразующие бактерии, участвующие в биовыщелачивании, являются аэробными [94]. Органические кислоты могут ускорять коррозию, если кислотная коррозия более активна, чем ингибирование кислородной коррозии, обусловленное аэробной кислотной биопленкой. Некоторые материалы, такие как нержавеющая сталь, устойчивы к кислородной коррозии благодаря своим тонким и плотным пассивирующим пленкам из оксида металла. Однако и такие минеральные пленки могут быть разрушены биопленками. Сообщалось, что уксусная кислота, продуцируемая Acetobacter aceti, ускоряет коррозию нержавеющей стали, разрушая защитную известковую пленку, образующуюся при катодной поляризации [93, 95]. Основными причинами внутренней коррозии газопроводов из углеродистой стали являются Clostridia и Butyribacteria [93, 95]. В общем случае образующиеся органические кислоты усиливают коррозию путем производства дополнительных катодных реагентов, связывания ионов металлов и разрушения пассивирующей пленки и препятствия пассивации, что в совокупности ускоряет растворение металла [96]. Ацетобактерии являются аэробными бактериями, которые могут окислять этанол в уксусную кислоту во время аэробной ферментации. Было обнаружено, что они ускоряют точечную коррозию стали и медных сплавов из-за образования уксусной кислоты в ее парообразной форме и при растворении в водном растворе [97]. Бактерии рода Acidithiobacillus увеличивают скорость коррозии металлов и сплавов путем продуцирования неорганических кислот ‒ продуктов их метаболизма, таких как серная кислота, при окислении тиосульфата [98].
Аэробные бактерии, продуцирующие слизь, обычно встречаются в морской среде. Биопленка, образованная этими бактериями, характеризуется пятнистым распределением на металлических поверхностях. Эти бактерии удаляют кислород из областей под биопленками посредством дыхания, что приводит к созданию участков с низкой концентрацией кислорода. Следовательно, эти области, как уже отмечалось, становятся анодными (относительно мест с большим количеством кислорода), и это приводит к локальной кислородной коррозии. Участки с менее плотной биопленкой или без нее и с более высокими концентрациями кислорода служат катодными участками для восстановления кислорода при потреблении электронов [99]. Этот процесс коррозии известен как выделение едких метаболитов.
АНАЭРОБНАЯ БИОКОРРОЗИЯ
Существует как минимум три различных типа анаэробной биокоррозии, основанных на анаэробном метаболизме [2]. Тип I вызван электрогенными микробами, такими как СВБ (и микробами, использующими молекулярный водород Н2 в качестве носителя электронов). Они атакуют углеродистую сталь, нержавеющую сталь и некоторые другие неблагородные металлы, являющиеся донорами электронов и имеющие достаточно отрицательные потенциалы восстановления. Процесс восстановления окислителя, такого как сульфат-анион, проходит с участием ферментного катализа в цитоплазме клетки микроба. Тип II вызывается ферментативными микробами, такими как кислотные бактерии, которые выделяют едкие метаболиты. Эти микробы действуют на поверхности металла внеклеточно. Ферментативный метаболизм не требует внешних акцепторов электронов, и сопровождается образованием большого количества органических и минеральных кислот. Восстановление протона при достаточно кислом pH может сочетаться с окислением железа с образованием термодинамически благоприятной окислительно-восстановительной реакции, которая кинетически не замедляется. Этот процесс ничем не отличается от воздействия органических кислот (например, уксусной кислоты) при обычной химической коррозии [94]. Механизмы биокоррозии I и II типов являются электрохимическими. Биокоррозия типа III может быть определена как микробная атака на органическое вещество, такое как полиуретан, с целью использования микробами органического углерода [100]. Этот тип неэлектрохимической коррозии более известен как биодеструкция. При этом микробы могут быть как аэробными, так и анаэробными. Кроме того, могут быть и другие типы биокоррозии, особенно в открытых водоемах с растворенным кислородом в качестве возможного акцептора электронов.
ДРУГИЕ МИКРООРГАНИЗМЫ, ВЫЗЫВАЮЩИЕ КОРРОЗИЮ
Огромную роль в процессе биокоррозии играют грибы (микологическая коррозия). Этот вид биокоррозии является частным случаем биоразрушения материалов [101]. Видовое многообразие грибов и их высокая приспособляемость к условиям обитания приводят к тому, что объем поврежденных ими материалов значительно превышает объем материалов, поврежденных бактериями. Известно, что в процессе роста грибы выделяют большое количество органических кислот (уксусную, щавелевую, лимонную, глутаровую и т.д.), вызывающих коррозию различных материалов. Например, гриб Serpula lacrymans выщелачивает кальций, серу, кремний и железо из минеральных материалов и штукатурки [102].
Грибы, относящиеся к родам Aspergillus, Penicillium, Chrysosporium merdarium, Talaromyces flavus, Saccharomyces, Paecilomyces parvus, Cladosporium herbarum выделяют как наиболее коррозионноопасные [103–105]. Aspergillus niger является одним из самых распространенных видов грибов [106]. Некоторые исследования показали, что A. niger может способствовать коррозии титана, углеродистой стали и магниевого сплава в эвтрофных средах [106–108]. Согласно некоторым исследованиям, щавелевая кислота, лимонная кислота и некоторые другие органические кислоты, производимые A. niger, могут вызывать коррозию металла [107, 109–112]. Однако до сих пор неясно, какая из них играет ключевую роль в процессе коррозии алюминиевых сплавов. Кроме того, коррозия алюминиевых сплавов, индуцируемая A. niger, еще более сильна, чем в растворе хлорида натрия [113]. В работе [114] изучено коррозионное поведение Al и Zn во влажной атмосфере в присутствии A. niger. Было показано, что A. niger действует либо как ускоритель коррозии, либо как ингибитор в зависимости от металла, который он колонизирует. В работе [106] показано, что A. niger значительно ускоряет коррозию магниевого сплава AZ31B на начальных стадиях, а затем ускорение ослабевает. Адсорбция A. niger на магниевом сплаве способствовала развитию точечной коррозии.
Другие микроорганизмы также могут вызывать коррозию. Например, водоросли могут производить кислород в биопленке в результате фотосинтеза в дневное время и преобразовать его в углекислый газ в ночное время. Они могут ускорять процесс коррозии, вызывая изменение металлических поверхностей в условиях растворенного кислорода [115]. В охлаждающей воде присутствуют три различных типа водорослей: сине-зеленые (цианобактерии), зеленые и диатомовые. Наиболее распространены следующие виды: Chlorella, Phomidium, Cyclotella [103, 116, 117].
В работах [118, 119] рассмотрены способы, с помощью которых водоросли могут способствовать коррозии. Водоросли способны изменять свою среду обитания путем изменения рН, концентрации кислорода в локальной зоне электролита, производя метаболиты, которые делают электролит более агрессивным для коррозии. Результаты показали, что фотосинтезирующие водоросли могут поднять рН выше 10 и, разлагаясь, могут понизить рН ниже, чем у окружающей морской воды.
При погружении в естественную морскую воду на поверхности металла образуются различные поверхностные пленки, в том числе слизь. Слизь – это вязкая секреция, вырабатываемая водорослями, которая распространяется по поверхности при их движении. Распространение слизи чаще всего неравномерно и способствует коррозии.
Поверхности, покрытые водорослями, создают благоприятные условия для роста бактерий [119]. Это косвенный механизм влияния водорослей на коррозию. СВБ относятся к числу бактерий, которые могут процветать в разлагающихся водорослях. Как уже отмечалось, СВБ усиливают коррозию различными способами ‒ продуцированием токсичного сероводородного газа, образованием сульфида железа, вызывающего точечную коррозию, и восстановлением сульфатов до коррозионного сульфида. Однако в ряде работ сообщается о полезной роли водорослей в предотвращении коррозии. В частности, в работе [120] было показано, что водоросли Hydroclathratus clathratus выступают ингибиторами кислотной коррозии мягкой стали.
Фактически, любые микроорганизмы, способные повредить минеральные пассивирующие слои на металлических поверхностях, могут вызвать коррозию. Этим микроорганизмам нет необходимости выделять окислитель самостоятельно, если там уже есть окислитель, такой как CO2 и кислород.
КОРРОЗИЯ КАМНЯ
Кроме металлов биокоррозии могут подвергаться и другие материалы. Биопленки на бетоне, камне и мраморе видны в виде цветного слизистого слоя или сухой корки. Микробные сообщества могут напрямую вызывать физическое и химическое разрушение исторических зданий и произведений искусства [121, 122]. Более того, слизистые биопленки удерживают влагу, которая может вызывать механическое напряжение в структуре материала во время циклов замораживания-оттаивания.
В коррозии камня участвуют разные группы микроорганизмов. Согласно данным целого ряда авторов, на поверхности каменных строительных материалов преобладают мицелиальные грибы родов Penicillium, Aspergillus, Trichoderma, Cephalosporium.
Биоповреждения в этом случае преимущественно сводятся к нарушению сцепления составляющих компонентов в результате воздействия минеральных или органических кислот, а также ферментов и вследствие химических реакций между цементным камнем и продуктами жизнедеятельности микроорганизмов [123].
На границе раздела камень-воздух присутствие света может привести к росту фототрофов, таких как водоросли и цианобактерии [124]. Грибы могут проникать в камень и производить внеклеточные ферменты и метаболиты, вызывающие химические и физические повреждения [125, 126]. Кислоты, продуцируемые бактериями, растворяют известковый камень [3].
Коррозия также может возникать на границе раздела камень-почва, включая фундаменты зданий и канализационные системы. Бетонные канализационные системы могут быть серьезно повреждены такими микроорганизмами, как Acidothiobacilli spp., которые производят серную кислоту, вступающую в реакцию с кальцитовым вяжущим материалом бетона [3, 127].
Коррозия карбонатного камня химически проста, однако общий процесс усложняется некоторыми факторами, связанными с пористостью материала ‒ наличием микроорганизмов, содержанием влаги и склонностью камня “дышать”.
Кроме того, карбонатный камень химически не однороден, а постоянно присутствующие переходные металлы и некарбонатные минералы усложняют картину коррозии. Несмотря на это, концептуальная картина коррозии довольно проста. В присутствии значительного количества атмосферного SO2 и высокой влажности (или присутствия воды) растворенный SO2 окисляется до ${\text{SO}}_{4}^{{2 - }}$ в растворе, а затем кальцит превращается в гипс. Последний, обладающий заметной растворимостью в воде, может легко вымываться в потоке. Кроме того, камень может подвергаться раскалыванию по мере ослабления цементирующей матрицы или по мере того, как сама ослабленная кристаллическая структура подвергается циклам смачивания и сушки, оттаивания и замораживания [128].
Если содержание серы в атмосфере минимально или вовсе отсутствует, микроорганизмы поселяются на камне и внутри него и генерируют оксалат-ионы. Полученные оксалаты кальция затем образуют слабый защитный поверхностный слой.
Попытки смягчить коррозию карбонатного камня сводятся к двум подходам: применение защитных средств для отталкивания воды и реакционноспособных соединений, а также пропитывание пор камня инертными материалами [129, 130]. Для сохранения каменных конструкций необходимы дальнейшие исследования защитных материалов. Для новых конструкций карбонатный камень следует выбирать только с полным знанием его физико-химических свойств, подверженности коррозии и возможностей применения защитных средств на начальном этапе и в течение всего срока службы [128].
МОРСКОЕ БИООБРАСТАНИЕ
Морское биообрастание является серьезной мировой проблемой, решение которой играет огромную роль как для морской экономики, так и для морской промышленности. Многолетние исследования привели к общему мнению о том, что морское биообрастание также как и МБК тесно связано с биопленкой и включает следующие этапы [32]: (1) на погруженной в морскую среду поверхности образуется первоначальная адсорбированная пленка; (2) бактерии и другие микроорганизмы прилипают к адсорбированной пленке и постепенно превращаются в биопленку, выделяя внеклеточные полимерные вещества, состоящие из белков и полисахаридов, используемые для фиксации микро- и макроорганизмов; (3) морские организмы, такие как диатомовые водоросли, споры личинок и микроводорослей, накапливаются на поверхностях материалов, поскольку биопленка может обеспечивать их питательными веществами; (4) личинки морских макроорганизмов, таких, например, как моллюски, оседают и растут на поверхностях материалов (рис. 5). Этот общий процесс обрастания иллюстрирует взаимосвязь между микроорганизмами и макрообрастателями [131]. Макрообрастание является основным заключительным результатом в этой схеме. Микроорганизмы в свою очередь способствуют биообрастанию за счет создания условий и накопления питательных веществ, которые они предоставляют для привлечения новых организмов. Активность микроорганизмов может регулировать образование макрообрастателей, в то время как их накопление, в свою очередь может обеспечить некоторую защиту от уничтожения микроорганизмов и биопленок. Однако следует иметь в виду, что в океане более 4000 организмов-обрастателей, и разные морские среды могут приводить к различным доминирующим организмам-обрастателям и различным биологическим взаимодействиям.

Между микробиологической коррозией и биообрастанием существует различие и сходство. Различие заключается в том, что:
• MБК ‒ это процесс коррозии, происходящий на микроуровне, в то время как биообрастание ‒ это процесс оседания и накопления обрастателей, происходящий на макроуровне;
• Организмы, связанные с МБК, представляют собой только различные микроорганизмы, в то время как организмы, связанные с биологическим обрастанием, включают как разные микроорганизмы, так и макрообрастатели ‒ растения и животные;
• MБК наносит непосредственный ущерб материалам, в то время как ущерб от биообрастания является более широким и сложным в различных областях.
Сходство между MБК и биообрастанием включает в себя следующие моменты:
• МБК и биообрастание начинаются с образования первоначально адсорбированной пленки на поверхности материала; эта стадия обратима, так как сорбированные клетки могут вернуться к планктонной форме существования.
• MБК и биологическое обрастание тесно связаны с биопленками, созданными морскими микроорганизмами, сорбированными первоначальной пленкой;
• близкое происхождение MБК и биологического обрастания объясняет сходные стратегии защиты (профилактики). Если метод может уничтожить сформированные биопленки и предотвратить образование новых биопленок, то он сможет устранить или предотвратить развитие MБК и биообрастание одновременно.
НАУЧНЫЕ ПОДХОДЫ В БОРЬБЕ С МИКРОБИОЛОГИЧЕСКОЙ КОРРОЗИЕЙ И БИООБРАСТАНИЕМ
Как уже отмечалось, биопленка является основным условием для возникновения МБК и биообрастания. В случае МБК биопленка непосредственно участвует в процессе коррозии. Для морского биообрастания биопленка является основным фактором, привлекающим большинство видов загрязняющих организмов для оседания и роста на поверхности материала. В результате, чтобы устранить MБК или морское биообрастание, необходимо контролировать активность микроорганизмов в биопленках или предотвращать образование биопленок и адгезию морских организмов. Одним из основных способов защиты материалов (металлы и сплавы, бетоны и др.) от биокоррозии и биообрастания являются защитные покрытия [132].
Для решения проблемы биообрастания, начиная с 1960-х годов, были коммерциализированы противообрастающие покрытия, содержащие трибутилолово (ТБО). Эти материалы характеризовались долговечностью при низкой себестоимости производства и были способны эффективно предотвращать обрастание корпуса в течение пяти лет. Однако оловосодержащие покрытия, выделяя вредные вещества в окружающую среду, обладали высокой токсичностью для морских организмов [29, 131, 133, 134]. Со временем в океан было выброшено достаточное количество соединений ТБО, причинивших необратимый ущерб окружающей среде [30, 135]. Эти неблагоприятные последствия привели к ограничениям на их применение во многих странах [136–138]. На основании рекомендаций Международной конвенции использование ТБО в качестве биоцидов в составе противообрастающих покрытий было запрещено с сентября 2008 года [29, 139].
Это привело к постепенному доминированию самополирующихся покрытий без добавления биоцидов. Кроме этого стали применять полимерные смолы на основе акриловых сополимеров, в которых ТБО был заменен на оксид меди (I) в качестве биоцида [140, 141]. Хотя Cu2O менее токсичен, чем соединения олова, он может отрицательно влиять на морскую среду. Массовое накопление ионов меди в морской среде связано с использованием медь-содержащих противообрастающих покрытий на судах [142, 143]. Кроме того, стоимость сырья для противообрастающих покрытий постоянно растет во всем мире [144]. Поэтому существует необходимость в разработке высокоэффективных и экологически чистых противообрастающих покрытий.
После запрета трибутилолова в противообрастающих красках было введено много альтернативных биоцидов для предотвращения заселения и роста морских организмов на корпусах судов. В настоящее время в качестве биоцидов для противообрастающих покрытий наиболее известны следующие соединения: Иргарол 1051 (N-2-метилтио-4-трет-бутиламино-6-циклопропиламино-s-триазин), диурон (1-(3,4-дихлорфенил)-3,3-диметилмочевина), пиритион цинка, дихлофлуанид (N,N-диметил-N '-фенил-N'-фтордихлорметилтиосульфамид), хлороталонил, “Seanine 211” (4,5-дихлор-2-n-октил-4-изотиазолин-3-он) и Эконея (тралопирил) (рис. 6).
Рис. 6.
Структуры биоцидов для противообрастающих покрытий: (а) Иргарол 1051; (б) диурон; (в) пиритион цинка; (г) дихлофлуанид; (д) хлороталонил; (е) “Seanine 211”; (ж) Эконея (тралопирил).

Одними из первых биоцидов, заменивших оловосодержащие соединения, стали Иргарол 1051 и Диурон, действующие как ингибиторы фотозинтеза. S-триазиновый биоцид Иргарол 1051 является высокоспецифичным и эффективным ингибитором фотосинтеза водорослей с очень низкой растворимостью в воде, что обеспечивает покрытиям хорошие противообрастающие свойства. Однако в настоящее время их применение в составах противообрастающих покрытий в ряде стран сильно ограничено. Считается, что Иргарол 1051 трудно разлагается в естественной морской среде с периодом полураспада от 100 до 350 дней [145–148]. Диурон менее устойчив в морской воде с периодом полураспада в 14 дней, однако при содержании в составах покрытий период полураспада значительно увеличивается [149]. Впервые о загрязнении окружающей среды Иргаролом 1051 сообщалось в 1993 г., когда биоцид (конц. 1700 нг/л) обнаружили в водах (Лазурный берег) Франции [145, 150]. Начиная с 1993 г., о содержании Иргарол 1051 в водах неоднократно сообщалось. Диурон крайне опасен для водной экосистемы, высокотоксичен для водорослей (в частности, зеленых водорослей Scenedesmus vacuolatus) и умеренно токсичен для таких водных растений, как ряска [151]. Одним из метаболитов диурона, который образуется при его разрушении в водной среде, является токсичный 3,4-дихлоранилин, обладающий генотоксическими свойствами. По некоторым данным, 3,4-дихлоранилин способен накапливаться в живых организмах. Другой метаболит диурона — 3,4-дихлорфенол. Это вещество обладает репродуктивной токсичностью, раздражает слизистые оболочки и поражает почки, печень и иммунную систему.
Некоторые европейские страны ограничили или полностью запретили применение этих биоцидов вследствие их неблагоприятного воздействия на окружающую среду. Использование диурона было запрещено в качестве активного компонента в составах противообрастающих покрытий в Нидерландах [152], Швеции [153] и наряду с Иргаролом ‒ в Великобритании [154, 155] и Финляндии [156]. Более того, в Швеции Иргарол разрешен только для судов длиной >25 м [157], но постепенно выводится из употребления [156]. Применение иргарола, диурона и дихлофлуанида также ограничено в Дании [153, 156, 158, 159].
Пиритион цинка (ZnPT) является одним из наиболее популярных биоцидов, применяемых в противообрастающих покрытиях. Биоцид представляет собой комплекс цинка с двумя лигандами пиритиона, в котором ионы Zn2+ связаны с атомами кислорода и серы. ZnPT широко используется в качестве альгицида, бактерицида и фунгицида [140, 160–162]. В основе его действия лежит разрушение транспортировочной способности мембраны путем блокирования протонного насоса, который подает энергию на транспортный механизм. ZnPT может быть введен в состав противообрастающего покрытия в качестве самостоятельного биоцида, но чаще его вводят в виде смеси с другими биоцидами, например, с дитиокарбаматом цинка, “Seanine 211” или тиоцианатом меди с концентрацией до 7% [163]. В водной среде ZnPT быстро разлагается, главным образом, путем фотолиза. Однако там, где ультрафиолетовый свет не может проникнуть из-за мутности воды или большой глубины, ZnPT может накапливаться в нерастворенном виде, непрерывно оказывая токсическое воздействие на морскую среду. На данный момент имеется недостаточно информации о биодеструкции и гидролизе ZnPT в морской среде. Кроме того, из-за его токсического воздействия на широкий спектр морских организмов рекомендуется контролировать использование ZnPT и тщательно проверять уровень ZnPT в окружающей среде [160].
Дихлофлуанид ‒ это галогенированное сульфонамидное производное из группы арильных дихлорфторметилтио-содержащих фунгицидов. Данный фунгицид действует как ингибитор тиолсодержащих ферментов путем образования дисульфидных мостиков [164]. Дихлофлуанид быстро подвергается гидролизу в воде с образованием N'-диметил-N-фенилсульфамида [165] с периодом полураспада в морской воде менее 20 часов [145, 148, 166]. Биоцид умеренно токсичен для беспозвоночных. Дихлофлуанид обладает меньшей токсичностью по сравнению с другими антиобрастающими агентами, хотя некоторые исследования выявили его токсические эффекты [167–169], такие как эмбриотоксичность у морского ежа, Glyptocidaris crenularis [170, 171]. К сожалению, отсутствуют данные о его токсичности для других водных видов.
Хлороталонил относится к фунгицидам класса хлорнитрилов. Токсичность этого пестицида для водных беспозвоночных до конца неясна. Некоторые исследования показывают, что хлороталонил наиболее токсичен на ранних стадиях развития для трех видов морских беспозвоночных: Paracentrotus lividus, Ciona intestinalis и Mytilus edulis, вызывая эмбриотоксичность, личиночное торможение и смертность [172]. Хлорталонил также токсичен для рыб, ЛД50 после 96-часового воздействия варьируется в интервале от 8.2 до 110 мкг/л, в зависимости от вида и условий воздействия. Он может накапливаться в тканях рыб [151].
Впервые биоцид “Sea-Nine 211” (DCOIT) был введен в качестве противообрастающего соединения в 1996 году [173]. В настоящее время “Sea-Nine 211” является одним из коммерческих противообрастающих агентов, широко используемых во всем мире. Производители биоцида “Sea-Nine 211” заявляют о его экологической безопасности главным образом из-за его заявленной быстрой деструкции в морской среде. Однако ряд исследований показывает, что кинетика разложения DCOIT зависит от морской воды, а также от таких факторов, как: температура, солнечный свет и рН. Это означает, что высвобождающийся из покрытия DCOIT не будет дезактивирован так быстро, как заявлено производителем [174].
Исследования токсичности данного биоцида показали, что водоросли, ракообразные и рыбы очень чувствительны к этому биоциду, их выживаемость начинает снижаться даже при низких концентрациях (от 2 до 4 мкг/л) [151]. “Sea-Nine 211” токсичен для широкого спектра водных организмов. Например, накопление “Sea-Nine 211” было обнаружено в водах Испании на уровне 3700 нг/л. Из-за различной скорости разложения и высокой токсичности применение DCOIT в качестве противообрастающего агента может нанести необратимый ущерб морской экосистеме. Судам, защитные покрытия которых содержат “Sea-Nine 211” и дихлофлуанид, запрещено заходить в воды некоторых европейских стран [153].
Тралопирил (Эконея) относительно недавно зарегистрирован в качестве биоцида для антиобрастающих покрытий (2014 г., [175]), став потенциальным новым загрязнителем окружающей среды. Тралопирил в качестве антиобрастающего агента рекомендован для борьбы с биообрастанием ракушками, гидроидами, мидиями, устрицами, полихетами и коммерциализирован под торговым названием Эконея (ECONEA ®, Janssen PMP, Бельгия). Согласно литературным данным тралопирил в морской среде очень быстро подвергается деструкции, его период полураспада составляет 3 часа при 25°С и 15 ч при 10°С соответственно [176, 177]. В связи с его недавним введением информации о его токсичности в литературе крайне мало. Известно, что тралопирил токсичен для морских ежей, а также для C. japonicus, P. depressus и для H. pulcherrimus. В работе [178] показано, что тралопирил является высокотоксичным и приводит к аномальному развитию личинок велигеров с ЛД50 3.1 мкг/л. Oliveira с соавторами [179] сообщили, что тралопирил значительно ингибирует количество АТФ и рост C. reinhardtii. Кроме того, тралопирил также высокотоксичен для эмбрионов данио-рерио со значением ЛД50 5 мкг/л и изменяет содержание белков, участвующих в энергетическом метаболизме, цитоскелете, дифференцировке клеток, делении клеток и сплайсинге мРНК [179, 180].
В связи с вышесказанным актуальным направлением является разработка и создание нетоксичных или малотоксичных биостойких покрытий, основой которых является полимерная матрица и различного рода нетоксичные добавки, препятствующие процессу биокоррозии и биообрастания. В последние годы, в связи с развитием нанотехнологий, появились новые возможности для создания материалов с уникальными мягкими биоцидными свойствами. Их преимуществом является экологичность, длительность действия, способность ингибировать развитие агрессивных микробных сообществ, не меняя свойств самого материала или улучшая их. В качестве мягких биоцидных добавок, не оказывающих разрушительного действия на окружающую среду, используются полимерные соединения, например, на основе гуанидина, хлорметильные производные ароматических углеродов с пиридином и др., фталоцианины, порфирины, аналоги хлорофилла, являющиеся фотосенсами, которые под действием солнечного света вырабатывают активные формы кислорода, угнетающие развитие бактерий и грибов [181–183]. Фотосенсибилизирующим действием обладает также диоксид титана в форме анатаза, водные суспензии которого в настоящее время активно апробируются для защиты камня [184–186].
В качестве мягких биоцидов перспективно использовать внутрикомплексные соединения гидроксиалкиламинов – атраны. Атраны характеризуются уникальной трициклической структурой, содержащей донорно-акцепторную трансаннулярную связь N → Э (рис. 7). Исследования в области атранов впервые широко начались в России под руководством академика М.Г. Воронкова. Фармакологические и биохимические исследования атранов на основе силатранов, герматранов, протатранов и гидрометаллатранов показали, что они являются малотоксичными веществами и обладают широким спектром полезного биологического действия (иммуномодулирующее, адаптогенное, противоопухолевое, противовоспалительное и др.) [187–191]. В работе [192] впервые показана возможность применения соединений класса атранов в качестве мягких биоцидов в составах защитных покрытий на основе силиконового каучука и полиметилфенилсилоксана. Высокую биостойкость проявили покрытия на основе полиметилфенилсилоксана, причем как с биоцидной добавкой, так и без нее. Развитие микроорганизмов на их поверхности было ограничено только каплями инокулята и локальными зонами по краю пластин. Однако покрытие с биоцидной добавкой на основе триэтаноламмониевой соли салициловой кислоты (3 мас. %) показало наиболее высокую биостойкость.
Рис. 7.
Основные представители класса атранов: (а) металлатраны (M = Si, Ge и др., X – OAlk, Alk, Hal и др.); (б) протатраны (X – анион протонной кислоты); (в) гидрометаллатраны (M = Co, Cu, Ni и др., X – анион протонной кислоты).

В работе [193] исследован процесс биообрастания лакокрасочных покрытий шести различных составов на основе сополимера винилхлорида с винилацетатом с добавкой эпоксидно-диановой смолы в естественных условиях Белого моря. В качестве мягких биоцидов были использованы соединения атранового ряда – протатраны и гидрометаллатраны с общим содержанием 2 мас. %. Однако процесс биообрастания был зафиксирован на всех исследуемых пластинах. Основу обрастания составляли прикрепленные формы, наиболее массовыми из них оказались сцифистомы (полипоидная стадия) медуз Aurelia aurita и Cyanea sp., мидии Mytilus edulis и гидроид Obelia longissima. Развитие биообрастания на поверхности покрытий авторы связали с недостаточной (низкой) концентрацией биоцидов и их выщелачиванием из покрытий.
Наиболее популярной тенденцией в настоящее время является сочетание как биацидных, так и противообрастающих средств с новыми полимерными матрицами, химическими веществами и инновационными технологиями, что, благодаря синергетическому действию приводит к уменьшению дозировки биоцидов и достижению лучшего бактерицидного эффекта. Бактерицидный синергист является одним из наиболее распространенных применяемых химических агентов, который может разрушать биопленки, превращая “сидячие” бактерии в планктонные и таким образом усиливая бактерицидный эффект традиционного биоцида [194, 195].
В случае морского биообрастания противообрастатель в основном применяется для контроля активности микроорганизмов в биопленках или предотвращения их адгезии и образования биопленок. Как правило, противообрастатель вносится в полимерные связующие, используемые в так называемых “антиадгезионных покрытиях”. Наиболее популярными связующими, используемыми в морской отрасли в настоящее время, являются самополирующиеся сополимеры, в основном акриловые полимеры, к молекулам которых крепятся молекулы противообрастателя [196]. В самополирующихся сополимерах боковые группы, такие, например, как силильные, могут подвергаться гидролизу с образованием гидрофильной поверхности, которая затем полируется потоком воды, вымывая таким образом “прилипших” обрастателей. При этом реагенты-противообрастатели высвобождаются, позволяя контролировать активность микроорганизмов в биопленках [197, 198]. Однако обновление поверхности происходит недостаточно быстро в отсутствие сильного потока воды, поэтому большинство самопополирующихся покрытий имеют плохую устойчивость к морским биообрастателям в статических условиях. Более того, медленная эрозия поверхности и постоянное водопоглощение могут вызвать набухание покрытий, что отрицательно сказывается на механических и противообрастающих характеристиках.
В работах [199–203] разработаны диблок-сополимеры трет-бутилдиметилсилилметакрилата и метилметакрилата с контролируемой микроструктурой для противообрастающих покрытий, которые демонстрируют более контролируемую скорость эрозии и противообрастающую эффективность, чем покрытия на основе традиционных сополимеров [200].
В работе [204] рассмотрены подходы формирования покрытий, к поверхности или матрице которых “привязан” противомикробный агент (биоцид). Один из основных методов получения антимикробной поверхности включает двухэтапный подход. На первом этапе поверхность подвергается обработке для получения функциональных групп, поддающихся химическому присоединению. На втором этапе биоцид вступает во взаимодействие с функциональными группами поверхности, что приводит к его ковалентному связыванию. В случае использования полимеров с функциональными группами дополнительная модификация поверхности не требуется. В некоторых случаях поверхностная функциональность создается в присутствии биоцида, и реакция происходит немедленно. В качестве основных методов модификации поверхности используются: плазменная обработка, плазменное осаждение, облучение и химические методы.
Указанные методы либо преобразуют существующие группы поверхностей в реактивные центры, либо вводят на поверхность новые функциональные группы. Природа функциональных групп зависит от используемого субстрата. Для металлов и полимеров на основе углеводородов, таких как полиолефины, обычно формируют кислородсодержащие центры, а для полиамидов и других азотсодержащих полимеров образуются азотсодержащие группы. Плазменная (т.е. тлеющий разряд) обработка поверхности приводит к возникновению свободных радикалов, гидроксильных, амино- и пероксидных функциональных групп. Плазменная обработка поверхности приводит к реакции на самых внешних молекулярных слоях поверхности. С помощью аргоновой плазмы были модифицированы поверхности силиконового каучука [205, 206], полиэтилентерефталата [207], целлюлозы [207] и другие для антимикробного связывания. Кроме того, полиэтилен был функционализирован таким же образом с помощью гелиевой плазменной обработки [208]. Плазма аргона и гелия генерирует на поверхности свободные радикалы. Эти реакционноспособные частицы могут непосредственно взаимодействовать с соответствующими материалами или превращаться в гидроксильные и пероксидные группы при воздействии воздуха или кислорода. Родственной формой плазменной обработки поверхности является плазменное осаждение. Этот метод вводит функциональные группы путем воздействия на поверхность ионизированных паров осаждаемого материала. Плазменное осаждение мономера пропаналя было использовано для создания тонкого полимерного покрытия с альдегидными поверхностными группами на поверхности контактной линзы из гидроксиэтилметакрилата [209].
Другим методом модификации поверхности является использование излучения или электронного пучка. Ионизирующее излучение обычно используется для создания свободных радикалов на поверхности, которые вступают в реакцию в присутствии соответствующих функциональных групп. Используя 60Co в качестве источника гамма-излучения, Hu с соавторами [210] модифицировали полиэфирную поверхность для дальнейшего присоединения биоцидов.
Хотя плазменная и лучевая обработка являются превосходными методами, они часто требуют сложного инструментария или специальных технологических возможностей для создания соответствующих поверхностей. Традиционные химические методы также могут быть использованы для модификации поверхности функциональными группами для последующего связывания антимикробных препаратов. Часто используется подход, основанный на окислении поверхностных групп, с использованием перекиси водорода, смеси серной кислоты с перекисью водорода и других реагентов. Кроме окисления часто применяется подход, основанный на гидролизе связей внутри этих материалов с использованием кислотных или щелочных реагентов [204].
Для создания функциональных групп также используется обработка поверхности пламенем и коронным разрядом. Оба метода работают по химическому механизму окисления. Обработка пламенем использует открытое пламя для окисления поверхностных групп, в то время как для обработки коронным разрядом (также известная, как воздушная плазма) используется высокое напряжение для ионизации воздуха в зазоре между электродом и подлежащей обработке поверхностью. Данные методы экономически эффективны и очень распространены для повышения смачиваемости, адгезии и поверхностной энергии полимерных материалов, особенно полиолефинов. Однако они неспецифичны в отношении типов получаемых полярных групп [204].
Среди первых противомикробных агентов были использованы катионные биоциды на основе четвертичных аммониевых солей, связанных с поверхностью алкоксисилана, успешно выпущенных на коммерческий рынок фирмой “Dow Corning”. Биоцид, представленный на рис. 8а, обладал широким противомикробным действием, которое не уменьшалось после многократных последовательных промывок, что указывало на его “привязку” к поверхности покрытия. Далее широкий ряд соединений со схожим строением были протестированы на противомикробную активность.

Было выдвинуто предположение, что антимикробный механизм действия катионных биоцидов происходит через разрушение клеточной мембраны [211, 212]. Предполагается, что происходит адсорбция отрицательно заряженных клеток на катионных поверхностях, что облегчает антимикробную активность последних.
Эпоксиды являются одним из распространенных классов соединений, используемых в лакокрасочной промышленности [204]. В работе [213] изучен противогрибковый характер эпоксидных смол на основе бисфенола А с привязанным карбендазимом, термически отвержденным изофорондиамином. В работе [214] в качестве биоцидов были использованы четвертичные аммониевые соли (рис. 8б, 8в), привязанные к эпоксидной матрице.
Silva с соавторами [215] показали эффективность противообрастающего состава на основе полиуретановой и силиконовой матрицы, к поверхности которой ковалентно прикреплены биоциды (Иргарол 1051 и Эконеа) с помощью изоцианатного линкера. Покрытия на основе полидиметилсилоксана показали наиболее хорошие противообрастающие свойства как с одним биоцидом (Эконеа), так и с двумя биоцидами с их общим содержанием не более 0.6 мас. %. Испытуемые образцы оставались практически чистыми после экспозиции в течение более одного года в Атлантическом океане (побережье Португалии).
Многие антимикробные соединения воздействуют на микроорганизмы специфическими биохимическими путями, которые требуют химического проникновения в клетку. Существенной проблемой при привязывании некатионных биоцидов к поверхностям является поддержание активности в привязанном состоянии. Привязывание биоцида, скорее всего, изменяет механизм его противомикробного действия. Кроме того, постоянной проблемой для всех присоединенных биоцидов является снижение активности, наблюдаемое при продолжающемся микробном заражении или обрастании. Слой мертвых клеток или белка быстро дезактивирует контактную активность.
В литературе известен подход инкапсулирования биоцидов или ингибиторов коррозии в различные инертные наноматериалы (наноконтейнеры), которые затем диспергируются в состав полимерной матрицы. Инкапсуляция биоцидов в нанополости является интересной и инновационной стратегией для контроля высвобождения биоцидов и снижения их концентрации. Этот подход позволяет сохранить биоцид в матрице, предотвращая его выщелачивание и обеспечивая при этом экологически безопасное и умеренное высвобождение. Как правило, наноматериалы с большим объемом пор и большой площадью поверхности используются для инкапсулирования соединений с применением либо метода физической адсорбции, либо физико-химического взаимодействия после стадии функционализации поверхности [216, 217]. При физической адсорбции наночастицы, например, SiO2 используются в качестве каркасов, на которых адсорбируется биологически активное соединение. Этот метод относительно прост и позволяет загружать наночастицы различными типами биоцидов в зависимости от растворителя. Однако имеет существенный недостаток ‒ быстрое выщелачивание биоцидов с поверхности [218, 219]. При использовании второго подхода, основанного на физико-химическом взаимодействии, поверхность наночастиц функционализируется для связывания с биоцидом. Этот подход позволяет увеличить время выщелачивания, однако является более сложным, трудоемким и специфичен для каждого класса биоцидов. В работе [220] рассмотрен подход инкапсуляции/инкорпорации экологически чистых биоцидов в инертные наносистемы (наноконтейнеры) на основе диоксида кремния. В работах [221, 222] этот подход был апробирован на примере коммерческого ингибитора коррозии ‒ 2-меркаптобензотиазола, инкапсулированного в нанокапсулы мезопористого диоксида кремния.
Из-за глобальной проблемы токсичности биоцидов исследования последних лет сосредоточены на экологически чистых технологиях, таких как применение гидрофильных и гидрофобных противообрастающих покрытий [223].
Гидрофильные противообрастающие покрытия не дают возможности или существенно задерживают прикрепление морских биообрастателей к корпусам морских судов за счет их супергладкой поверхности. Для придания гидрофильности поверхности в работах [224–226] использовали хитозан, которым обрабатывали наночастицы, например, такие как оксиды меди(I) и цинка. Покрытия с введенными хитозан-модифицированными наночастицами показали превосходную противообрастающую эффективность в течение 30 суток без вреда для окружающей среды. В работе [227] противообрастающее действие полиуретанового покрытия было улучшено введением в их состав наночастиц оксида цинка, модифицированного полианилином, за счет повышения гидрофильности и смачивания, что привело к значительному уменьшению биопленки на поверхности по сравнению с немодифицированным полиуретановым покрытием. Для повышения гидрофильности покрытий часто применяется полиэтиленгликоль (ПЭГ). Поверхность ПЭГ характеризуется устойчивостью к адсорбции белков и антиадгезионными свойствами к морским обрастающим организмам [228]. ПЭГ является нейтральным гидрофильным полимером, состоящим из повторяющихся оксиэтиленовых групп (−CH2CH2O–). Благодаря своей уникальной структуре ПЭГ может участвовать в образовании водородных связей с водой, что придает ему высокую степень гидрофильности и хорошую растворимость в воде. ПЭГ обычно используется в качестве гидрофильного сегмента амфифильных полимеров. В работе [229] получены ПЭГ-модифицированные наночастицы SiO2, которые были введены в акрил-полиуретановые покрытия, что позволило снизить краевой угол смачивания до 38.7°.
В последнее время применение гидрогелей в противообрастающих покрытиях привлекает все большее внимание исследователей. Гидрогель представляет собой сеть сшитых полимерных цепей, которые являются гидрофильными. Трехмерное твердое состояние является результатом того, что гидрофильные полимерные цепи удерживаются вместе поперечными связями.
Как правило, гидрофобные покрытия содержат фторсодержащие и силиконовые соединения, которые характеризуются высоким углом смачивания и низкой поверхностной энергией. Осевшие микро- и макрообрастатели непрочно прикрепляются к поверхности таких покрытий и удаляются механически при движении судна [230]. Гидрофобные покрытия представляют собой экологически чистую альтернативу традиционным противообрастающим покрытиям с высоким сроком службы. Гидрофобные и супергидрофобные материалы обладают рядом уникальных свойств – водонепроницаемостью, стойкостью к коррозии, устойчивостью к биообрастанию, к неорганическим, а в ряде случаев и к органическим загрязнениям. Вблизи гидрофобной поверхности таких материалов облегчено скольжение жидкого потока [231]. Для получения материалов с высокими краевыми углами необходимо использовать совместное влияние шероховатости поверхности и химической структуры. В настоящее время для придания гидрофобности поверхности активно используются следующие методы: полимеризация покрытия из раствора с образованием губчатой фазы; плазменное травление поверхности полимеров; химическое осаждение из паров упорядоченных структур с последующей обработкой гидрофобными материалами; электроосаждение и электрохимическое осаждение наночастиц и пленок с последующей обработкой гидрофобными материалами, применение темплатных методов для создания шероховатости с последующим удалением темплата и обработкой гидрофобными материалами и др.
Однако основным недостатком гидрофобных и супергидрофобных покрытий являются процессы старения и деградации, которые приводят к потере гидрофобности. Одна из причин потери гидрофобности покрытий при эксплуатации на открытом воздухе связана с атмосферными загрязнениями. Как правило, в результате осаждения на поверхности пыли и химических веществ органической природы значительно усиливается ее гидрофильность [231]. В работе [232] показано, что под воздействием морской волны и взвешенных в ней примесей шероховатость супергидрофобного покрытия нарушалось, и краевой угол уменьшался от 151° до 70° через 35 дней эксперимента.
Для борьбы с биообрастанием поверхностей может быть использовано температурно переключаемое смачивание, наблюдаемое в ряде систем, имеющих нижнюю критическую температуру растворения [231]. Наиболее изученным полимером, обладающим этим свойством, является поли(N-изопропилацетамид). Переход смачивания связывают с изменением конформации молекул полимера, привитых к поверхности. В работе [233] показано, что такой конформационный переход при повышении температуры вызывает изменение угла смачивания поверхности с 63° до 93°. Путем нанесения текстуры в виде канавок микрометрового размера можно добиться перехода от супергидрофобности к супергидрофильности с очень малым гистерезисом смачивания. Система выдерживает многократное переключение режимов смачивания без деградации покрытия.
Разработка покрытий c динамической поверхностью является еще одним подходом к получению противообрастающих покрытий. Динамическая поверхность относится к изменяющейся поверхности, которая постоянно обновляется в морской воде и, следовательно, уменьшает адгезию биообрастателей [234, 235]. Известны покрытия с динамической поверхностью на основе полиэфир-полиуретановой матрицы, модифицированной биоразлагаемыми полимерами, такими как, полиэфир, полиэфир-акрилат [223]. Такие биоразлагаемые полимеры использовались в качестве линкеров, которые контролировали скорость высвобождения экологически безопасных антиобрастающих агентов и демонстрировали отличное противообрастающее действие с регулируемой возобновляемостью [236, 237].
Механизм высвобождения антиобрастающих веществ включает две стадии: биодеструкция и эрозия (разрушение с поверхности). Под воздействием факторов окружающей среды (тепло, свет, хим. вещества) полимер подвергается деструкции с изменением свойств, таких как прочность на растяжение, цвет, форма и т.д. [238, 239]. Процессы физической эрозии можно представить как гетерогенные или гомогенные. При гетерогенной эрозии (или поверхностной эрозии) полимер разрушается только на поверхности и при этом сохраняет свою физическую целостность при деструкции. Большинство полимеров подвергаются гомогенной эрозии, что означает, что гидролиз происходит равномерно по всей полимерной матрице [240, 241].
Авторы работы [242] обнаружили, что полиуретан на основе сополимера капролактона и гликолида демонстрирует высокие скорости разложения, которые возрастают с увеличением содержания гликолида. Испытание в морской воде в течение 3 мес. для полиуретана с 10 мол. % гликолида показало наилучшие противообрастающие свойства без какого-либо противообрастающего вещества. Это указывает на то, что разлагаемый полимер сам по себе эффективен в противообрастании.
В работе [196] также отмечается, что разлагаемый полимер эффективнее для противообрастающих покрытий, чем традиционный самополирующийся сополимер. Его постоянная скорость обновления поверхности в морской воде более стабильно выделяет противообрастатель, что может продлить срок службы покрытий и уменьшить загрязнение окружающей среды. Можно заключить, что сочетание эффективного противообрастателя с разлагаемым полимерным связующим с динамической поверхностью может стать потенциальным методом предотвращения морского биообрастания.
Деструкция полимера создает изменяющуюся динамическую поверхность, которая предотвращает постоянную бактериальную адгезию из-за снижения адгезионной прочности. Процесс биодеструкции полимера происходит на протяжении всего погружения в морскую воду, поскольку всегда происходит гидролитическая и ферментативная деструкция [236]. С другой стороны, биоразлагаемые полимеры могут подвергаться ферментативному и гидролитическому расщеплению основной полимерной цепи, в результате чего образуются олигомеры или небольшие молекулы, которые через определенный период времени превращаются в углекислый газ и воду. Следует отметить, что биоразлагаемые участки полимеров представляют собой очень мелкие частицы и поэтому не оказывают негативного воздействия на окружающую среду [223].
Таким образом, как МБК, так и биообрастание в морской среде являются следствием адгезии и роста морских микроорганизмов на поверхности материалов, и образование биопленок играет важную роль в возникновении и протекании этих процессов. Исследования показали, что биопленку трудно удалить с поверхности материала. Для уничтожения “сидячих” бактерий, растущих в биопленках, необходимо использовать высокие концентрации биоцидов, которые значительно выше, чем для бактерий в планктонном состоянии [243, 244].
ВНЕШНЕЕ ЭЛЕКТРИЧЕСКОЕ ПОЛЕ В БОРЬБЕ С БИОКОРРОЗИЕЙ
В работе [245] впервые было высказано предположение, что внеклеточные полимерные вещества (ВПВ), состоящие из белков и полисахаридов и являющиеся метаболитами микроорганизмов, защищающие бактерии от антибактериальных препаратов, участвуют в связывании биоцидов до того, как они достигнут клеток-мишеней. Это вызвано тем, что экзополисахарид, содержащийся в ВПВ, заряжен и обладает ионообменными свойствами. Основываясь на этой гипотезе, авторы работы [246] предположили, что разрушение зарядов на ВПВ может значительно облегчить проникновение биоцидов в клетки-мишени. Было также обнаружено, что некоторые промышленные биоциды могут оказывать усиленное действие против биопленок P. aeruginosa в слабом электрическом поле с низкой плотностью тока. Однако природа механизмов остается до конца неясной, но может быть результатом электропорации, электрофореза, ионтофореза и др. Авторы работы [247] также исследовали совместное влияние электрического поля и биоцида на разрушение биопленок СВБ. Результаты показали, что дополнительное электрическое поле практически не влияет на процесс формирования биопленок, но при этом повреждает структуру образовавшихся биопленок и способствует как массопереносу биоцидов внутрь биопленок, так и десорбции ионов кальция и магния из биопленок. Указанные закономерности приводят к возникновению эффективного синергетического эффекта внешнего электрического поля с биоцидами. Учитывая положительное влияние внешнего электрического поля на разрушение биопленок, некоторые электроактивные материалы, которые могут сами создавать микроэлектрическое поле (например, пьезоэлектрические материалы), можно рассматривать как потенциальные материалы для предотвращения МБК и биологического обрастания.
ПРОВОДЯЩИЕ ПОЛИМЕРЫ
Ввиду электрической активности биопленок на поверхностях материалов в морской среде проводящие полимеры считаются потенциальными материалами для предотвращения морской коррозии и биообрастания. Исследования антикоррозионных свойств проводящих полимеров начались с работ [248, 249], в которых впервые обнаружили, что электроосажденная пленка из полианилина на нержавеющей стали может значительно снизить скорость коррозии стали в растворе серной кислоты. В работах [250–255] изучена коррозионная стойкость различных проводящих полимеров, таких как полианилин, полипиррол, политиофен и их производных. Большинство этих исследований было сосредоточено на традиционных механизмах коррозионной защиты металлов, таких как формирование пассивирующего слоя на поверхности металла, увеличение потенциала коррозии металла и снижение скорости коррозии. В последнее время все больше исследователей стали обращать внимание на возможности использования проводящих полимеров для предотвращения МБК и биообрастания. В связи с этим были проведены дополнительные исследования и выдвинуты следующие концепции:
а) Регулирование значений pH проводящего полимерного покрытия для его стабилизации в кислотном диапазоне с целью предотвращения адгезии и роста микроорганизмов и морских организмов, приспособленных к щелочной морской воде на поверхности материала [256].
б) Проводящие полимеры можно использовать в качестве анода, а части металлов, которые контактируют с морской водой ‒ в качестве катода. Когда между этими двумя электродами подается слабый ток, поверхностная морская вода электролизуется до гипохлорита натрия и затем образует ионную мембрану, которая может повредить клеточные ткани организмов-обрастателей. Следует отметить, что концентрация гипохлорита натрия в морской воде достаточно низкая, поэтому его образование не оказывает существенного влияния на окружающую среду [256, 257].
в) Проводящие полимеры с проводимостью выше 109 См/см можно применять в качестве основной матрицы покрытия без использования тока [258]. В работе [259] такая возможность была продемонстрирована на примере полианилина. Было показано, что полианилин обладает хорошими противообрастающими свойствами в морской среде без применения дополнительного электрического тока. Противообрастающий эффект полианилина усиливается при использовании биоцидов, таких как оксид меди(I) или дихлордифенилтрихлорэтан (синергетический эффект). Недавние исследования [260] показали, что проводящий полимер полипиррол под действием электрического тока способен изменять гидрофобность своей поверхности. Это указывает на то, что полипиррол может создавать электрически управляемую амфифильную поверхность для предотвращения адгезии морских микроорганизмов.
Представленные исследования продемонстрировали, что проводящие полимеры являются перспективными полимерными матрицами для разработки антикоррозионных и противообрастающих покрытий.
ЗАКЛЮЧЕНИЕ
Микробиологическая коррозия и биообрастание материалов являются двумя основными причинами морской коррозии, вызывающей повреждение и выход из строя оборудования и конструкций, обслуживаемых в морской среде. Стратегии предотвращения МБК и биообрастания в основном сосредоточены на контроле активности микроорганизмов в биопленках, их адгезии и формировании биопленок. Новые материалы и технологии в сочетании с традиционными биоцидами или противообрастающими средствами за счет синергетического действия, позволяют снизить их дозировку и одновременно достичь лучшего бактерицидного эффекта. С другой стороны, для используемых в настоящее время противообрастающих веществ отсутствует четкое понимание токсикологического фона, что требует проведения дополнительных исследований для изучения их неблагоприятных эффектов и механизмов действия на морские организмы, особенно после продолжительного воздействия. Кроме того, поскольку противообрастающие средства и биоциды обычно используются одновременно, их комбинированное действие остается неизученным и представляет потенциальный риск для здоровья морской экосистемы. Следующая неопределенность, вызывающая особое беспокойство, заключается в том, что большинство природоохранных ведомств не включили противообрастающие средства и биоциды в регулярную программу мониторинга для изучения их присутствия в морской среде. Если бы мы получили полное представление о деструкции, загрязнении и токсичности этих соединений, то стало бы возможно четко контролировать и регулировать их применение. Это, в свою очередь, привело бы к продвижению противообрастающей промышленности вперед и, в конечном итоге, позволило бы найти оптимальные противообрастающие вещества, являющиеся одновременно эффективными и экологически безопасными.
Для разработки высокоэффективной противообрастающей системы выбор биоцидного нанокомпозита оказывает существенное влияние на гидрофобность/гидрофильность покрытия. По сравнению с традиционными биоцидами нанокомпозитный биоцид высвобождается в меньшем количестве и демонстрирует отличные противообрастающие характеристики. Кроме того, подход, основанный на использовании биоразлагаемого полимера в сочетании с экологически безопасным противообрастающим веществом, демонстрирует длительную защиту от биообратания, а также позволяет контролировать высвобождение противообрастающего вещества за счет постоянной возобновляемости поверхности.
Тенденция развития методов борьбы с МБК и биообрастанием заключается в поиске методов и материалов, которые обладают высокой эффективностью, длительным сроком службы, простым процессом реализации, низкой стоимостью и являются безопасными для окружающей среды. Научные подходы, представленные в этом обзоре, продемонстрировали перспективы борьбы с биокоррозией и биообрастанием. Можно надеяться, что синергетическое действие химических веществ, материалов и инновационных технологий будет в будущем очень важным направлением исследований в области морской защиты от коррозии и обрастания.
Перспективным направлением современного материаловедения является разработка многофункциональных покрытий с динамической поверхностью, а также экологически безопасных биоцидов с контролируемой скоростью высвобождения, для обеспечения эффективной защиты морской техники от главной проблемы современного материаловедения – биологического обрастания.
Список литературы
Gaines H.A. // J. Engineer. Ind. Chem. 1910. V. 2. P. 128–130.
Gu. T. // J. Microbial. Biochem. Technol. 2012. V. 4. P. 4.
Kip N., van Veen J.A. // The ISME Journal. 2015. V. 9. P. 542–551.
Андреюк Е.И., Билай В.И., Коваль Э.З., Козлова И.А. Микробная коррозия и ее возбудители. Киев: Наук. думка, 1980. 288 с.
Jia R., Unsal T., Xu D. et al. // International biodeterioration and biodegradation. 2019. V. 137. P. 42–58.
Пехташева Е.Л., Неверов А.Н., Заиков Г.Е. и др. // Вестник Казанского технологического университета. 2012. Т. 15. № 5. С. 131–133.
Горленко М.В. Некоторые биологические аспекты биодеструкции материалов и изделий / Биоповреждения в строительстве. М., 1984. С. 9–17.
Соломатов В.И., Ерофеев В.Т., Смирнов В.Ф. и др. Биологическое сопротивление материалов. Саранск, 2001. 196 с.
Коваль Э.З. Микодеструкторы промышленных материалов / Коваль Э.З., Сидоренко Л.П. Киев: Наук. думка, 1989. 192 с.
Zuo R. // Appl. Microbiol. Biotechnol. 2007. V. 76. 1245–1253.
Вигдорович В.И., Шель Н.В., Крылова А.Г. // Вестник Тамбовского университета. Серия: Естественные и технические науки. 2001. Т. 6. № 3. С. 279–289.
Семенов С.А., Гумаргалиева К.З., Заиков Г.Е. // Вестник МИТХТ им. М.В. Ломоносова. 2008. Т. 3. № 2. С. 1–21.
Алехова Т.А., Александрова А.В., Загустина Н.А. и др. // Микология и фитопатология. 2009. Т. 43. № 5. С. 377.
Михайлов А.А., Стрекалов П.В. // Коррозия: материалы, защита. 2006. № 3. С. 213.
Карпов В.А., Ковальчук Ю.Л., Харченко У.В., Беленева И.А. // Коррозия: материалы, защита. 2011. № 3. С. 11–18.
Харченко У.В., Беленева И.А., Ковальчук Ю.Л., Карпов В.А. // Коррозия: материалы, защита. 2010. № 12. С. 30–34.
Андреюк Е.И., Билай В.И., Коваль Э.З., Козлова И.А. Микробная коррозия и ее возбудители. Киев: Наук. думка, 1980. 286 с.
Ильичев В.Д. Биоповреждения. М.: Высш. шк., 1987. 352 с.
Герасименко А.А. // Защита металлов. 1998. № 2. С. 192–207.
Панова О.А., Великанова Л.Л., Тимонин В.А. // Микология и фитопатология. 1982. Т. 16. № 6. С. 514–518.
Vaiday R.U., Butt D.P., Hersman L.E., Zurek A.K. // Corrosion. 1997. V. 53. P. 136–141.
Костицына И.В., Паршуков В.П., Бирюков А.И., Тюрин А.Г. // Вестник Южно-Уральского государственного университета. Серия: Химия. 2011. № 12(229). С. 54–57.
Фрелунд Б., Шмидт Х. // Водоочистка. Водоподготовка. Водоснабжение. 2009. № 3. С. 40–42.
Пантелеева А.Р., Андреева Ю.В., Егорова С.В. и др. // Практика противокоррозионной защиты. 2008. № 3. С. 40–43.
Семенова И.В., Флорианович Г.М., Хорошилов А.В. Коррозия и защита от коррозии. М.: Физматлит, 2002, 335 с.
Melchers R.E. // Bioelectrochemistry. 2014. V. 97. P. 89–96.
Li Y., Ning C. // Bioactive Materials. 2019. V. 4. P. 189–195.
Колесникова Н.Н., Луканина Ю.К., Хватов А.В. и др. // Вестник Казанского технологического университета. 2013. Т. 16. № 1. С. 170–174.
Lejars M., Margaillan A., Bressy C. // Chem. Rev. 2012. V. 112. P. 4347–4390.
Selim M.S., Shenashen M.A., El-Safty S.A. et al. // Prog. Mater. Sci. 2017. V. 87. P. 1–32.
Grozea C.M., Walker G.C. // Soft Matter. 2009. V. 5. P. 4088–4100.
Lindholdt A., Dam-Johansen K., Olsen S.M. et al. // J. Coat. Technol. Res. 2015. V. 12. P. 415–444.
Liu H.W., Gu T.Y., Asif M. et al. // Corros. Sci. 2017. V. 114. P. 102–111.
Chen B., Qin S., Chen L. et al. // Corros. Sci. Prot. Technol. 2014. V. 26. P. 499–504.
Castaneda H., Benetton X.D. // Corros. Sci. 2008. V. 50. P. 1169–1183.
Videla H.A., Herrera L.K. // Int. Microbiol. 2005. V. 8. P. 169–180.
Enning D., Garrelfs J. // Appl. Environ. Microbiol. 2014. V. 80. P. 1226–1236.
Heidelberg J.F., Seshadri R., Haveman S.A. et al. // Nat. Biotechnol. 2004. V. 22. P. 554–559.
Chamritski I.G., Burns G.R., Webster B.J., Laycock N.J. // Corrosion. 2004. V. 60. P. 658–669.
Barton L.L., Tomei F.A. Chapter 1. Characteristics and activities of sulfate-reducing bacteria / In: Barton L.L. (Ed.) Sulfate-reducing Bacteria // Springer, Boston, MA, 1995. P. 1–32.
Dinh H.T., Kuever J., Mussmann M. et al. // Nature. 2004. V. 427. P. 829–832.
Till B.A., Weathers L.J., Alvarez P.J.J. // Environmental Science & Technology. 1998. V. 32. P. 634–639.
Linhardt P. // Mater. Corros. 2010. V. 61. P. 1034–1039.
Rajendran A. // Corrosion Sci. 2007. V. 49. P. 2694–2710.
Li Y.C., Xu D.K., Chen C.F. et al. // J. Mater. Sci. Technol. 2018. V. 34. P. 1713–1718.
Makhlouf A.S.H., Botello M.A. Chapter 1. Failure of the metallic structures due to microbiologically induced corrosion and the techniques for protection / In: Handbook of Materials Failure Analysis. 2018. P. 1–18.
Fink J.K. Chapter 5. Bacteria control / In: Petroleum Engineer’s Guide to Oil Field Chemicals and Fluids // Gulf Professional Pub, Waltham, MA. 2012. P. 185–216.
Javaherdashti R. Microbiologically Influenced Corrosion: An Engineering Insight / Springer, London, 2008.
Anandkumar B., George R.P., Maruthamuthu S. et al. // Corrosion Reviews. 2016. V. 34. P. 41–63.
Hernandez M.E., Newman D.K. // Cell. Mol. Life Sci. 2001. V. 58. P. 1562–1571.
Zhang P., Xu D.K., Li Y. et al. // Bioelectrochemistry. 2015. V. 101. P. 14–21.
Du Z., Li H., Gu T. // Biotechnol. Adv. 2007. V. 25. P. 464–482.
Грибанькова А.А., Мямина М.А., Белоглазов С.М. // Вестник Балтийского федерального университета им. И. Канта. 2011. № 7. С. 23–29.
King R.A., Miller J.D.A. // Nature. 1971. V. 233. P. 491–492.
Bothe H., Ferguson S.J., Newton, W.E. Biology of Nitrogen Cycle. Elsevier Science, Amsterdam, 2007.
Kutvonen H., Rajala P., Carpen L., Bomberg M. // Front. Microbiol. 2015. V. 6. Article 1079.
Luque-Almagro V.M., Gates A.J., Moreno-Vivian C. et al. // Biochem. Soc. Trans. 2011. V. 39. P. 1838–1843.
Kielemoes J., De Boever P., Verstraete W. // Environ. Sci. Technol. 2000. V. 34. № 4. P. 663–671.
Huang C.P., Wang H.W., Chiu P.C. // Water Res. 1998. V. 32. P. 2257–2264.
Zawaideh L.L., Zhang T.C. // Water Sci. Technol. 1998. V. 38. P. 107–115.
Chew F.C., Zhang T.C. // Water Sci. Technol. 1998. V. 38. P. 135–142.
Van Hecke K., Van Cleemput O., Baert L. // Environ. Pollut. 1990. V. 63. P. 261–274.
Van Hecke K., Van Cleemput O., Baert L. Theoretical aspects on chemodenitrification of nitrate in groundwater / In Calvet R. (ed.), Nitrates-Agriculture-Eau; Intern. Symposium, Paris, France, 7–8 November 1990. P. 215–220.
Iino T., Ito K., Wakai S. et al. // Applied and Environmental Microbiology. 2015. V. 81. № 5. P. 1839–1846.
Uchiyama T., Ito K., Mori K. et al. // Appl. Environ. Microbiol. 2010. V. 76. № 6. P. 1783–1788.
Ghiorse W.C. // Annu. Rev. Microbiol. 1984. V. 38. P. 515–550.
Harder E.C. Iron-Depositing Bacteria and Their Geological Relations / Washington, DC: US Geol. Survey. Professional Paper 113, 1919.
Emerson D., Fleming E.J., McBeth J. M. // Annu. Rev. Microbiol. 2010. V. 64. P. 561–583.
Ehrlich H.L., Ingledew W.J., Salerno J.C. Iron- and manganese-oxidizing bacteria / In: ed. Shivley J.M., Barton L.L., Variations in Autotrophic Life // Academic Press, New York. 1991. P. 147–170.
Roden E.E., Sobolev D., Glazer B., Luther G.W.I. // Geomicrobiol. J. 2004. V. 21. P. 379–391.
Emerson D. // Biofouling. 2018. V. 34. P. 989–1000.
Mumford A.C., Adaktylou I.J., Emerson D. // Appl. Environ. Microbiol. 2016. V. 82. P. 6799–6807.
Tebo B.M., Johnson H.A., McCarthy J.K., Templeton A.S. // 2005. Trends Microbiol. 2005. V. 13. P. 421–428.
Martinez-Ruiz E.B., Cooper M., Fastner J., Szewzyk U. // Chemosphere. 2020. V. 238. № 124625.
Dubiel M., Hsu C.H., Chien C.C. et al. // Appl. Environ. Microbiol. 2002. V. 68. P. 1440–1445.
Tebo B.M., Bargar J.R., Clement B.G. et al. // Annu. Rev. Earth Planet Sci. 2004. V. 32. P. 287–328.
Hennebel T., De Gusseme B., Boon N., Verstraete W. // Trends Biotechnol. 2009. V. 27. P. 90–98.
Nealson K.H. // Prokaryotes. 2006. V. 5. P. 222–231.
Stumm W., Morgan J.J. Aquatic Chemistry: An Introduction Emphasizing Chemical Equilibria in Natural Waters / 2nd Edition, John Wiley & Sons Ltd., New York, 1981.
Ehrlich H.L. Geomicrobiology. Marcel Dekker, New York. 1981. P. 165–249.
Jones D., Amy P. // Corrosion. 2002. V. 58. P. 638–645.
Rao T., Sairam T., Viswanathan B., Nair K. // Corros. Sci. 2000. V. 42. P. 1417–1431.
Yuan S., Liang B., Zhao Y., Pehkonen S. // Corros. Sci. 2013. V. 74. P. 353–366.
Lenhart T.R., Duncan K.E., Beech I.B. et al. // Biofouling. 2014. V. 30. P. 823–835.
Huber B., Herzog B., Drewes J.E. et al. // BMC Microbiology. 2016. V. 16. № 153.
Roberts D.J., Nica D., Zuo G., Davis J.L. // Int. Biodeter. Biodegr. 2002. V. 49. № 4. P. 227–234.
Bielefeldt A., Gutierrez-Padilla M.G.D., Ovtchinnikov S. et al. // J. Environ. Eng. 2009. V. 136. № 7. P. 731–738.
Diercks M., Sand W., Bock E. // Experientia. 1991. V. 47. № 6. P. 514–516.
Wei S., Jiang Z., Liu H. et al. // Braz. J. Microbiol. 2013. V. 44. № 4. P. 1001–1007.
O’Connell M., Mcnally C., Richardson M.G. // Cem. Concr. Compos. 2010. V. 32. № 7. P. 479–485.
Wells T., Melchers R.E. // Cem. Concr. Res. 2015. V. 77. P. 82–96.
Alabbas F. M., Mishra B. Microbiologically Influenced Corrosion of Pipelines in the Oil & Gas Industry // In book: Proceedings of the 8th Pacific Rim International Congress on Advanced Materials and Processing. 2013. P. 3441–3448.
Little B., Lee J. Microbiologically Influenced Corrosion / Wiley-Interscience A John Wiley & Sons: Hoboken, NJ, USA, 2007.
Gu T., Rastegar S.O., Mousavi S.M. et al. // Bioresour.Technol. 2018. V. 261. P. 428–440.
Little B., Wagner P., Mansfeld F. // Electrochimica acta. 1991. V. 37. № 12. P. 2194–2185.
Wolfgang S. // International Biodeterioration and Biodegradation. 1997. V. 40. № 2. P. 183–190.
Sowards J.W., Mansfield E. // Corrosion Sci. 2014. V. 87. P. 460–471.
Dierksen D., Kühner P., Kappler A., Nickel K.G. // J. Eur. Ceram. Soc. 2011. V. 31. P. 1177–1185.
Skovhus T.L., Enning D., Lee J.S. (Eds.) Microbiologically Influenced Corrosion in the Upstream Oil and Gas Industry / CRC Press, Boca Raton, Florida, 2017.
Joosten M.W., Kolts J., Hembree J.W. // Corrosion. 2002. Paper № 02294. (Houston, TX: NACE, 2002).
Заикина Н.А., Дуганова Н.В. // Микология и фитопатология. 1975. Т. 9. № 4. С. 303–306.
Сазанова К.В., Щипарев С.М., Власов Д.Ю. // Микробиология, 2014. Т. 83. № 5. С. 525–533.
Kokilaramani S., Al-Ansari M.M., Rajasekar A. et al. // Chemosphere. 2021. V. 265. Paper № 129075.
Little B., Ray R. // Corrosion Reviews. 2001. V. 19. P. 401–418.
Binkauskiene E., Bucinskiene D., Lugauskas A. // Long-time corrosion of metals and profiles of fungi on their surface in outdoor environments in Lithuania / In: Prasad R. (eds) Mycoremediation and Environmental Sustainability. Fungal Biology // Springer, Cham. 2017.
Qu Q., Wang L., Li L. et al. // Corrosion Science. 2015. V. 98. P. 249–259.
Zhang T., Wang J., Zhang G., H. Liu H. // Corrosion Science. 2020. V. 176. Paper № 108930.
Zhang D., Zhou F., Xiao K. et al. // J. Mater. Eng. Perform. 2015. V. 24. P. 2688–2698.
Bohlmann J.T., Cameselle C., Nunez M.J., Lema J.M. // Bioprocess Eng. 1998. V. 19. P. 337–342.
Ozdal M., Kurbanoglu E.B. // Waste Biomass Valorization. 2018. V. 10. P. 1–10.
Lu F., Ping K., Wen L. et al. // Process Biochem. 2015. V. 50. P. 1342–1348.
Wang J., Xiong F., Liu H. et al. // Bioelectrochemistry. 2019. V. 129. P. 10–17.
Dai X., Wang H., Ju L.K. et al. // Int. Biodeterior. Biodegrad. 2016. V. 115. P. 1–10.
Juzeliunas E., Ramanauskas R., Lugauskas A. et al. // Corros. Sci. 2007. V. 49. P. 4098–4112.
Landoulsi J., Cooksey K.E., Dupres V. // Biofouling. 2011. V. 27. P. 1105–1124.
Olivia M., Moheimani N., Javaherdashti R. et al. // Adv. Mat. Res. 2013. V. 626. P. 861–866.
Samimi A. // International Journal of Basic and Applied science. 2013. V. 1. P. 705–715.
Allwright H., Enshaei H. // J. Basic. Appl. Sci. Res. 2016. V. 6. P. 28–37.
Edyvean R.G.J., Terry L.A. Chapter 15. Recent Investigations into the Effects of Algae on Corrosion / In: Evans L.V., Hoagland K.D. (eds) Algal Biofouling // Studies in Environmental Science. 1986. V. 28. P. 211–229.
Kamal C., Sethuraman M.G. // Research on Chemical Intermediates. 2012. V. 39. P. 3813–3828.
Gaylarde C.C., Morton L.H.G., Loh K., Shirakawa M.A. // Int. Biodeter. Biodegrad. 2011. V. 65. P. 1189–1198.
Vlasov D.Yu., Panova E.G., Zelenskaya M.S. et al. Chapter 6. Changes of Granite Rapakivi under the Biofouling Influence / In: Rene M., Aiello G., Bahariya G.E. (eds.), Geochemistry // IntechOpen, 2020.
Светлов Д.А., Качалов А.Н. // Транспортные сооружения. 2019. Т. 6. № 4. С. 1–19.
Lamenti G., Tiano P., Tomaselli L. // J. Appl. Phycol. 2000. V. 12. P. 427–433.
Warscheid T., Braams J. // Int. Biodeter. Biodegrad. 2000. V. 46. P. 343–368.
Sazanova K., Osmolovskaya N., Schiparev S. et al. // Current Microbiology. 2015. V. 70. № 4. P. 520–527.
Crispim C.A., Gaylarde C.C. // Microbial. Ecol. 2005. V. 49. P. 1–9.
Graedel T.E. // J. Electrochemical Society. 2000. V. 147. P. 1006–1009.
Amoroso G.G., Fassina V. Stone Decay and Conservation: Atmospheric Pollution, Cleaning, Consolidation and Protection (Materials Science Monographs) / Elsevier Science Ltd. 1983. P. 299-432.
Wendler E. / In: Saving our Architectural Heritage, Baer N.S. and Snethlage R., (Eds.) // John Wiley, Chichester, U.K. 1997. P. 181–196.
Han X., Wu J., Zhang X. et al. // J. Materials Science & Technology. 2021. V. 61. P. 46–62.
Vlasov D.Yu., Parfenov V.A., Zelenskaya M.S. et al. Methods of Monument Protection from Damage and Their Performance / In: Frank-Kamenetskaya O.V., Vlasov D.Yu., Rytikova V.V., The Effect of the Environment on Saint Petersburg’s Cultural Heritage. Results of Monitoring the Historical Necropolis Monuments // Springer. 2019. P. 161–178.
Ulaeto S.B., Rajan R., Pancrecious J.K. et al. // Prog. Org. Coat. 2017. V. 111. P. 294–314.
Batista-Andrade J.A., Caldas S.S., Batista R.M. et al. // Environ. Pollut. 2018. V. 234. P. 243–252.
Ma C., Zhang W., Zhang G., Qian P. // ACS Sustainable Chem. Eng. 2017. V. 5. P. 6304–6309.
Murai R., Takahashi S., Tanabe S., Takeuchi I. // Mar. Pollut. Bull. 2005. V. 51. P. 940–949.
Gall S.C., Thompson R.C. // Mar. Pollut. Bull. 2015. V. 92. P. 170–179.
Lahbib Y., Abidli S., Trigui-El Menif N. // Mar. Pollut. Bull. 2018. V. 128. P. 17–23.
Detty M.R., Ciriminna R., Bright F.V., Pagliaro M. // Acc. Chem. Res. 2014. V. 47. P. 678–687.
Yebra D.M., Kiil S., Dam-Johansen K. // Prog. Org. Coat. 2004. V. 50. P. 75–104.
Rath S.K., Chavan J.G., Ghorpade T.K. et al. // J. Coat. Technol. Res. 15 (2018). 185–198.
Ytreberg E., Karlsson J., Eklund B. // Sci. Total Environ. 2010. V. 408. P. 2459–2466.
Koppel D.J., Gissi F., Adams M.S. et al. // Environ. Pollut. 2017. V. 228. P. 211–221.
Yang W.W., Li J., Zhou P. et al. // Chem. Eng. J. 2017. V. 327. P. 849–854.
Thomas K. Chapter 20. The use of broad-spectrum organic biocides in marine antifouling paints / In: Advances in Marine Antifouling Coatings and Technologies. // Woodhead Publishing. 2009. P. 522–553.
Scarlett A., Donkin P., Fileman T.W., Morris R.J. // Mar. Pollut. Bull. 1999. V. 38. P. 687–691.
Hall JR L.W., Giddings J.M., Solomon K.R., Balcomb R. // Critical Reviews in Toxicology 1999. V. 29. P. 367–437.
Thomas K.V., Mchugh M., Waldock M. // Sci. Total Environ. 2002. V. 293. P. 117–127.
Thomas K.V., Mchugh M., Hilton M., Waldock M. // Environ. Pollut. 2003. V. 123. P. 153–161.
Readman J.W., Kwong L.L.W., Grondin D. et al. // Environmental Science and Technology. 1993. V. 27. P. 1940–1942.
Amara I., Miled W., Slama R.B., Ladhari N. // Environmental Toxicology and Pharmacology. 2018. V. 57. P. 115–130.
Bannink, A.D. //Water Sci. Technol. 2004. V. 49. P. 173–181.
Price A.R.G., Readman, J.W. Booster biocide antifoulants: is history repeating itself? / In: Late Lessons from Early Warnings: Science, Precaution, Innovation. Part B – Emerging Lessons from Ecosystems // The European Environment Agency. Publications office of the European Union. Luxembourg. Part 12. 2013. P. 297–310.
United Kingdom Health and Safety Executive (UK HSE) Pesticides News Letter № 49 // The Biocides and Pesticides Assessment Unit, Health and Safety Executive, Magdalen House, Stanley Precinct, Bootle, Merseyside UK. 2000
Bowman J.C., Readman J.W., Zhou J.L. // Mar. Pollut. Bull. 2003. V. 46. P. 444–451.
Background information. Reassessment of antifouling paints // European Chemicals Agency (ECHA), Accessed 27.12.16.
Konstantinou I.K., Albanis T.A. // Environ. Int. 2004. V. 30. P. 235–248.
Fact sheet № 24: Anti-fouling bottom paint // Danish Environmental Protection Agency. Ministry of the Environment, København, Denmark. 2008.
Ansanelli G., Manzo S., Parrella L. et al. // Regional Studies in Marine Science. 2017. V. 16. P. 254–266.
Soon Z.Y., Jung J.-H., Jang M. et al. // Water Air Soil Pollut. 2019. V. 230. Article № 310.
Bao V.W., Leung K.M., Qiu J.W., Lam M.H. // Mar. Pollut. Bull. 2011. V. 62. P. 1147–1151.
Dafforn K.A., Lewis J.A., Johnston E.L. // Mar. Pollut. Bull. 2011. V. 62. P. 453–465.
Cima F., Ballarin L. // Toxicology & Pharmacology. 2015. V. 169. P. 16–24.
Johansson P., Eriksson K., Axelsson L., Blanck H. // Arch. Environ. Contam. Toxicol. 2012. V. 63. P. 365–377.
Hamwijk C., Schouten A., Foekema E.M. et al. // Chemosphere. 2005. V. 60. P. 1316–1324.
Callow M.E., Finlay J.A. // Biofouling. 1995. V. 9. P. 153–165.
Wang H., Li Y., Huang H. et al. // Environ. Toxicol. Chem. 2011. V. 30. P. 692–703.
Lee S.E., Won H.S., Lee Y.W. // Bull. Environ. Contam. Toxicol. 2010. V. 85. P. 538–543.
Lee S., Chung J., Won H. et al. // J. Hazard. Mater. 2011. V. 185. P. 1318–1325.
Xu X., Wang X., Li Y. et al. // Hum. Exp. Toxicol. 2011. V. 30. P. 1009–1021.
Guardiola F.A., Cuesta A., Meseguer J., Esteban M.A. // Int. J. Mol. Sci. 2012. V. 13. P. 1541–1560.
Bellas J. // Sci. Total Environ. 2006. V. 367. P. 573–585.
Cima F., Bragadin M., Ballarin L. // Aquat. Toxicol. 2008. V. 86. P. 299–312.
Chen L., Lam J.C.W. // J. Environmental Sciences. V. 61. 2017. P. 68–79.
Regulation (EU) № 528/2012 of the European Parliament and of the Council of 22 May 2012 concerning the making available on the market and use of biocidal products, European Commission. 2012. OJ L 167. P. 1–123.
Kempen T. Efficacy, chemistry and environmental fate of tralopyril, a nonmetal antifouling agent // In: European Coatings Conference “Marine Coatings III” // Berlin, 28 February 2011.
Oliveira I.B., Schonenberger R., Barroso C.M., Suter M.J.-F. // Chemosphere. 2016. V. 145. P. 445–449.
Oliveira I.B., Beiras R., Thomas K.V. et al. // Ecotoxicology. 2014. V. 23. P. 1336–1344.
Oliveira I.B., Groh K.J., Schonenberger R. et al. // Aquat. Toxicol. 2017. V. 191. P. 164–174.
Chen X., Teng M., Zhang J. // Science of the Total Environment. 2020. V. 746. Article № 141860.
Arunrangsi T., Raethong S., Songsrirote K. // Songklanakarin Journal of Science and Technology. 2013. V. 35. P. 303–308.
Tomaselli L., Lamenti G., Tiano P. // Ann. Microbiol. 2002. V. 52. P. 197–206.
Ali H., van Lier J.E. Porphyrins and Phthalocyanines as Photosensitizers and Radiosensitizers / In: Kadish K.M., Smith K.M., Guilard R. (Eds.) Handbook of porphyrin science // Singapore: World Scientific Publishing Co. Pte. Ltd. 2010. V. 4. P. 1–119.
Crupi V., Fazio B., Gessini A. et al. // Construction and Building Materials. 2018. V. 166. P. 464–471.
Ruffolo S.A., La Russa M.F. // Front. Mater. 2019. V. 6. Article № 147.
La Russa M.F., Macchia A., Ruffolo S.A. et al. // International Biodeterioration & Biodegradation. 2014. V. 96. P. 87–96.
Воронков М.Г., Расулов М.М. // ХФЖ. 2007. Т. 41. № 1. С. 3–7. [Voronkov M.G., Rasulov M.M. // Pharm. Chem. J. 2007. V. 41. P. 1–6.]
Гарабаджиу А.В., Воронков М.Г., Няникова Г.Г. и др. // Докл. АН. 2011. Т. 439. № 6. С. 838–840.
Воронков М.Г., Колесникова О.П., Расулов М.М., Мирскова А.Н. // ХФЖ. 2007. Т. 41. № 5. С. 13–17. [Voronkov M.G., Kolesnikova O.P., Rasulov M.M., Mirskova A.N. // Pharm. Chem. J. V. 2007. V. 41. № 5. P. 244–248.]
Воронков М.Г., Барышок В.П. // Вест. Росс. акад. наук. 2010. Т. 80. № 11. С. 985–992. [Voronkov M.G., Baryshok V.P. // Her. Russ. Acad. Sci. 2010. V. 80. P. 514–521.]
Kondratenko Yu.A., Ugolkov V.L., Vlasov D.Yu., Kochina T.A. // Mendeleev Commun. 2020. V. 30. P. 639–641.
Кондратенко Ю.А., Власов Д.Ю., Буслаев Г.С. и др. // ФХС. 2019. Т. 45. № 5. С. 469–477. [Kondratenko Yu.A., Vlasov D.Yu., Buslaev G.S. et al. // Glass Phys. Chem. 2019. V. 45. № 5. P. 372–378.]
Шилова О.А., Халаман В.В., Комендантов А.Ю. и др. // ФХС. 2021. Т. 47. № 2. С. 209–228. [Shilova O.A., Khalaman V.V., Komendantov Yu. et al. // Glass Phys. Chem. 2020. V. 46. № 6. P. 620–634.]
Roberts D., Rittschof D., Holm E., Schmidt A.R. // J. Exp. Mar. Biol. Ecol. 1991. V. 150. P. 203–221.
Liu H.F., Huang L., Liu T., Yulong H.U. // J. Chin. Soc. Corros. Prot. 2009. V. 29. P. 154–160.
Wen J., Zhao K., Gu T.Y. et al. // Int. Biodeterior. Biodegrad. 2009. V. 63. P. 1102–1106.
Xu H.J., Liu Y. // J. Membr. Sci. 2011. V. 376. P. 266–274.
Сагайдак А.И. / В сборнике трудов конференции: “Повышение износостойкости и долговечности машин и механизмов на водном транспорте”. Труды V Международного симпозиума по транспортной триботехнике “Транстрибо-2013”. 2013. С. 114–118.
Xu D.K., Li Y., Gu T.Y. // Mater. Corros. 2014. V. 65. P. 837–845.
Xu D.K., Li Y., Gu T.Y. // World J. Microbiol. Biotechnol. 2012. V. 28. P. 3067–3074.
Xie Q.Y., Pan J.S., Ma C.F., Zhang G.Z. // Soft Matter. 2019. V. 15. P. 1087–1107.
Omae I. // Chem. Rev. 2003. V. 103. P. 3431–3448.
Bressy C., Nguyen M.N., Tanguy B. et al. // Polym. Degrad. Stabil. 2010. V. 95. P. 1260–1268.
Kugel A., Stafslien S., Chisholm B.J. // Prog. Org. Coat. 2011. V. 72. P. 222–252.
Gottenbos B., van der Mei H.C., Klatter F. et al. // Biomaterials. 2002. V. 23. P. 1417–1423.
Oosterhof J.J.H., Buijssen K.J.D.A., Busscher H.J. et al. // Appl. Environ. Microbiol. 2006. V. 72. P. 3673–3677.
Cen L., Neoh K.G., Kang E.T. // Langmuir. 2003. V. 19. P. 10295–10303.
Thome J., Hollander A., Jaeger W. et al. // Surf. Coat. Technol. 2003. V. 174–175. P. 584–587.
Zhu H., Kumar A., Ozkan J. et al. // Optometry and Vision Science. 2008. V. 85. P. 292–300.
Hu S.G., Jou C.H., Yang M.C. // J. Appl. Polym. Sci. 2002. V. 86. P. 2977–2983.
Tashiro T. // Macromol. Mater. Eng. 2001. V. 286. P. 63–87.
Klibanov A.M. // J. Mater. Chem. 2007. V. 17. P. 2479–2482.
Park E.-S., Lee H.-J., Park H.Y. et al. // J. Appl. Polym. Sci. 2001. V. 80. P. 728–736.
Stovicek P. / US Patent 5084096A. United States. 1992.
Silva E.R., Ferreira O., Ramalho P.A. et al. // Science of the Total Environment. 2019. V. 650. P. 2499–2511.
Chan A.C., Cadena M.B., Townley H.E. et al. // J. R. Soc. Interface. 2017. V. 14. Article № 20160650.
Popat A., Liu J., Hu Q. et al. // Nanoscale. 2012. V. 4. P. 970–975.
Zheng Z., Huang X., Schenderlein M. et al. // Adv. Funct. Mater. 2013. V. 23. P. 3307–3314.
Michailidis M., Sorzabal-Bellido I., Adamidou E.A. et al. // ACS Appl. Mater. Interfaces. 2017. V. 9. P. 38364–38372.
Ruggiero L., Bartoli F., Fidanza M.R. et al. // Applied Surface Science. 2020. V. 514. Article № 145908.
Ruggiero L., Bartolomeo E.D., Gasperi T. et al. // J. Alloy. Compd. 2019. V. 798. P. 144–148.
Maia F., Tedim J., Lisenkov A.D. et al. // Nanoscale. 2012. V. 4. P. 1287–1298.
Ali A., Jamil M.I., Jiang J. et al. // J. Polymer Research. 2020. V. 27. Article N. 85.
Abiraman T., Ramanathan E., Kavitha G. et al. // Ultrason Sonochem. 2017. V. 34. P. 781–791.
Abiraman T., Balasubramanian S. // Ind. Eng. Chem. Res. 2017. V. 56. P. 1498–1508.
Al-Naamani L., Dobretsov S., Dutta J., Burgess J.G. // Chemosphere. 2017. V. 168. P. 408–417.
Mooss V.A., Hamza F., Zinjarde S.S., Athawale A.A. // Chem. Eng. J. 2018. V. 359. P. 1400–1410.
Sun X., Li Q., Guo Z. et al. // ACS Appl. Mater. Interfaces. 2019. V. 11. P. 21323–21333.
Lin B., Zhou S. // Prog. Org. Coat. 2017. V. 106. P. 145–154.
Дринберг А.С., Козлов Г.В., Машляковский Л.Н. и др. // Известия СПбГТИ(ТУ). 2018. № 46(72). С. 76–80.
Бойнович Л.Б., Емельяненко А.М. // Успехи химии. 2008. Т. 77. № 7. С. 619–638. [Boinovich L.B., Emelyanenko A.M. // Russ. Chem. Rev. 2008. V. 77. № 7. P. 583–600.]
Уколов А.И., Попова Т.Н., Кулиш А.В. // В сборнике: Актуальные проблемы биоразнообразия и природопользования. Материалы II Национальной научно-практической конференции, посвященной 20-летию кафедры экологии моря ФГБОУ ВО “КГМТУ”. 2019. С. 649–654.
Sun T., Wang G., Feng L. et al. // Angew. Chem. Int. Ed. 2004. V. 43. P. 357–360.
Kyrikou I., Briassoulis D. // J. Polym. Environ. 2007. V. 15. P. 125–150.
Gross R.A., Kalra B. // Science. 2002. V. 297. P. 803–807.
Xie Q., Xie Q., Pan J. et al. // Appl. Mater. Interfaces. 2018. V. 10. P. 11213–11220.
Chen Y., Liu Z., Han S. et al. // J. Appl. Polym. Sci. 2016. V. 133. Article № 43667.
Luckachan G.E., Pillai C.K.S. // J. Polym. Environ. 2011. V. 19. P. 637–676.
Shaik M.R., Korsapati M., Panati D. // Int. J. Pharma Sci. 2012. V. 2. P. 112–116.
Gunatillake P., Mayadunne R., Adhikari R. // Biotechnol. Annual Rev. 2006. V. 12. P. 301–347.
Vainionp S., Rokkanen P. // Prog. Polym. Sci. 1989. V. 14. P. 679–716.
Nguyen M.N., Bressy C., Margaillan A. // J. Polym. Sci. Part A: Polym. Chem. 2005. V. 43. P. 5680–5689.
LeChevallier M.W., Cawthon C.D., Lee R.G. // Appl. Environ. Microbiol. 1988. V. 54. P. 2492–2499.
Wright J.B., Ruseska I., Athar M.A. et al. // Infect. Control Hosp. Epidemiol. 1989. V. 10. P. 408–415.
Costerton J.W. // Dev. Ind. Microbiol. 1985. V. 26. P. 249–261.
Blenkinsopp S.A., Costerton J.W. // Appl. Environ. Microbiol. 1992. V. 58. P. 3770–3773.
Liu J., Zheng J.S., Xu L.M. // Corros. Sci. Prot. Technol. 2002. V. 14. P. 23–26.
Deberry D.W. // J. Electrochem. Soc. 1984. V. 131. P. C302 .
Deberry D.W. // J. Electrochem. Soc. 1985. V. 132. P. 1022–1026.
Armelin E., Oliver R., Liesa F. et al. // Prog. Org. Coat. 2007. V. 59. P. 46–52.
Ocampo C., Armelin E., Liesa F. et al. // Prog. Org. Coat. 2005. V. 53. P. 217–224.
Wessling B., Posdorfer J. // Electrochim. Acta. 1999. V. 44. P. 2139–2140.
Zhong L., Xiao S., Hu J. et al. // Corros. Sci. 2006. V. 48. P. 3960–3968.
Ocon P., Cristobal A.B., Herrasti P., Fatas E. // Corros. Sci. 2005. V. 47. P. 649–660.
Tuken T., Yazici B., Erbil M. // Prog. Org. Coat. 2004. V. 51. P. 205–212.
Zheng H., Ye Y. // Corrosion Science and Protection Technology. 2013. V. 25. P. 429–432
Xie Z.P., Wang J.J., Huang C.S., Ye Z.J. // Material Development and Application. 2011. V. 26. P. 85–88.
Zhou C.L. // Chinese Coatings. 1998. № 6. P. 9–10.
Wang X.-H., Li J., Zhang J.-Y. et al. // Synthetic Metals. 1999. V. 102. P. 1377–1380.
Zhou Z., Li W., He T. et al. // J. Mater. Sci. Technol. 2016. V. 32. P. 950–955.
Дополнительные материалы отсутствуют.
Инструменты
Физикохимия поверхности и защита материалов