

Зоологический журнал, 2020, T. 99, № 11, стр. 1307-1320
Гостальный аспект территориальной экспансии микроба чумы Yersinia pestis из популяций монгольского сурка-тарбагана (Marmota sibirica)
В. В. Сунцов *
Институт проблем экологии и эволюции имени А.Н. Северцова РАН
119071 Москва, Россия
* E-mail: vvsuntsov@rambler.ru
Поступила в редакцию 28.04.2019
После доработки 19.12.2019
Принята к публикации 19.01.2020
Аннотация
На основе экологических исследований предложена концепция синхронного политопного видообразования микроба чумы Yersinia pestis из психрофильного сапрозообионтного микроба псевдотуберкулеза Y. pseudotuberculosis O:1b в Центральной Азии в трех географических популяциях монгольского сурка-тарбагана (Marmota sibirica): восточной, центральной и западной. В восточной части ареала (Хэнтэй, Маньчжурия), заселенной подвидом M. sibirica sibirica, исходным стал геновариант микроба чумы 2.ANT3; в центральной части (Хангай), где обитает подвид M. sibirica caliginosus, – 3.ANT2; на западе Монголии и в Тыве в популяциях пока не описанного подвида монгольского сурка (M. sibirica ssp.) отмечен геновариант 4.ANT1. Территориальная экспансия Y. pestis из районов видообразования, сопровождавшаяся формированием производных геновариантов во вновь осваиваемых паразитарных системах грызун-блоха, проходила по трем маршрутам и имела различные масштабы. Восточный фронт экспансии регионального масштаба охватил популяции даурского суслика (Spermophilus dauricus, 2.ANT3), монгольской песчанки (Meriones unguiculatus, 2.MED3), полевки Брандта (Lasiopodomys brandti, 0.PE4m) и алашанского суслика (Spermophilus alaschanicus, 2.MED3) в восточной Монголии, Забайкалье и в ряде провинций восточного Китая. Центральный фронт локального масштаба охватил лишь симпатрические популяции длиннохвостого суслика (S. undulatus, 3.ANT2) и восточную часть географической популяции монгольской пищухи, представленной подвидом Ochotona pallasi pricei (геновариант 0.PE5, ulegeica). Западный фронт экспансии Y. pestis стартовал из западной части ареала монгольского сурка (геновариант 4.ANT1) и охватил популяции многих видов норовых грызунов (сурков, сусликов, песчанок, полевок) и пищух, образовав множество геновариантов (биовары и “филогенетические линии” 0.ANT, 0.PE, 1.ORI, 1.IN, 2.MED и другие) на обширных пространствах горной и равнинной Азии, от Монгольского Алтая и Тибетского нагорья на востоке до Кавказа и Ближнего Востока на западе и от северного Прибалхашья, северного Приаралья и северного Прикаспия на севере до юга Индостана и Юннаня на юге. Экологическая концепция территориальной экспансии микроба чумы во многом не совпадает с последовательностью внутривидовой диверсификации Y. pestis, декларируемой молекулярно-генетическим (МГ) подходом. Жестко детерминированных МГ-филогенетических ветвей 0, 1, 2, 3 и 4 и филогенетических линий 0.ANT, 0.PE, 1.ORI, 1.IN, 2.MED не существует. Возникновение того или иного геноварианта определялось условиями циркуляции микроба в местной (локальной) гостальной среде, т.е. в местных очагах чумы.
Чума – особо опасная инфекция, циркулирующая в естественной природе, главным образом в популяциях норовых грызунов (Rodentia) и пищух (Lagomorpha, Ochotona), – продолжает оставаться актуальной проблемой здравоохранения многих стран мира. Эта проблема последовательно решается по мере накопления знаний об этиологии болезни и возбудителе – микробе Yersinia pestis. В частности, достижению цели в немалой мере способствуют знания о происхождении и эволюции этого микроба.
Классическая теория природной очаговости чумы, идея которой была высказана академиком Д.К. Заболотным еще на рубеже XIX–XX веков и которая достигла полноценного развития ко второй половине XX века, в качестве исходных хозяев возбудителя болезни предполагала сурков, в частности монгольского (Marmota sibirica) (Ралль, 1965; Wu Lien-The et al., 1936). В то время закрепилось представление о том, что исходные сурочьи очаги сформировались в глубокой древности, возможно в олигоцене–плиоцене Старого или Нового Света. По биохимическим признакам были выделены биовары возбудителя: античный (antique, ANT), средневековый (mediaevalis, MED) и восточный (orientalis, ORI). Биовар ANT, характерный для сурочьих очагов, считали наиболее древним. Но на рубеже нового тысячелетия мнение о глубокой древности чумы резко поменялось. Проблема происхождения, филогении и систематики микроба чумы стала прерогативой молекулярной генетики (МГ). МГ-методами было показано, что исходная популяция Y. pestis сформировалась из клона психрофильного энтеропатогенного псевдотуберкулезного микроба Y. pseudotuberculosis O:1b в Азии в эволюционном масштабе времени недавно, не ранее 30 тыс. лет назад, предположительно в популяциях сурков или полевок (Achtman et al., 1999, 2004; Skurnik et al., 2000; Morelli et al., 2010; Cui et al., 2013). Некоторые оценки времени расхождения псевдотуберкулезного и чумного микробов, приведенные в недавних работах, показывают, что это событие могло случиться в более отдаленные сроки, до 80 тыс. лет назад (Rasmussen et al., 2015).
МГ-исследователи полагают, что исходная популяция Y. pestis в процессе территориальной экспансии из района видообразования дала начало наиболее древней филогенетической ветви (линии) 0.ANT, в основании которой отделились ветви так называемых “полевково-пищуховых” форм Pestoides (подвидов, геновариантов, филогрупп) 0.PE1 – 0.PE10. В дальнейшем исходная ветвь 0.ANT дала начало ветвям 1.ANT, 2.ANT, 3.ANT и 4.ANT. Из первой ветви по мере дальнейшей экспансии микроба выделились более молодые ветви, представленные биоварами orientalis (1.ORI) и intermedium (1.IN), из второй ветви – биовар mediaevalis (2.MED). Все филогенетичесие ветви включают филогруппы (геноварианты), которые характеризуют конкретные очаги чумы или их взаимосвязанные группы (мегаочаги). Например, ветвь 0.ANT разделилась на субветви (группы, кластеры, геноварианты) 0.ANT1, 0.ANT2, 0.ANT3 и 0.ANT5, которые характеризуют очаги Тянь-Шаня. Ветвь 2.MED разделилась на четыре группы: 2.MED0, 2.MED1, 2.MED2 и 2.MED3, отмеченные, соответственно, в очагах Кавказа, Средней Азии, Джунгарии и Синцзяне и на равнинах северо-восточного Китая (Cui et al., 2013; Kutyrev et al., 2018; Zhang et al., 2018). Различными биохимическими и МГ-методами раскрыто богатое внутривидовое фенотипическое и генетическое разнообразие чумного микроба, выявлены родственные связи внутривидовых форм, построены многочисленные филогенетические схемы, иллюстрирующие дивергенцию чумного микроба от псевдотуберкулезного и отражающие последовательность формирования внутривидовой структуры Y. pestis по мере освоения новых гостальных экологических ниш (Вариабельность…, 2009; Achtman et al., 2004; Anisimov et al., 2004; Zhou et al., 2004, 2004a, 2004b; Zhou, Yang, 2009; Morelli et al., 2010; Cui et al., 2013; Kutyrev et al., 2018; Demeure et al., 2019). Географическая локализация геновариантов позволяет подойти к реконструкции внутривидовой филогеографии микроба чумы. Тем не менее, на тривиальные вопросы – где, когда, при каких обстоятельствах и каким образом осуществлялось преобразование клона псевдотуберкулезного микроба в популяцию возбудителя чумы, как проходило освоение микробом чумы континентов Старого и Нового Света – однозначного ответа МГ-достижения не дают. Причина этого кроется в пока еще недостаточном объеме накопленных МГ-характеристик микроба из разных очагов мира и несовершенстве МГ-методов филогенетических построений, а также в игнорировании или недооценке МГ-подходом биологической сути проблемы. В МГ-филогениях нет места полноценной экологической (в широком смысле), биогеографической, зоолого-паразитологической, популяционно-генетической, гостально-векторной характеристике представителей выявленных филогенетических групп. МГ-характеристика геновариантов чумного микроба, именуемых “древними” или “молодыми”, не убедительна и экологически не валидна. Например, по МГ-представлениям геновариант 2.MED0 является относительно древним, базовым в филогенетической линии 2.MED (Носов и др., 2016), а при экологическом подходе он видится одним из наиболее молодых, его ареал максимально удален от района видообразования возбудителя чумы.
Вопросы видообразования и эволюции популяций – это прерогатива, прежде всего, экологии. Популяционная экология и популяционная генетика лежат в основе отточенных многолетними исследованиями законов и закономерностей эволюционной теории, позволяющих охарактеризовать экологические механизмы видообразовательных и микроэволюционных процессов на уровне популяций, фенотипов и генотипов и привязать их к конкретным биогеоценозам (среде обитания), чего лишен МГ-подход. Для коррекции созданных разными методами и заметно не сходных между собой МГ-филогений возбудителя чумы мы предложили экологический подход, позволяющий встроить МГ-данные в канву природных событий, инициировавших появление чумного микроба (Сунцов, Сунцова, 2000, 2006). Согласно экологическому сценарию, микроб Y. pestis дивергировал от клона психрофильного псевдотуберкулезного микроба Y. pseudotuberculosis O:1b в континуальной гетерогенной (гетеротермной и гетероиммунной) среде – паразитарной системе монгольский сурок-тарбаган–сурочья блоха (Marmota sibirica–Oropsylla silantiewi) – в условиях последнего максимального (сартанского) похолодания климата Центральной Азии 22–15 тыс. лет назад. При этом формирование чумного микроба как нового вида прошло чрезвычайно быстро путем популяционно-генетической макроэволюции (т.е. был совершен быстрый переход исходной(ых) популяции(й) в принципиально иную экологическую нишу и адаптивную зону) по “квантовому” принципу (Сунцов, Сунцова, 2008; Сунцов, 2015, 2018, 2020). Его дальнейшая естественная азиатская экспансия из района видообразования проходила по принципу “масляного пятна”. В освоении микробом чумы Африканского континента и Нового Света решающую роль сыграли антропогенные факторы, в частности коммерческая и военная активность и развитие быстроходного морского пароходства (рис. 1) (Сунцов и др., 2011, с. 245).
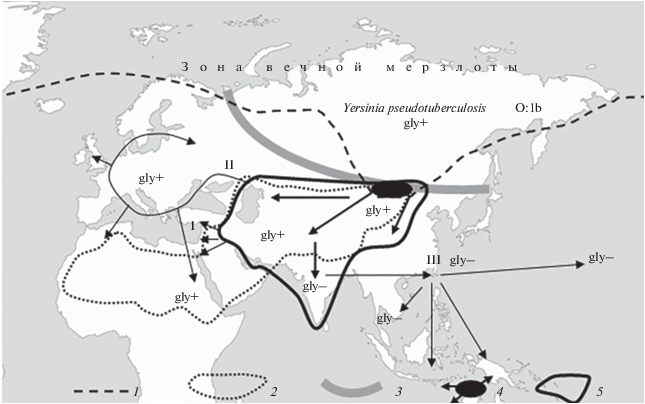
Рис. 1.
Район происхождения микроба чумы Yersinia pestis в ареале монгольского сурка (Marmota sibirica) в Центральной Азии и его естественная и антропогенная мировая экспансия: 1 – южная граница зоны вечной мерзлоты; 2 – Сахаро-Гобийская аридная зона; 3 – граница доминантного распространения псевдотуберкулезного микроба 1-го серотипа; 4 – район видообразования микроба чумы и направления его естественной экспансии в позднем плейстоцене–голоцене; 5 – ареал первичных природных очагов чумы в Азии; I – первая пандемия, “чума Юстиниана”; II – вторая пандемия, “Черная смерть”; III – третья пандемия, современная. gly+ (gly–) – биохимический маркер фенотипа микроба: способность (неспособность) штаммов ферментировать глицерин.
В предлагаемой статье мы предприняли попытку консолидировать МГ-данные по генетическому разнообразию микроба чумы с экологическими представлениями о его происхождении и внутривидовой диверсификации по мере экспансии из района видообразования в Центральной Азии. Несмотря на методологическое несовершенство как МГ, так и экологического подходов, примененных для реконструкции путей экспансии чумного микроба, такой синтез будет полезен в качестве нуль-гипотезы для оптимизации дальнейших исследований. МГ-исследователей он нацелит на поиск надежных маркеров, позволяющих выявить прямых предков и потомков среди уже описанных геновариантов (статус “древних” или “молодых” геновариантов, констатируемых МГ-методами, без экологических обоснований вызывает глубокое сомнение), а экологам предоставит убедительные филогенетические линии микроба для реконструкции/корректировки действительных маршрутов его азиатской и мировой экспансии.
Видообразование Y. pestis: моно- или политопическое?
Мы показали, что основной средой видообразования микроба чумы являются популяции сурка-тарбагана в Центральной Азии (Сунцов, Сунцова, 2000, 2006). В последние 30 тыс. лет (это датировка видообразования Y. pestis методом молекулярных часов, по: Achtman et al., 1999, 2004; Morelli et al., 2010) ареал сурка-тарбагана не подвергался радикальным изменениям. Под влиянием максимального сартанского похолодания (22–15 тыс. лет назад) и наступившего после него голоценового климатического оптимума границы ареала смещались незначительно (Ербаева, 1970; Бибиков, 1989; Erbaeva, 2003). Более заметным было изменение вертикальных границ распространения тарбагана, менявших кружево ареала. Во время максимального похолодания и развития локальных ледников на горных вершинах юга Сибири и Центральной Азии поселения монгольского сурка смещались вниз в межгорные долины, в настоящее время покрытые сухими степями и не заселенные сурком (Дмитриев, 2006). Поэтому можно полагать, что в наиболее холодное время позднего плейстоцена–голоцена ареал сурка-тарбагана был более консолидирован, менее расчленен на отдельные альпийские и горностепные популяции (Бибиков, 1989).
Ареал сурка-тарбагана включает три относительно изолированные географические популяции: восточную (а), центральную (б) и западную (в) (рис. 2). Подвид M. sibirica sibirica занимает восточную часть ареала: Хэнтэй, Забайкалье, Маньчжурию. M. sibirica caliginosus обитает в центральных районах Монголии, с большой численностью заселяет Хангай. На западе Монголии и в Тыве на Хархира-Монгун-Тайгинском горном узле предполагается наличие третьего, пока не описанного подвида M. sibirica ssp. (Банников, 1954; Brandler, 2018). В каждой из этих трех географических популяций имеются очаги чумы, в которых представлен один доминирующий (major) геновариант микроба, соответственно, 2. ANT3, 3.ANT2 и 4.ANT1 и несколько не доминирующих (minor), редких и единично представленных геновариантов (Павлова и др., 2012; Куклева и др., 2015; Ерошенко и др., 2018; Балахонов и др., 2019; Riehm et al., 2012; Kutyrev et al., 2018). Полагают, что стабильное существование очагов поддерживают только доминирующие геноварианты (Li et al., 2008; Xu et al., 2018; Zhang et al., 2018). Поэтому выяснение филогенетических связей доминирующих геновариантов представляется наиболее важным. Из описанных более 30 геновариантов (Morelli et al., 2010; Cui et al., 2013; Kutyrev et al., 2018) к основным можно отнести 15 следующих: 0.PE2 (Кавказ: основной хозяин – обыкновенная полевка (Microtus arvalis)); 0.PE4h (Гиссар: арчевая полевка (Microtus carruthersi)); 0.PE4a (Алтай, Баян-Улгий: монгольская пищуха (Ochotona pallasi)); 0.PE4m (cеверо-восточный Китай: полевка Брандта (Lasiopodomys brandti)); 0.PE5 (западная и юго-западная Монголия: монгольская пищуха); 0.PE10 (Тибет: цинхайская полевка (L. fuscus)); 2.MED1 (Джунгария, Средняя Азия, Прикаспий, Кавказ, Иран: большая (Rhombomys opimus) и другие песчанки, малый (Spermophilus pygmaeus) и другие суслики); 2.MED2 (Синцзян, Памир, Кунь-Лунь: красный (M. caudata) и гималайский (M. himalayana) сурки); 2.MED3 (северо-восточный и центральный Китай: монгольская песчанка (Meriones unguiculatus), алашанский суслик (S. alaschanicus)); 0.ANT1 (восточный Тянь-Шань: алтайский сурок (Marmota baibacina), длиннохвостый суслик (S. undulatus)); 0.ANT3 (Тянь-Шань, Памир, Гиндукуш: алтайский и красный сурки, длиннохвостый суслик); 4.ANT1 (Алтай, Тыва, западная Монголия: алтайский и монгольский (M. sibirica) сурки, длиннохвостый суслик); 2.ANT3 (Хэнтэй, Забайкалье, Маньчжурия: монгольский сурок, даурский суслик (S. dauricus)); 3.ANT2 (Хангай: монгольский сурок, длиннохвостый суслик); 1.IN2 (Тибет: гималайский сурок); 1.IN3 (Юннань: полевая мышь Шевриера (Apodemus chevrieri), юннаньская красная полевка (Eothenomys miletus)).
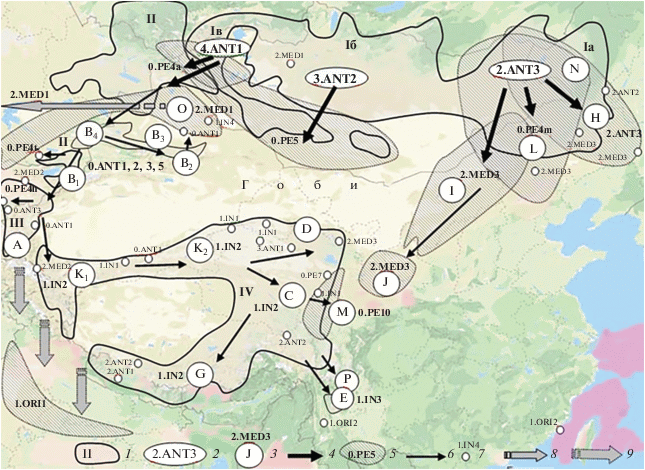
Рис. 2.
Естественная экспансия микроба чумы Yersinia pestis из трех географических популяций монгольского сурка-тарбагана (Marmota sibirica) по двум основным маршрутам: восточному (Хэнтэйскому) и западному (Алтайскому). 1 – восстановленные ареалы сурков: монгольского тарбагана (Iа, Iб, Iв – три географические популяции), алтайского (II), красного (III), гималайского (IV); 2 – геноварианты возбудителя чумы в очагах, расположенных в разных географических популяциях монгольского сурка; 3 – очаги чумы и циркулирующие в них основные (major, доминантные) геноварианты возбудителя на территории Китая, обозначения очагов от А до Р по: Zhou et al., 2004; 4 – переход возбудителя чумы из популяций сурка-тарбагана в симпатрические популяции хозяев других видов; 5 – очаги чумы с основными хозяевами, не относящимися к роду Marmota; 6 – естественное распространение микроба чумы из западной и восточной географических популяций монгольского сурка-тарбагана; 7 – места обнаружения неосновных (minor, единично встречающихся) геновариантов микроба чумы в очагах Монголии и Китая; 8 – распространение возбудителя чумы геноварианта 2.MED1 из джунгарской популяции большой песчанки в Среднюю Азию; 9 – предположительное направление перехода возбудителя чумы из популяций красного и/или гималайского сурков в популяции индийской песчанки с формированием геноварианта 1.ORI1 на Индостане. Распространение геновариантов показано по: Куклева и др., 2015; Носов и др., 2016; Ерошенко и др., 2018; Cui et al., 2008, 2013; Li et al., 2008, 2009, 2013; Zhang et al., 2009; Morelli et al., 2010; Riehm et al., 2012; Kutyrev et al., 2018; Zhang et al., 2018.
Нельзя отрицать, что исходные наиболее древние в соответствии с экологическим сценарием доминантные геноварианты 2.ANT3, 3.ANT2 и 4.ANT1 могли сформироваться одновременно в уже состоявшихся географических популяциях монгольского сурка. В теории эволюции такое явление называют множественным появлением (multiple origin) нового вида, или политопным видо- формообразованием (polytopic origin), которое достаточно широко распространено в природе (Heads, 1985; Williams, Ebach, 2008). Определяющими факторами и необходимыми условиями подобного явления считают близкое родство исходных (предковых) популяций и однонаправленное действие отбора (Гродницкий, 2002; Schluter, Nagel, 1995). Что касается нашей экологической модели происхождения возбудителя чумы, то его отделение от псевдотуберкулезного микроба O:1b серотипа можно связать с тремя вышеупомянутыми географическими популяциями сурка-тарбагана.
Индуктором видообразования микроба чумы стала факультативная гематофагия личинок сурочьей блохи O. silantiewi (Сунцов, Сунцова, 2000, 2006). Личиночный паразитизм был вызван обширным сартанским похолоданием и глубоким промерзанием грунта в Южной Сибири и Центральной Азии, а с ним условия для видообразования микроба чумы могли сформироваться в разных географических популяциях монгольского сурка синхронно. Тогда вид Y. pestis мог образоваться политопически и синхронно в трех различных популяциях псевдотуберкулезного микроба, соответствующих географическим популяциям сурка-тарбагана, и начальная экспансия этих топических форм (геновариантов) могла идти самостоятельными путями. Большое генотипическое разнообразие псевдотуберкулезного микроба хорошо известно (Евсеева и др., 2015). Отсюда вывод: при разработке консолидированной филогенетической системы Y. pestis не следует отвергать вероятность возникновения трех параллельных филогенетических линий, стартовавших из трех популяций псевдотуберкулезного микроба, свойственных географическим популяциям (а, б, в) монгольского сурка и различающихся генофондами: Y. pseudotuberculosis O:1ba → Y. pestis 2.ANT3; Y. pseudotuberculosis O:1bб → Y. pestis 3.ANT2; Y. pseudotuberculosis O:1bв → Y. pestis 4.ANT1 (рис. 2).
Три направления экспансии Y. pestis из географических популяций монгольского сурка
На основании анализа МГ-признаков псевдотуберкулезного и чумного микробов Zhou et al. (2004) пришли к выводу, что экспансия возбудителя чумы на территорию Китая проходила по двум маршрутам: восточному из южной Сибири и Дальнего Востока и западному из Центральной (Средней) Азии. Эти маршруты сближались южнее Гоби, но не сливались. В работе этих авторов Монголия осталась “белым пятном”. Накопленные за последующие годы МГ-данные, в совокупности с экологическими, позволяют обозначить стартовые позиции этих маршрутов именно в Монголии и конкретизировать их детали.
Восточный фронт (Хэнтэйский). МГ-методами многократно и с большой убедительностью показано, что представители филогенетической ветви 2.MED по нуклеотидной структуре наиболее близки к возбудителю из ветви 2.ANT и с большой вероятностью являются его производными (Одиноков и др., 2013; Achtman et al., 2004; Zhou et al., 2004; Achtman, 2008, 2012; Li et al., 2009; Riehm et al., 2012; Cui et al., 2013). Как покажем ниже, этот факт находит наглядное подтверждение в экологическом сценарии экспансии микроба чумы.
В Хэнтэйской географической популяции сурка-тарбагана сформировался очаг чумы с доминантным геновариантом 2.ANT3 (рис. 2). Этот геновариант in situ перешел в симпатрические популяции даурского суслика, оказался достаточно устойчивым при циркуляции в совмещенных поселениях сурка и суслика и, распространяясь далее на территорию северо-восточного Китая, образовал моногостальный сусликовый очаг H, при этом (пока?) не изменил своих свойств. Поэтому в настоящее время в восточных районах Монголии, в Забайкалье, Маньчжурии и Внутренней Монголии имеются природные очаги чумы с геновариантом 2.ANT3 в популяциях монгольского сурка, даурского суслика и их симпатрических популяциях (Павлова и др., 2012; Куклева и др., 2015; Cui et al., 2013).
В Хэнтэйском сурочьем очаге чумы популяции сурка-тарбагана пространственно совмещены с популяциями полевки Брандта, которая является основным хозяином возбудителя чумы во Внутренней Монголии (очаг L). В этом очаге доминирует полевковый геновариант возбудителя 0.PE4m (Cui et al., 2013). МГ-анализ показывает, что этот геновариант возник из биовара antique раньше, чем биовары mediaevalis и orientalis (Achtman et al., 2004; Tong et al., 2005; Cui et al., 2013). Эти данные соответствуют экологическому сценарию азиатской экспансии возбудителя чумы, согласно которому в Хэнтэйском сурочьем природном очаге чумы геновариант 2.ANT3 in situ перешел в популяции полевки Брандта, симпатрические с популяциями монгольского сурка. При дальнейшем распространении возбудителя во Внутреннюю Монголию, где сурок-тарбаган либо отсутствует, либо малочислен, микроб в новой гостальной среде сформировал моногостальный полевковый очаг L с геновариантом 0.PE4m (рис. 2).
В Хэнтэйском сурочьем очаге чумы популяции сурка-тарбагана находятся в состоянии симпатрии также с популяциями монгольской песчанки – основного хозяина возбудителя чумы в китайских северо-западных провинциях Гирин, Шэнси, Хэбей, во Внутренней Монголии, автономном округе Нинся (Cui et al., 2013). Геновариант 2.ANT3 in situ перешел из популяций монгольского сурка на Хэнтэе в симпатрические с ними популяции монгольской песчанки и далее распространился за пределы ареала сурка, образовав моногостальный песчанковый очаг I с геновариантом 2.MED3, встречающимся на востоке Монголии и в прилежащих районах Китая (рис. 2). Здесь же имеются симпатрические популяции монгольской песчанки и даурского суслика, поэтому не удивительно, что из организма последнего и его блох был изолирован как не доминантный песчанковый геновариант 2.MED3 (Cui et al., 2013).
Обширный ареал даурского суслика от Забайкалья простирается далеко на юг до лессового плато в провинциях Гансю и Нинся. В южной части ареала обитает алашанский суслик (некоторые авторы считают его подвидом даурского суслика S. dauricus alaschanicus). В популяциях этого суслика возник очаг чумы (J) с геновариантом 2.MED3, перешедшим в не измененном виде из соседнего песчаночьего очага (I) (Zhou et al., 2004) (рис. 2).
Единично геновариант 2.MED3, как не доминантный, встречен на востоке Цинхайского (Qinghai) плато, расположенного на северо-востоке Тибетского нагорья (Cui et al., 2013). Это место самого западного обитания алашанского суслика и обнаружения геноварианта 2.MED3. Подобные случаи обнаружения не типичных для конкретных очагов геновариантов относят к дальним заносам (Zhang et al., 2018).
Итак, напрашивается вывод, что формирование геновариантов чумного микроба 2.MED3 и 0.PE4m прошло напрямую от сурочьего геноварианта 2.ANT3 на восточном фронте экспансии из Хэнтэйской популяции монгольского сурка, и они не имеют прямых родственных связей с другими геновариантами в кластерах 2.MED и 0.PE. Восточная экспансия возбудителя чумы, начавшаяся в Хэнтэйской популяции монгольского сурка, завершилась на южной и юго-восточной границах ареала алашанского суслика, как это показано китайскими авторами (Zhou et al., 2004). Очаги чумы вдоль восточного маршрута экспансии микроба до наших дней остаются вполне активными, регулярно отмечаются случаи заболевания людей (Ben-Ari et al., 2012).
Центральный фронт (Хангайский). В сурочьем очаге чумы, сформировавшемся в географической популяции сурка-тарбагана на Хангае, доминирует эндемичный для Монголии геновариант 3.ANT2 (Куклева и др., 2015; Riehm et al., 2012) (рис. 2). Повсеместно поселения сурка здесь совмещены с поселениями длиннохвостого суслика. На юге Хангайского нагорья поселения монгольского сурка на обширной территории совмещены с плотными поселениями монгольской пищухи, которая также многочисленна южнее в сухих низкогорных степях, где сурок отсутствует.
В популяциях пищухи циркулирует возбудитель чумы геноварианта 0.PE5, эндемичный для Монголии и отнесенный к подвиду Y. pestis ulegeica (Anisimov et al., 2004; Kutyrev et al., 2018). В этом случае нет сомнения, что исходный сурочий геновариант микроба 3.ANT2 проник в популяции монгольской пищухи, где преобразовался в геновариант 0.PE5. Южная граница ареала монгольской пищухи проходит по северным окраинам безжизненной пустыни Гоби (Smith, Xie, 2008), поэтому дальнейшее распространение чумы по центральному фронту на юг должно было натолкнуться на географический барьер. Насколько непреодолимым был этот барьер на рубеже плейстоцена и голоцена и позднее, пока не ясно. С большой уверенностью можно предположить, что эндемичный для Монголии пищуховый геновариант чумного микроба 0.PE5 является непосредственным потомком сурочьего геноварианта 3.ANT2 и относится к категории наиболее древних производных геновариантов (подобно полевковому геноварианту 0.PE4m на восточном фронте экспансии).
Западный фронт (Алтайский). Если восточная и центральная экспансии чумного микроба из Хэнтэйской и Хангайской географических популяций монгольского сурка-тарбагана завершились лишь региональным и локальным распространением возбудителя в восточных провинциях Китая и в пределах Монголии, то западный фронт открыл магистральный путь к естественной азиатско-европейской территориальной экспансии, охватившей обширные горные поднятия Алтая, Тянь-Шаня, Памира, Тибета, Ирана, Кавказа. В зону экспансии попали прилегающие равнинные степи и пустыни западного Китая, Турана, Ближнего Востока, Индостана. Развитие общества человека, его хозяйственная активность уже в постантичные времена привели к антропогенному выносу возбудителя из природных очагов Ближнего Востока (?), Средней Азии и Индостана в существовавшие там населенные пункты, к распространению инфекции среди людей во многих странах Азии и Европы и как апофеоза чумы – к возникновению трех известных апокалиптических пандемий: “юстиниановой” чумы в VI–VIII веках, “Черной смерти” в XIV–XVIII веках и современной, начавшейся в середине XIX века.
В очагах чумы западной Монголии и Тывы отмечен геновариант возбудителя 4.ANT1 (Куклева и др., 2015; Ерошенко и др., 2018; Балахонов и др., 2019; Kutyrev et al., 2018) (рис. 2). Повсеместно поселения монгольского сурка здесь совмещены с поселениями длиннохвостого суслика. При этом, сходно с ситуацией на Хэнтэе, в Забайкалье и на Хангае, в популяциях сусликов и сурков доминирует один и тот же геновариант.
К западной границе ареала тарбагана на Монгольском Алтае и в Тыве примыкает восточная граница ареала алтайского сурка. Отсюда ареал этого вида простирается на юго-запад до центрального Тянь-Шаня, где южная граница его ареала смыкается с северной границей ареала красного сурка. В свою очередь, юго-восточная граница красного сурка на западных отрогах хребтов Кунь-Луня и Гималаев в китайской провинции Синцзян смыкается с западной границей ареала гималайского сурка. Таким образом, цепь горных систем, связывающая Алтай и Тибетское нагорье, заселена четырьмя видами сурков: монгольским тарбаганом, алтайским, красным и гималайским. На границах ареалов встречаются совместные поселения соседствующих видов. В популяциях всех четырех видов сурков поддерживаются горные очаги чумы, возбудителя которой относят в основном к сурочьему биовару antique (ANT и близкий к нему IN), но геноварианты в разных очагах различные.
В Горном Алтае, Тыве и западной Монголии в популяциях алтайского и монгольского сурков и длиннохвостого суслика описан единый мегаочаг чумы с возбудителем 4.ANT1 (Ерошенко и др., 2018; Балахонов и др., 2019; Корзун и др., 2019; Kutyrev et al., 2018). В Горном Алтае в популяциях монгольской пищухи, соседствующих или симпатричных с поселениями сурков и суслика, циркулирует “пищуховый” геновариант 0.PE4a (Pestoides, altaica), надо полагать, производный от исходного 4.ANT1. Если это так, то подвиды Y. pestis altaica (0.PE4a) и Y. pestis ulegeica (0.PE5) имеют самостоятельные ареалы, перекрывающиеся в центральной части единой вытянутой с востока на запад географической популяции монгольской пищухи O. pallasi pricei (Anisimov et al., 2004). При этом подвиды altaica и ulegeica не связаны прямым родством, т.е. возникли из разных предковых геновариантов – 4.ANT1 и 3.ANT2, соответственно, и осваивали географическую популяцию монгольской пищухи с разных сторон – с востока и с запада.
На Тянь-Шане, заселенном алтайским сурком и длиннохвостым сусликом, имеется несколько очагов чумы (В1, В2, В3, В4) в популяциях алтайского сурка в Китае и два очага, Сарыджазский и Верхненарынский, в Киргизии. Возбудителей, циркулирующих в этих очагах, относят к считающейся наиболее древней эволюционной ветви 0.ANT (Zhou et al., 2004; Wang et al., 2006; Cui et al., 2013; Eroshenko et al., 2017; Sariyeva et al., 2019), в то время как экологический сценарий предполагает, что тянь-шанские сурочьи очаги возникли по мере расселения микроба из популяций монгольского сурка, т.е. все геноварианты тянь-шанских сурочьих очагов группы 0.ANT произошли, с наибольшей вероятностью, от 4.ANT1. Географическое распространение геновариантов 0.ANT1– 0.ANT5 остается пока слабо изученным (Cui et al., 2013; Kutyrev et al., 2018).
Из тянь-шанских сурочьих очагов в процессе экспансии в южном направлении возбудитель чумы распространился в популяции красного сурка на Памире, Гиндукуше, в западных частях Кунь-Луня и Гималаев, где образовал очаги (А) с геновариантами 0.ANT3 и 2.MED2 и несколькими минорными (Сui et al., 2013). Из этих очагов далее возбудитель чумы внедрился в популяции гималайского сурка, где образовал 5 относительно изолированных очагов (K1, K2, C, D, G) с двумя доминирующими геновариантами: 1.IN2, сохранившим основные признаки биовара antique, и 2.MED2 – типичным mediaevalis (Cui et al., 2013; Xu et al., 2018). Очевидных географических и гостальных связей геноварианта 2.MED2 c геновариантами 2.MED0, 2.MED1 и 2.MED3, которые позволили бы характеризовать консолидированную филогенетическую линию 2.MED, обнаружить не удается (рис. 2).
Три очага, производных от сурочьих, возникли на востоке и юго-востоке Тибетского нагорья в популяциях цинхайской полевки (очаг М, геновариант 0.PE10=0.PE4b), полевой мыши Шевриера (Apodemus сhevrieri), азиатской полевой мыши (A. spesiosus) и юннаньской красной полевки (Eothenomys miletus) (очаги Е и Р, геновариант 1.IN3). Эти очаги стали юго-восточным пределом тибетской экспансии чумного микроба. Предположения китайских авторов (Xu et al., 2018) о формировании в Юннани ответственной за 3-ю пандемию филогруппы 1.ORI2 в популяциях желтобрюхой крысы (Rattus flavipectus) из сурочьего геноварианта 1.IN2, как увидим ниже, экологически не валидны.
В процессе распространения микроба чумы в популяциях сурков в горных районах Азии, очаги возникали также на прилежащих к ним средне- и низкогорных и равнинных территориях с разными видами хозяев, разной динамикой численности животных, разными экологическими характеристиками. Поэтому здесь возникли очаги, производные от сурочьих, с разными геновариантами возбудителя: 2.MED2, 2.ANT1, 2.ANT2, 0.PE4h, 0.PE4t, 1.IN4 и другими. Наибольший интерес среди них представляют 2.MED1 и 1.ORI1, которые в настоящее время доминируют в очагах западной и южной Азии.
Происхождение геноварианта 2.MED1
Геновариант 2.MED1 характерен для горных и равнинных сусличьих и песчаночьих очагов на Кавказе, Иранском нагорье, в полупустынях и пустынях Средней Азии, Приаралья, Прибалхашья, Прикаспия. Согласно МГ-заключениям, этот геновариант составляет две относительно обособленные ветви: Кавказско-Каспийскую и Среднеазиатско-Китайскую (Носов и др., 2016; Kutyrev et al., 2018). Китайские исследователи (Li et al., 2009; Cui et al., 2013) полагают, что исходной была Китайско-Среднеазиатская ветвь, из Китая возбудитель чумы филогруппы 2.MED1 через Джунгарские ворота распространился в Туран, Среднюю Азию, в Иран, на Кавказ (рис. 2). Исследования последних лет подтверждают эту версию (Zhang et al., 2018).
Основным хозяином возбудителя в пустынных песчаночьих очагах Китая (Джунгария) и Средней Азии служит большая песчанка. Джунгарский песчаночий очаг, в котором циркулирует геновариант 2.MED1, находится между горными цепями Горного Алтая, Монгольского Алтая и Тянь-Шаня, где расположены сурочьи и сусличьи очаги с геновариантами микроба 3.ANT2, 4.ANT1, 0.ANT1, 0.ANT2, 0.ANT3, 0.ANT5. В джунгарских популяциях большой песчанки обнаружены также как не доминантные геноварианты 0.ANT1 и 1.IN4 (Zhang et al., 2018). Ареал большой песчанки заходит на территорию юго-западной Монголии. В Южно-Гобийском аймаке имеется малоизученный очаг чумы, где основной носитель – большая песчанка, а основной переносчик – блоха Xenopsylla skrjabini (Чинболд и др., 1980; Вержуцкий, Адъясурен, 2019). Несколько севернее известны очаги в популяциях монгольской пищухи (геновариант 0.PE5). Геновариант 2.MED1 как не доминантный обнаружен в Дзабханском аймаке Монголии в сурково-сусличиьих очагах Хангая (Куклева и др., 2015). Наиболее вероятно, геновариант 2.MED1 в джунгарских популяциях большой песчанки произошел или от геноварианта 0.ANT1 из тянь-шанских сурочьих очагов, или от 4.ANT1 из алтайских сурочьих очагов. В любом случае, изначально геновариант 2.MED1 возник на западном маршруте экспансии микроба чумы из популяций монгольского сурка-тарбагана и сформировался независимо и параллельно с геновариантом 2.MED3 восточного маршрута экспансии. Таким образом, геноварианты 2.MED1 и 2.MED3 хотя и включены в один МГ-кластер 2MED, но порождены различными мутациями (Павлова и др., 2012) и не представляют единую филогенетическую ветвь 2.MED.
Происхождение биовара orientalis (1.ORI)
Изучение структуры генома Y. pestis привело к выводу, что биовар orientalis возник напрямую от antique (Skurnik et al., 2000; Zhou et al., 2004). МГ-методами показано, что микроб чумы геноварианта 1.IN2, имеющего признаки биоваров antique и Pestoides (Rha+), циркулирующий в популяциях гималайского сурка на Тибете, в процессе распространения в юго-восточном направлении перешел в популяции дикоживущих мышей (A. chevrieri и A. speciosus) и полевок (E. miletus), где в китайской провинции Юннань возникли природные очаги E и P с возбудителем геноварианта 1.IN3 (Li et al., 2009; Cui et al., 2013; Wang et al., 2018). Еще в древности хозяйственная деятельность человека в этой провинции привела к возникновению многочисленных популяций синантропной желтобрюхой крысы (R. flavipectus). Как полагают МГ-исследователи, возбудитель чумы из природных популяций мышевидных грызунов в Юннане проник в дикие и синантропные популяции желтобрюхой крысы, образовав синантропические очаги F, где приобрел свойства биовара orientalis (геновариант 1.ORI2) (Achtman et al., 2004; Zhou et al., 2004; Ben-Ari et al., 2012). Биовар orientalis действительно стал виновником 3-й пандемии, начавшейся в Гонконге в 1894 г.
Эта стройная концепция, казалось бы, бесспорно обоснованная МГ-данными, с зоолого-паразитологических позиций видится не обоснованной, не валидной. На желтобрюхой крысе, наземно-древесном обитателе горных областей с гумидным и субгумидным климатом, не устраивающем глубоких сложных нор, в природе нет специфических для нее блох, и в ее диких популяциях не могли сформироваться природные очаги чумы. В китайской провинции Юннань и соседнем Вьетнаме существуют только синантропические или антропургические крысиные очаги в населенных пунктах и их ближайшем окружении, а переносчиком чумы в них стала космополитная блоха африканского происхождения Xenopsylla cheopis (Сунцов и др., 2011; Yin et al., 2011). Ее основной хозяин – нильская травяная мышь (Arvicanthis niloticus), обитающая в африканской Сахели. X. cheopis была разнесена по миру из африканской Сахели с синантропными крысами лишь в середине-конце XIX века (Traub, 1972). В то же время хорошо известно, что биовар orientalis чумного микроба является эндемичным для природных популяций индийской песчанки (Tatera indica) и ее блохи X. astia на Индостане (Sharif, 1951; Kingston et al., 2009). Эволюционный переход возбудителя чумы геноварианта 1.IN2 из популяций гималайского (красного?) сурка в популяции индийской песчанки мог произойти где-то в предгорьях Гиндукуша, западного Кунь-Луня, западных Гималаев, т.е. в районах соприкосновения или близкого схождения границ ареалов сурков и индийской песчанки в местах наиболее вероятного прямого норового контакта сурков и песчанок или опосредованного контакта через других многочисленных норовых грызунов (рис. 2). Мы предположили, что причиной изменения биохимических свойств микроба в популяциях индийской песчанки и преобразования биовара antiqua в биовар orientalis стали физиологические свойства организма индийской песчанки. В отличие от всех других видов основных хозяев микроба чумы в природных очагах Азии, T. indica имеет афротропические корни, обитает в теплом климате и не накапливает жир (источник метаболического глицерина), как это свойственно зимнеспящим животным и не зимнеспящим, обитающим в более холодных климатических условиях (Козлов, 1979; Сунцов, Сунцова, 2006; Сунцов, 2014). Дефицит метаболического глицерина в тканях индийской песчанки может объяснить возникновение у чумного микроба признака gly– (неспособность ферментировать глицерин).
Из природных очагов чумы в популяциях индийской песчанки возбудитель чумы в условиях антропогенных изменений перешел в популяции синантропной черной крысы (R. rattus) и на Индостане образовал многочисленные синантропические очаги (Sharif, 1951; May, 1952; Seal, 1969; Kingston et al., 2009). С большой вероятностью можно полагать, и на то имеются некоторые исторические свидетельства, что именно из индостанских очагов через территорию современной Мьянмы биовар orientalis к 1850-м годам проник торговыми и военными маршрутами на юг Китая, в Юннань и далее в Гонконг, где в популяциях синантропной желтобрюхой крысы и паразитирующей на ней космополитной блохи X. cheopis сформировались синантропические крысиные очаги, ставшие источником 3-й пандемии.
Итак, как рабочую гипотезу можем предложить следующий маршрут территориальной экспансии чумного микроба, завершившийся формированием геноварианта 1.ORI1. Выйдя из западной Монголии, биовар 4.ANT сначала распространился в поселениях алтайского сурка на Алтае, далее дошел до Тянь-Шаня, где в настоящее время циркулируют геноварианты 0.ANT 1, 2, 3, 5 и 1.IN4 в популяциях сурков, сусликов и песчанок (Cui et al., 2013; Kutyrev et al., 2018; Zhang et al., 2018) (рис. 2). В процессе естественной экспансии в поселения гималайского сурка некоторые из этих геновариантов преобразовались в варианты, специфические для его новых хозяев, т.е. в 1.IN1 и 1.IN2. По МГ-маркерам биовар orientalis наиболее близок к геновариантам группы 1.IN (Li et al., 2013; Zhang et al., 2018), что позволяет предполагать проникновение микроба чумы из популяций сурков в популяции индийской песчанки именно в районе совместного или соседствующего обитания гималайского сурка и индийской песчанки.
Статус “полевковых” подвидов Y. pestis
Некоторые МГ-исследователи прямым потомком псевдотуберкулезного микроба и наиболее древним микробом чумы считают полевковый подвид Y. pestis microtus (Платонов и др., 2013; Li et al., 2009; Xu et al., 2018). Характерным его свойством является способность ферментировать рамнозу (Rha+). Этим свойством обладают возбудители чумы из очагов Кавказа (хозяева обыкновенная полевка, полевая мышь), Гиссарского хребта (арчевая полевка), Таласского хребта (узкочерепная и серебристая полевки), Горного Алтая (монгольская пищуха), Баян-Улэгейского аймака Монголии (монгольская пищуха), северо-восточного Китая (полевка Брандта), восточного Тибета (цинхайская полевка). Этим свойством обладают также возбудители биовара intermedium (1.IN) из сурочьих очагов на Тибетском нагорье и Тянь-Шане и из песчаночьих очагов в Джунгарии (Li et al., 2009; Xu et al., 2018; Zhang et al., 2018). В целом, признак Rha+ нельзя считать исключительным для популяций полевок и пищух (Вариабельность…, 2009).
Li et al. (2008, 2009), чтобы убедить в филогенетическом единстве полевковых геновариантов, высказывают идею о существовании двух волн азиатской экспансии чумы в поселения грызунов и пищух. Первая волна связана с широким распространением исходного “полевкового” подвида в Азии после его отделения от предкового псевдотуберкулезного микроба с последующей инсуляризацией первичного метаочага и образования разрозненных удаленных друг от друга более мелких очагов. Вторую волну экспансии образовали возникшие из полевковых очагов эпидемически более опасные очаги с основными хозяевами сусликами, сурками, песчанками (Rasmussen et al., 2015). Однако мнению о филогенетическом единстве полевковых очагов имеются серьезные возражения (Вариабельность …, 2009; Achtman et al., 2004; Tong et al., 2005). Согласно экологическому сценарию, единой филогенетической ветви 0.PE с субветвями 0.PE1–0.PE10 не существует. Геноварианты этой квазиветви возникали по ходу экспансии Y. pestis из популяций монгольского сурка самостоятельно на разных географических территориях, в разное время и из разных предковых геновариантов, образовав очаги с основными далеко не родственными хозяевами – полевками и пищухами.
Уникальные геноварианты
Среди множества описанных геновариантов два относят к уникальным, представленным лишь одним или несколькими (штаммами): 0.PE3 (подвид angolica) и 0.PE7 (подвид tibetica) (Kutyrev et al., 2018). Первый возбудитель выделен от больного человека в Анголе, второй – от больного человека и сибирского тушканчика на Тибетском нагорье. Эти геноварианты требуют отдельного внимания, так как их относят к наиболее древним, корневым на филогенетическом древе Y. pestis (Eppinger et al., 2010; Morelli et al., 2010; Cui et al., 2013). Получается, что МГ-методы указывают на происхождение чумы по меньшей мере в двух далеко расположенных друг от друга районах мира. Следовательно, МГ-подход в этом отношении не совершенен, разграничение геновариантов и филогенетических линий на “древние” и “молодые” не убедительно. Согласно экологическому сценарию, эти геноварианты в силу их уникальности не могут характеризовать цельные популяции и филогенетические ветви, их пока следует рассматривать как единичные генотипы (мутации), требующие дополнительных исследований.
Филогения Y. pestis: нуль-гипотеза
Итак, видообразование микроба чумы Y. pestis могло проходить в трех географических популяциях монгольского сурка-тарбагана, в которых, по нашему пониманию, циркулировали возбудители псевдотуберкулеза с несколько различающимися МГ-характеристиками. Политопное видообразование привело к синхронному возникновению трех геновариантов чумного микроба: 2.ANT3, 3.ANT2 и 4.ANT1. Биогеоценотическая обстановка в каждой из географических популяций сурка-тарбагана предопределила различную филогенетическую судьбу этих трех геновариантов, распространившихся по трем разномасштабным фронтам.
При расползании из мест видообразования по принципу “масляного пятна”, нередко в условиях эволюционного “бутылочного горлышка”, микроб чумы попадал в гостально-векторную среду с разным соотношением в ней основных и дополнительных хозяев, с разной численностью и популяционной динамикой хозяев и переносчиков, различающимися физиологическими характеристиками хозяев и переносчиков. Это разнообразие новой среды обитания определило появление популяционных адаптаций микроба чумы, проявляющихся на уровне структуры генома. В конце концов, возникли дискретные очаги чумы с геновариантами, имеющими генетические метки в каждом очаге или даже на отдельных участках одного очага. Филогению Y. pestis на основе описанных геновариантов и с учетом экологических обстоятельств внутривидовой эволюции можно представить в следующем виде (рис. 3).
Рис. 3.
Экологическая интерпретация филогении Yersinia pestis (отображены только доминантные геноварианты). Распространение геновариантов, помещенных в бокс, недостаточно изучено.
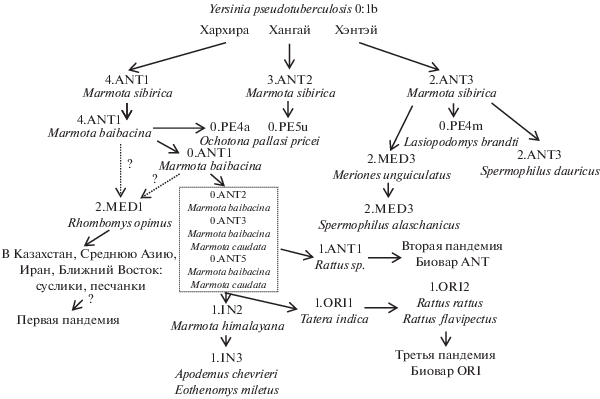
Таким образом, филогенетические деревья при МГ- и экологическом подходах явно различаются. Но более богатое биологическое содержание экологического подхода позволяет принять экологический сценарий как нуль-гипотезу для будущего эколого-молекулярно-генетического синтеза в проблеме происхождения и эволюции возбудителя чумы.
ВЫВОДЫ
1. Существовало 3 маршрута территориальной экспансии микроба из популяций монгольского сурка: центральный (Хангайский), имевший локальный масштаб, ограниченный центром и юго-западом Монголии; восточный (Хэнтэйский), имевший региональный масштаб, относительно короткий, ограниченный северо-востоком и востоком Китая; и западный (Алтайский), ставший проспектом для естественной азиатско-европейской и антропогенной мировой экспансии.
2. Реальность МГ-обоснованных филогенетических ветвей 0, 1, 2, 3 и 4 и филогенетических линий 0.ANT, 0.PE, 1.ORI, 1.IN, 2.MED и других микроба Y. pestis, отражающих степень родства геновариантов с предковым псевдотуберкулезным микробом и между собой, не находит поддержки в экологическом сценарии происхождения и мировой экспансии чумы. Возникновение того или иного геноварианта в большой мере определялось условиями циркуляции микроба в местной (локальной) гостальной среде, т.е. в местных очагах, сформировавшихся в процессе автономной территориальной экспансии чумного микроба из трех географическх популяций монгольского сурка.
Список литературы
Балахонов С.В., Ярыгина М.Б., Гладких А.С., Миронова Л.В., Феранчук С.И. и др., 2019. Молекулярно-генетическая характеристика штаммов Yersinia pestis, выделенных на монгольской территории трансграничного Сайлюгемского природного очага чумы // Проблемы особо опасных инфекций. № 3. С. 34–42.
Банников А.Г., 1954. Млекопитающие Монгольской Народной Республики. М.: Изд-во АН СССР. 670 с.
Бибиков Д.И., 1989. Сурки. М.: ВО “Агропромиздат”. 255 с.
Вариабельность возбудителя чумы и проблемы его диагностики, 2009. Ред. Ломов Ю.М. Ростов-на-Дону: ООО “Антей”. 534 с.
Вержуцкий Д.Б., Адъясурен З., 2019. Природные очаги чумы в Монголии: аннотированный список // Байкальский зоологический журнал. № 2 (25). С. 92–103.
Гродницкий Д.Л., 2002. Две теории биологической эволюции. 2-е изд. Саратов: Научная книга. 160 с.
Дмитриев П.П., 2006. Млекопитающие в степных экосистемах Внутренней Азии // Труды совместной Российско-Монгольской комплексной биологической экспедиции. Т. XLVIII. Москва. 224 с.
Евсеева В.В., Платонов М.Е., Дентовская С.В., Анисимов А.П., 2015. Типирование Yersinia pseudotuberculosis c помощью мультилокусного анализа вариабельного числа тандемных повторов // Проблемы особо опасных инфекций. № 4. С. 55–57.
Ербаева М.А., 1970. История антропогеновой фауны зайцеобразных и грызунов Селенгинского среднегорья. М.: Наукa. 132 с.
Ерошенко Г.А., Попов Н.В., Краснов Я.М., Никифоров К.А., Кузнецов А.А. и др., 2018. Природный мегаочаг основного подвида Yersinia pestis античного биовара филогенетической ветви 4.ANT в Горном Алтае // Проблемы особо опасных инфекций. № 2. С. 49–56.
Козлов М.П., 1979. Чума (природная очаговость, эпизоотология, эпидемические проявления). М.: Медицина. 191 с.
Корзун В.М., Балахонов С.В., Денисов А.В., Рождественский Е.Н., Токмакова Е.Г. и др., 2019. Эпизоотическая ситуация в монгольской части трансграничного Сайлюгемского природного очага чумы в 2018 г. // Проблемы особо опасных инфекций. № 2. С. 79–86.
Куклева Л.М., Шавина Н.Ю., Одиноков Г.Н., Оглодин Е.Г., Носов Н.Ю. и др., 2015. Анализ разнообразия и определение геновариантов штаммов возбудителя чумы из очагов Монголии // Генетика. Т. 51. № 3. С. 298–305.
Носов Н.Ю., Оглодин Е.Г., Краснов Я.М., Куклева Л.М., Шавина Н.Ю. и др., 2016. Филогенетический анализ штаммов Yersinia pestis средневекового биовара из природных очагов чумы Российской Федерации и сопредельных стран // Проблемы особо опасных инфекций. № 2. С. 75–78.
Одиноков Г.Н., Ерошенко Г.А., Краснов Я.М., Куклева Л.М., Черкасов А.В. и др., 2013. Анализ полногеномной последовательности штаммов Yersinia pestis на основе ступенчатого 680-SNP алгоритма // Проблемы особо опасных инфекций. № 3. С. 49–54.
Павлова А.И., Ерошенко Г.А., Одиноков Г.Н., Куклева Л.М., Шавина Н.Ю. и др., 2012. Анализ генетической изменчивости штаммов Yersinia pestis средневекового биовара из природных очагов чумы России и Монголии // Проблемы особо опасных инфекций. № 114. С. 49–53.
Платонов М.Е., Евсеева В.В., Дентовская С.В., Анисимов А.П., 2013. Молекулярное типирование Yersinia pestis // Молекулярная генетика, микробиология и вирусология. № 2. С. 2–12.
Ралль Ю.М.,1965. Природная очаговость и эпизоотология чумы. М.: Медицина. 363 с.
Сунцов В.В., 2014. Происхождение и мировая экспансия микроба чумы Yersinia pestis: фактор изоляции // Успехи современной биологии. Т. 134. № 4. С. 411–425.
Сунцов В.В., 2015. Происхождение микроба чумы Yersinia pestis: концепция популяционно-генетической макроэволюции в переходной среде // Журнал общей биологии. Т. 76. № 4. С. 310–318.
Сунцов В.В., 2018. Квантовое видообразование микроба чумы Yersinia pestis в гетероиммунной среде – популяциях гибернирующих сурков-тарбаганов (Marmota sibirica) // Сибирский экологический журнал. № 4. С. 381–396.
Сунцов В.В., 2020. Перспективы синтеза молекулярно-генетического и экологического подходов к проблеме видообразования микроба чумы Yersinia pestis // Успехи современной биологии. Т. 140. № 1. С. 43–57.
Сунцов В.В., Сунцова Н.И., 2000. Экологические аспекты эволюции микроба чумы Yersinia pestis и генезис природных очагов // Известия РАН. Серия биологическая. № 6. С. 645–657.
Сунцов В.В., Сунцова Н.И., 2006. Чума. Происхождение и эволюция эпизоотической системы (экологические, географические и социальные аспекты). М.: Товарищество научных изданий КМК. 247 с.
Сунцов В.В., Сунцова Н.И., 2008. Макро- и микроэволюция в проблеме происхождения и мировой экспансии возбудителя чумы Yersinia pestis // Известия РАН. Серия биологическая. Вып. 4. С. 389–395.
Сунцов В.В., Сунцова Н.И., Румак В.С., Данг Туан Дат, Хоанг Ань Тует, Лыонг Тхи Мо, 2011. Структура и генезис эпизоотических систем “грызун–блоха– микроб Yersinia pestis” в ценозах Вьетнама, включая территории экоцида // Ред. Румак В.С. Окружающая среда и здоровье человека в загрязненных диоксинами регионах Вьетнама. М.: Товарищество научных изданий КМК. С. 202–258.
Чинболд Л., Зонов Г.Б., Очиров Ю.Д., Логачев А.И., Жигмэд С., 1980. Новые данные эпизоотологической характеристики природного очага чумы Южно-Гобийского аймака МНР // Проблемы природной очаговости чумы. Тезисы докладов IV советско-монгольской конференции специалистов противочумных учреждений. Часть 1. Иркутск. С. 74–75.
Achtman M., 2008. Evolution, Population structure, and Phylogeography of Genetically Monomorphic Bacterial Pathogens // Ann. Rev. Microbiol. V. 62. P. 53–70.
Achtman M., 2012. Insights from genomic comparisons of genetically monomorphic bacterial pathogens // Phil. Trans. R. Soc. № 367. P. 860–867.
Achtman M., Zurth K., Morelli G., Torrea G., Guiyoule A., Carniel E., 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis // PNAS. Nov. 23. V. 96. № 24. P. 14 043–14 048.
Achtman M., Morelli G., Zhu P., Wirth T., Diehl I., et al., 2004. Microevolution and history of the plague bacillus, Yersinia pestis // PNAS. V. 101. № 51. P. 17 837–17 842.
Anisimov A.P., Lindler L.E., Pier G.B., 2004. Intraspecific diversity of Yersinia pestis // Clin. Microbiol. Rev. V. 17. № 2. P. 434–464.
Ben-Ari T., Neerinckx S., Agier L., Cazelles B., Xu L., et al., 2012. Identification of Chinese plague foci from long-term epidemiological data // PNAS. V. 109. № 21. P. 8196–8201.
Brandler O., 2018. Molecular diversity and taxonomy in marmots // Proceed. 7th Intern. Conf. on the Genus Marmota “Marmots of the old and new world”. Ulan-Bator. P. 97–104.
Cui Y., Li Y., Gorge O., Platonov M., Yan Y. et al., 2008. Insight into Microevolution of Yersinia pestis by Clustered Regullarly Interspaced Short Palindromic Repeats // PloS ONE. V. 3. № 7. e2652.
Cui Y., Yu C., Yan Y., Li D., Li Y., et al., 2013. Historical variations in mutation rate in an epidemic pathogen, Yersinia pestis // PNAS. V. 110. № 2. P. 577–582.
Demeure C.E., Dussurget O., Fiol G.M., Le Guern A.-S., Savin C., et al., 2019. Yersinia pestis and plague: an updated view on evolution, virulence determinants, immune subversion, vaccination, and diagnostics // Genes Immunity. doi.org/https://doi.org/10.1038/s41435-019-0065-0
Eppinger M., Worsham P.L., Nikolich M.P., Riley D.R., Sebastian Y., et al., 2010. Genome Sequence of the Deep-Rooted Yersinia pestis Strain Angola Reveals New Insights into the Evolution and Pangenome of the Plague Bacterium // Jour. Bacteriol. V. 192. № 6. P. 1685–1699.
Erbaeva M.A., 2003. History, evolutionary development and systematics of marmots (Rodentia, Sciuridae) in Transbaikalia // Rus. Journ. Theriol. V. 2. № 1. P. 33–42.
Eroshenko G.A., Nosov N.Y., Krasnov Y.M., Oglodin Y.G., Kukleva L.M., et al., 2017. Yersinia pestis strains of ancient phylogenetic branch 0.ANT are widely spread in the highmountain plague foci of Kyrgyzstan // PLoS ONE. V. 12. № 10. e0187230.
Heads M., 1985. On the Nature of Ancestors // Syst. Zool. V. 34. № 2. P. 205–215.
Kingston J.J., Tupeja U., Kapil M., Murali H.S., Batra H.V., 2009. Genotyping of Indian Yersinia pestis strains by MLVA and repetitive DNA sequence based PCRs // Antonie van Leeuwenhhoek. V. 96. № 3. P. 303–312.
Kutyrev V.V., Eroshenko G.A., Motin V.L., Nosov N.Y., Krasnov J.M., et al., 2018. Phylogeny and Classification of Yersinia pestis Through the Lens of Strains from the Plague Foci of Commonwealth of Independent States // Front. Microbiol. V. 9. Art. 1106. P. 1–11.
Li Y., Dai E., Cui Y., Zhang Y., Li M., Zhang Y., et al., 2008. Different region analysis for genotyping Yersinia pestis isolates from China // PLoS One. V. 3. № 5. E2166. P. 1–10.
Li U., Cui Y., Hauck Y., Platonov M.E., Dai E., et al., 2009. Genotyping and Phylogenetic Analysis of Yersinia pestis by MLVA: Insights into the Worldwide Expansion of Central Asia Plague Foci // PLoS ONE. V. 4. № 6. e6000.
Li Y., Cui Y., Cui B. Yan Y., Yang X., et al., 2013. Features of Variable Number of Tandem Repeats in Yersinia pestis and the Development of a Hierarchial Genotyping Scheme // PLoS ONE. V. 8. № 6. e66567. P. 1–10.
May J.M., 1952. Map of the World Distribution of Plague // Geographical Review. V. 42. № 4. P. 628.
Morelli G., Song Y., Mazzoni C.J., Eppinger M., Roumagnac P., et al., 2010. Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity // Nat. Genet. V. 42. № 12. P. 1140–1143.
Rasmussen S., Allentoft M.E., Nielsen K., Orlando L., Sikora M., et al., 2015. Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago // Cell. V. 163. № 3. P. 571–582.
Riehm J.M., Vergnaud G., Kiefer D., Damdindorj T., Dashdavaa O., et al., 2012. Yersinia pestis Lineages in Mongolia // PLoS ONE. V. 7. № 2. e30624
Sariyeva G., Bazarkanova G., Maimulov R., Abdikarimov S., Kurmanov B., et al., 2019. Marmots and Yersinia pestis Strains in Two Plague Endemic Areas of Tien Shan Mountains // Front. Vet. Sci. V. 6. № 207. P. 1–14.
Schluter D., Nagel L.M., 1995. Parallel Speciation by Natural Selection // Amer. Natur. V. 146. № 2. P. 292–301.
Seal S.C., 1969. Epidemiological Studies of Plague in India // Bull. Wld. Hlth. Org. V. 23. P. 283–292.
Sharif M., 1951. Spread of Plague in the Southern and Central Divisions of Bombay Province and Plague Endemic Centres in Indo-Pakistan Subcontinent // Bull. Wld. Hlth. Org. V. 4. P. 75–109.
Skurnik M., Peippo A., Ervela E., 2000. Characterization of the O-antigen gene cluster of Yersinia pseudotuberculosis and the cryptic O-antigen gene cluster of Yersinia pestis shows that the plague bacillus is most closely related to and has evolved from Y.pseudotuberculosis serotype O:1b // Molec. Microbiol. V. 37. № 2. P. 316–330.
Smith A.T., Xie Y., 2008. A Guide to the Mammals of China. Ed. W. Sung. Princeton-Oxford: Princeton Univ. Press. 544 p.
Tong Z., Zhou D., Song Y., Zhang L., Pei D., et al., 2005. Pseudogene accumulation might promote the adaptive microevolution of Yersinia pestis // J. Med. Microbiol. V. 54. № 3. P. 259–268.
Traub R., 1972. Notes of fleas and the ecology of plague // J. Med. Entomol. V. 9. № 6. P. 603.
Wang X., Zhou D., Qin L., Dai E., Zhang J., et al., 2006. Genomic comparison of Yersinia pestis and Yersinia pseudotuberculosis by combination of suppression subtractive hybridization and DNA microarray // Arch. Microbiol. V. 186. P. 151–159.
Wang P., Shi L., Zhang F., Guo Y., Zhang Z.,et al., 2018. Ten years of surveillance of the Yulong plague focus in China and the molecular typing and source tracing of the isolates // PLoS Negl. Trop. Dis. V. 12. № 3. e0006352
Williams D.M., Ebach M.C., 2008. Foundations of Systematics and Biogeography. N.-Y.: Springer S.B. Media. 308 p.
Wu Lien Teh, Chun J.W.H., Pollitzer R., Wu C.Y., 1936. Plague: a manual for medical and public health workers. Shanghai. Merc. Press. 547 p.
Xu X., Cui Y., Xin X., Yang X., Yang X., et al., 2018. Genetic diversity and spatial-temporal distribution of Yersinia pestis in Qinghai Plateau, China // PLoS Negl. Trop. Dis. V. 12. № 6. E0006579. P. 1–17.
Yin J., Geater A., Chongsuvivatwong V., Dong X.-Q., Du C.-H., Zhong Y.-H., 2011. Predictors for Abundance of Host Flea and Floor Flea in Households of Villages with Endemic Commensal Rodent Plague, Yunnan Province, China // PLoS Negl. Trop. Dis. V. 5. № 3. e997.
Zhang X., Hai R., Wei J., Cui Z., Zhang E., et al., 2009. MLVA distribution characteristics of Yersinia pestis in China and the correlation analysis // BMC Microbiology. V. 9. № 205. P. 1–8.
Zhang Y., Luo T., Yang C., Yue X., Guo R., et al., 2018. Phenotypic and Molecular Genetic Characteristics of Yersinia pestis at an Emerging Natural Plague Focus, Junggar Basin, China //Am. J. Trop. Med. Hyg. V. 98. № 1. P. 231–237.
Zhou D., Han Y., Huang P., Yang R., 2004. Comparative and evolutionary genomics of Yersinia pestis // Microbes and Inf. № 6. P. 1226–1234.
Zhou D., Han Y., Song Y., Tong Z., Wang J., et al., 2004a. DNA Microarray Analysis of Genome Dynamics in Yersinia pestis: Insights into Bacterial Genome Microevolution and Niche Adaptation // J. Bacteriol. V. 186. № 15. P. 5138–5146.
Zhou D., Tong Z., Song Y., Han Y., Pei D., et al., 2004b. Genetics of Metabolic Variations between Yersinia pestis Biovars and the Proposal of a New Biovar, microtus // J. Bacteriol. V. 186. № 15. P. 5147–5152.
Zhou D., Yang R., 2009. Molecular Darwinian Evolution of Virulence in Yersinia pestis // Infect. Immun. V. 77. № 6. P. 2242–2250.
Дополнительные материалы отсутствуют.
Инструменты
Зоологический журнал