

Зоологический журнал, 2020, T. 99, № 11, стр. 1232-1241
Недоучет последствий биологических инвазий в филогеографических реконструкциях на примере Daphnia magna (Crustacea, Cladocera)
Д. П. Карабанов a, *, Е. И. Беккер b, **, А. А. Котов b, ***
a Институт биологии внутренних вод им. И.Д. Папанина РАН
152742 пос. Борок, Россия
b Институт проблем экологии и эволюции
им. А.Н. Северцова РАН
119071 Москва, Россия
* E-mail: dk@ibiw.ru
** E-mail: evbekker@ya.ru
*** E-mail: alexey-a-kotov@yandex.ru
Поступила в редакцию 25.04.2020
После доработки 27.04.2020
Принята к публикации 10.05.2020
Аннотация
На примере филогеографического анализа Daphnia magna Straus 1820 (Crustacea, Cladocera) изучено влияние на результаты этого анализа недоучета последствий биологических инвазий, а именно, контаминации общего пула гаплотипов чужеродными последовательностями. Предложен алгоритм выявления последних.
За прошедшие двадцать лет после выхода классического обобщающего труда Джона Ависа (Avise, 2000) филогеография как метод изучения пространственного распределения генеалогических линий из частного подхода превратилась в самостоятельную дисциплину со своей сложной и строго обоснованной методологией и детально разработанной теоретической базой. Филогеографический подход показал свою высокую эффективность при изучении различных аспектов эволюционной истории животных (Avise et al., 2016). Несмотря на достигнутые успехи, филогеография как биологическая дисциплина все еще находится на стадии становления, и исследователям известен ряд методологических проблем, требующих своего решения в будущем.
Для математической обработки совокупности генетических и биогеографических данных Алан Темплетон (Templeton, 1998, 2004) разработал алгоритм “филогеографического анализа вложенных клад” (NCPA), основанный на первичной реконструкции сетей гаплотипов с последующим вычислением географических и генетических расстояний между вариантами, их объединением в более крупные агломерации и определением исторических процессов, способных привести к наблюдаемым современным паттернам (Templeton, 2004). Хотя для целей восстановления филогении подход пересчета генетических и географических дистанций между группами гаплотипов порождает (во многом – обоснованную) критику (Panchal, Beaumont, 2010), в целом его полезность для реконструкции истории расселения и формирования популяционно-генетической структуры у животных не вызывает сомнений (Williford et al., 2016).
Современный биогеографический подход, апеллирующий к различным моделям расселения–вымирания–кладогенеза, также вносит заметный вклад в филогеографические реконструкции (Ree, Smith, 2008; Zink et al., 2000). Однако и этот анализ имеет свои ограничения. Например, проблемы вызывает соотнесение времени геологических событий (образования гор, изменения русла рек для создания географической изоляции, и т.д.) и времени молекулярной дивергенции (прежде всего определение скорости накопления нуклеотидных замен в “молекулярных часах”). Для достижения этого соответствия даже было предложено проводить “калибровку” филогенетических деревьев на основании крупных геологических событий (Ho et al., 2015). Но в целом восстановленная филогения высших таксонов обычно хорошо соотносится с палеонтологическими и палеогеографическими данными на больших временны́х интервалах (Xiang, Soltis, 2001).
Наконец, имеется еще один филогеографический подход, направленный на изучение общего генетического разнообразия какой-либо группы через различные коэффициенты подразделенности (FST, GST, PST и др.). Этот подход основан на множественном сравнении между матрицами генетических различий и матрицами географических расстояний, например, через тест Мантеля (Diniz-Filho et al., 2013). Однако есть серьезные основания сомневаться в достоверности выводов, полученных в исследованиях с применением традиционных индексов меры сходства (Jost, 2008). К тому же, как правило, изучение популяционно-генетической структуры проводится на высокополиморфных микросателлитных локусах, имеющих значительные ограничения для оценки популяционной дифференциации (Горбачев, 2011).
Все эти подходы призваны проанализировать исторические процессы формирования ареала некоего таксона. Однако они практически не учитывают важнейший модус формирования биоразнообразия в новейшей истории – деятельность человека, резко увеличившую вероятность случайного или преднамеренного вселения чужеродных видов в новые местообитания (Элтон, 1960; Дгебуадзе и др., 2018). Перенос видов за пределы исторических ареалов может создать значительную неопределенность и даже вовсе сделать невозможными реконструкции естественных процессов происхождения тех или иных популяций. Вычленение чужеродного компонента, особенно для широко распространенных видов, требует скрупулезного сравнения различных современных и музейных образцов из нативных и новых частей ареала, при этом не гарантируя абсолютной уверенности в выявлении центров происхождения, коридоров и векторов инвазии (Dormontt et al., 2014).
В настоящее время ветвистоусые ракообразные (Crustacea, Cladocera), являющиеся общепризнанной модельной группой для эволюционных, генетических, физиологических, палеолимнологических исследований (Lampert, 2011; Smirnov, 2017), стали также модельной группой для изучения биологических инвазий. К кладоцерам относится несколько хорошо известных вселенцев, в том числе тех, которые значительно видоизменили подвергнувшиеся инвазиям экосистемы (Berg, Garton, 1994; Frisch et al., 2013) или потенциально способны их видоизменить в будущем (Karabanov et al., 2018). Но во многих случаях вселение чужеродных видов ракообразных может быть не столь разрушительно для экосистем и может оставаться незамеченным до проведения специальных генетических работ. Такие работы показывают, что ныне чужеродные таксоны и гаплотипы большинства широко распространенных групп видов планктонных кладоцер находятся в несвойственных для них регионах (Ishida, Taylor, 2007; Mergeay et al., 2005; Kotov et al., 2020). Для нектонных (Taylor et al., 2020) и бентосных (Sharma, Kotov, 2015) видов таких случаев описано меньше, что однозначно объясняется значительно меньшим вниманием генетиков к непланктонным кладоцерам по сравнению с планктонными (особенно принадлежащими к роду Daphnia O.F. Müller).
До начала генетического исследования по некой группе можно лишь сделать некоторые предварительные предположения о наличии в общем пуле исследуемых особей, таковых, находящихся в неаборигенных для них регионах. В большинстве случаев нет никаких документальных свидетельств прошедших инвазий, если они не имели катастрофических последствий. При этом произошедшее недавно вселение (в том числе, трансконтинентальное) чужеродных гаплотипов, если оно не будет распознано каким-либо методом, может сильно исказить результаты филогеографического анализа.
Daphnia magna Straus 1820 (Cladocera, Anomopoda, Daphniidae) – обычный в Северной Евразии таксон ветвистоусых ракообразных, которому было посвящено несколько филогеографических исследований (Fields et al., 2018; De Gelas, De Meester, 2005). В ходе наших работ по филогенетическому анализу, на протяжении практически всего современного ареала было выявлено несколько филогенетических линий, объединяемых в две большие группы гаплотипов – “A” (европейско-западносибирская) и “B” (сибирско-беренгийская), одновременно были получены свидетельства антропогенного вселения на американский континент представителей европейской митохондриальной линии (Bekker et al., 2018). Представленная работа является прямым продолжением этого исследования. Она специально посвящена изучению влияния наличия в общем пуле особей, обитающих в результате биологической инвазии на неаборигенных территориях, на результаты филогеографического анализа и выработке формального алгоритма их распознания.
МАТЕРИАЛЫ И МЕТОДЫ
Материалом для исследования послужили нуклеотидные последовательности 5'-участка гена субъединицы I цитохромоксидазы-c митохондриальной ДНК (COI), полученные авторами ранее для различных популяций D. magna (Bekker et al., 2018), а также все доступные на текущий момент записи для этого таксона из международной базы данных GenBank (NCBI). В анализ включили только последовательности филогенетической линии “A”, в то время как линия “B” была рассмотрена как внешняя группа.
Общая схема обработки данных включала несколько этапов, подробно разобранных далее. Вначале были определены общая структура взаимоотношений между группами гаплотипов и их географическое распределение. На этом этапе использовались алгоритмы восстановления сетей гаплотипов и анализ NCPA.
Определение “звездчатых структур” (групп с основным центральным гаплотипом и совокупности производных гаплотипов, отличающихся на одну или несколько нуклеотидных замен) и их редукцию до сколлапсированной последовательности проводили согласно Форстеру с соавторами (Forster et al., 2001), принимая во внимание то, что сокращение входящих данных при биогеографических исследованиях – довольно распространенный в практике филогеографического анализа метод (Williford et al., 2016). Визуализацию генетических связей провели путем построения сетей гаплотипов (математического графа с вершинами в виде последовательностей, связанных ребрами сообразно числу “эволюционных шагов”, как правило – доле нуклеотидных замен) в программе SplitsTree v.5.1 (Huson, Bryant, 2006) с использованием алгоритма расщепления графа, что значительно облегчает визуальную интерпретацию (Huson et al., 2011).
Для проведения теста Мантеля использовали программу PASSaGE v.2.0 (Rosenberg, Anderson, 2011). Матрицу генетических “простых” p-дистанций построили в MEGA-X (Kumar et al., 2018). Матрицу географических расстояний построили средствами PASSaGE исходя из географических координат каждой исследованной популяции. Тест выполнили с 1000 перестановками, корреляция между значением теста и дистанцией получена средствами PASSaGE.
Филогеографический анализ вложенных клад NCPA включал нескольких частично автоматизированных этапов. Входящую сеть гаплотипов построили в программе TSC v.1.23 (Clement et al., 2000). Вычисление дистанций выполнили в программе GeoDis v.2.6 (Posada et al., 2000) с коррекцией Данна-Сидака на множественные сравнения, что существенно повышает точность филогеографического вывода методом NCPA (Templeton, 2009).
Восстановление филогении провели в пакете BEAST2 v.2.6 (Bouckaert et al., 2019). Было запущено четыре независимые цепи MCMC в 100M поколений, с отбором каждого 10k дерева. Эффективность MCMC контролировали по всем доступным параметрам с помощью универсального анализа log-файлов BEAST2 в пакете Tracer v.1.7 (Rambaut et al., 2018) c оценкой эффективного размера выборки (ESS) для всех параметров выше 300. Консенсусное дерево, основанное на максимальной достоверности клады (MCC), построили в программе TreeAnnotator v.2.6 (Drummond et al., 2012) с игнорированием первых 50% деревьев. Проекцию филогенетического дерева на географическую карту провели в пакете BEAST2 GEO_SPHERE (Bouckaert, 2016).
Для биогеографической реконструкции в качестве исходных файлов для пакета RASP4 v.4.2 (Yu et al., 2015) из полученного набора деревьев BEAST2 случайно отобрали 100 деревьев, а также итоговое конденсированное дерево. Для тестирования филогеографических моделей использовали пакет BioGeoBEARS (Matzke, 2013), интегрированный в RASP4. Исходные данные выбирали исходя из представленности по всем основным географическим локациям для филогенетической линии “A”. Объективные ограничения вычислительных мощностей позволили взять в анализ только 27 последовательностей, относящиеся к девяти большим географическим локациям, что в сочетании с четырьмя потенциальными ареалами обеспечило достаточную достоверность реконструкции. Всего в BioGeoBEARS протестировали шесть биогеографических моделей (стандартных дисперсионно–викариантных и с поправкой +J на событие-видообразование), оцененных по взвешенному критерию AICc (Matzke, 2014). В качестве лучшей модели с наименьшим значением AICc была выбрана модель расселения–вымирания–кладогенеза (DEC) с вероятностью скачкообразного расселения (+J). Следует отметить, что версия модели DEC для RASP4 экспортирует вероятность всех возможных биогеографических сценариев, оцененных в данном узле, что позволяют всесторонне оценить степень неопределенности наследуемого состояния в биогеографических реконструкциях (Yu et al., 2015). Поправка +J учитывает вероятность происхождения новых линий непосредственно колонизацией “основателем” без промежуточного существования широко распространенного предка (Clark et al., 2008).
В качестве альтернативного метода, основанного на стохастическом подходе, для описания эволюции ареала как непрерывного процесса, использовали реализованную в RASP4 модель BayArea (Landis et al., 2013). В качестве входящих файлов для этого анализа использовали тот же набор деревьев, что и для анализа DEC, а также географические координаты локаций. Анализ выполнили на 10M поколений MCMC, с отбором каждого 10k дерева и игнорированием первых 20% поколений. Провели четыре независимых запуска BayArea для увеличения эффективных размеров выборки, а также в качестве сравнительного теста на отклонения в поведении разных MCMC. Результаты тестов объединили и визуализировали через модуль комбинации результатов в RASP4.
Информация о последовательностях, все входящие файлы и результаты вычислений, доступны в репозитарии Open Science Framework (https:// osf.io/8ec3m/).
РЕЗУЛЬТАТЫ
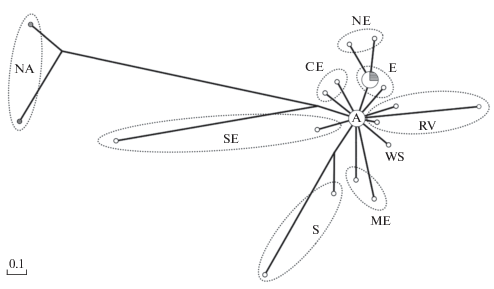
В расщепленной сети гаплотипов (рис. 1) можно выделить несколько кластеров, формирующихся, преимущественно, по географическому принципу. Хотя, как правило, нельзя однозначно определить соответствие того или иного гаплотипа конкретной локации, наблюдается некоторая общая географическая приуроченность гаплогрупп, что отмечалось и ранее (De Gelas, De Meester, 2005). Кроме широко распространенной центральной группы гаплотипов “A”, можно выделить ряд производных групп, преимущественно приуроченных к определенным территориям. Однако типичные “европейские” гаплотипы найдены и в Северной Америке (США и Мексика; указаны штриховкой на рис. 1). При этом на данном континенте имеются свои, генетически сильно дифференцированные представители (группа NA на рис. 1, соответствующая кладе “A2” по: Bekker et al., 2018).
Рис. 1.
Расщепленная сеть гаплотипов (split decomposition) после преобразования “звездчатых структур” (star contraction). Показаны крупные группы гаплотипов относящихся к кладе “A” (по: Bekker et al., 2018), сгруппированные преимущественно по большим географическим зонам: NA – Северная Америка; NE – Северная Европа; CE – Центральная Европа; E – разные выборки из Европы, в том числе России; RV – выборки из бассейна Волги; SE – Южная Европа; S – Южная Европа и Северная Африка; ME – выборки из Большого Ближнего Востока; WS – Западная Сибирь; A – центральный пул гаплотипов, широко распространённый на Европейском континенте. Штриховкой выделены последовательности из Западного полушария. Указана генетическая p-дистанция.
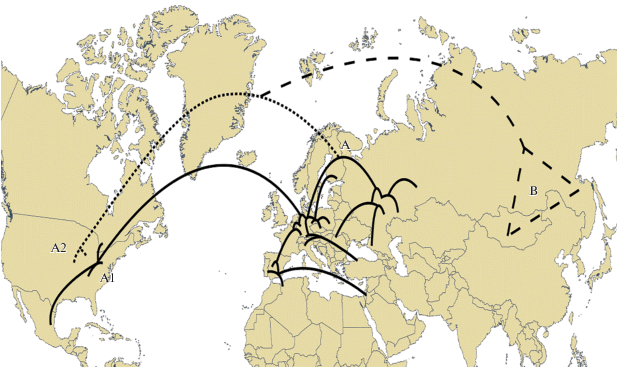
Более наглядно представить парадоксальность генетического ландшафта D. magna можно путем проецирования филогенетического дерева дафний на географическую карту (рис. 2). Предполагая естественные процессы формирования современного ареала D. magna, приходится допускать, что филогенетическая линия “A1” характеризуется огромной пространственной протяженностью и существенной изоляцией от других клад более высокого уровня, но при этом обладает низкой генетической изменчивостью на всем ареале. Не удивительно, что в случае принятия подобной гипотезы процессы выделения прочих клад не могут быть определены, а результат биогеографической реконструкции становится неубедительным (NCPA вывод IO, см. дополнительные материалы https://osf.io/8ec3m/).
Рис. 2.
Проекция филогенетического дерева на географическую карту для D. magna в Северном полушарии. Показана только топология восстановленной филогении. Сплошная линия – популяции Европы (и инвазийные популяции Америки) – филогенетическая линия “A1”; прерывистая – филогенетическая линия “А2”, исторически обитающая в Северной Америке; пунктир – внешняя группа (филогенетическая линия “B”).
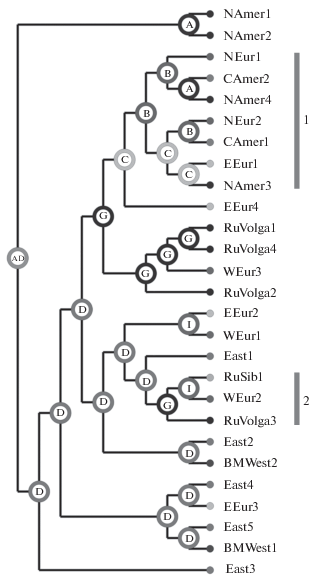
Результаты комбинирования реконструкции DEC+J с байесовой реализацией BayArea показаны рис. 3. На большей части современного ареала D. magna выделяются несколько групп с общим вероятным предком. Однако есть две ветви (ветви 1 и 2 на рис. 3), внутри которых не представляется возможным подтвердить достоверность отнесения события ни к кладогенезу, ни к дисперсии (подробную статистику см. на https://osf.io/ 8ec3m/). В случае ветви 1 неопределенность явным образом объясняется контаминацией нативных выборок из Европы чужеродными выборками из Северной Америки (см. далее). При этом аборигенные последовательности из Северной Америки достоверно формируют отдельную ветвь с наиболее вероятным процессом кладогенеза (см. дополнительные материалы https://osf.io/ 8ec3m/). Можно с высокой степенью уверенности утверждать, что неопределенность, возникшая для ветви 1 (рис. 3), связана с ложным дисперсионным событием, возникшим благодаря деятельности человека, а именно – биологической инвазией европейских представителей D. magna на американский континент.
Рис. 3.
Предполагаемая биогеографическая история на консенсусном дереве по локусу COI мтДНК для основных географических зон ареала D. magna. Комбинированный результат с моделью DEC + J и реконструкции BayArea. Показана только топология дерева. Круговые диаграммы в каждом узле показывают вероятности нахождения альтернативных предков, наиболее вероятное состояние отображается буквой в центре. Выделены ветви с неопределенностью выбора между дисперсионным и викариантным процессом (см. обсуждение в тексте).
ОБСУЖДЕНИЕ
Применение двух “идеологически независимых” методов филогеографической реконструкции (анализ сетей гаплотипов и филогенетических деревьев) привело к схожим результатам, и мы можем с большой долей вероятности интерполировать полученные данные на реальные природные процессы.
В исследовании, посвященном преимущественно западноевропейским популяциям D. magna (De Gelas, De Meester, 2005), была отмечена некоторая корреляция распределения гаплотипов мтДНК с географическим положением выборок. Однако применение того же анализа NCPA для современного набора последовательностей со всего ареала показало заметное рассогласование между генетическими и географическими расстояниями. Это выражается в отсутствии значимых различий в дистанциях конечных (Dc) и вложенных (Dn) клад и провале χ2 теста как для клады низшего порядка, так и для объединяющей клады для группы “A” вплоть до уровня континентов, когда разделяются лишь группы “A1” и “A2” (см. дополнительные материалы https://osf.io/8ec3m/).
При интерпретации анализа с учетом “странных” (чрезвычайно широко распространенных, рис. 2) клад возникает парадоксальный вывод о формировании генетической структуры вида как результата ограниченного потока и рассеивания генов (NCPA вывод RG), в совокупности с возможными процессами расселения на большие расстояния (вывод LDC) и в промежуточных областях (или как результата исторических процессов, за которым следует исчезновение промежуточных популяций, вывод PF).
Однако подобная неопределенность может возникнуть и для нативных популяций, когда очень широко расселены животные одной гаплогруппы. Примером этого может служить ветвь 2 (рис. 3). Здесь животные с гаплотипами одной группы расселены по нескольким географическим зонам – от Западной Европы до Сибири. У нас нет данных, опровергающих нативный статус этих выборок, однако и здесь нельзя вовсе исключить возможность образования некоторых популяций в результате переноса покоящихся яиц кладоцер в ходе множественных миграций человека в историческое время, при том что в самое последнее время вероятность инвазии повысилась катастрофически. Отметим, что с “биоинформатической” точки зрения разница между процессами исторического саморасселения видов и биологических инвазий – только в скоростях экспансии и накопления генетических различий в новых популяциях. Поэтому любое расширение ареала автоматически будет определяться как дисперсионное событие.
Конечно, нельзя вовсе исключить формирования атипичной популяционно-генетической структуры, отображенной на рис. 2, но в таком случае придется принять предположение либо о крайне быстром транс-континентальном расселении дафний (что в голоцене навряд ли было возможно), либо о викариантном пути формирования подобного ареала, связанного с крайне древним сценарием: мезозойским распадом и дрейфом континентов. Однако и филогеографические реконструкции для европейских популяций D. magna (De Gelas, De Meester, 2005), и восстановление филогении с демографическими тестами для популяций Евразии в целом (Bekker et al., 2018), и палеонтологические данные (Kotov and Taylor, 2011) не поддерживают ни предположение о быстрой экспансии, ни предположение о столь древнем происхождении этих линий дафний, несмотря на значительную древность таксонов кладоцер различного ранга (Kotov, Korovchinsky, 2006; Kotov, Taylor, 2011; Smirnov, 1992). Для другого широко распространенного в Голарктике ветвистоусого ракообразного – Polyphemus pediculus (Linnaeus 1758) (Cladocera, Onychopoda) – было убедительно показано, что популяции генетически отличаются друг от друга тем значительнее, чем больше географическое расстояние между ними, а вся экспансия этого вида в Голарктике протекала в четвертичном периоде (Xu et al., 2009).
Прояснить ситуацию с демографическими процессами для D. magna помогает анализ времени дивергенции тех американских популяций, естественный (неинвазионный) статус которых не вызывает сомнений (клада “А2” по: Bekker et al., 2018). Отделение этой клады от предковых евроазиатских форм предположительно произошло порядка 20 млн лет назад, примерно в тот же период, что и разделение между “европейскими” и “азиатскими” филогенетическими линиями “A” и “B” у D. magna. Это как раз вполне можно объяснить существованием Берингийского перешейка, соединявшего Евразию и Северную Америку в единый суперконтинент в миоцене. Подобная генетическая близость ряда популяций Европы и Северной Америки наблюдается и у P. pediculus (Xu et al., 2009), только она связана с более современной, познечетвертичной, эпохой формирования ареала этого таксона.
Конечно, может возникнуть справедливое замечание, что при анализе распространения и генетической изменчивости D. magna в “берингийской” группе популяций “B” (предположительно имеющей эволюционный возраст, сравнимый со временем существования клады “А2”) наблюдается значительное генетическое разнообразие в отличие от “европейских” дафний. Для объяснения таких различий по естественным причинам можно выдвинуть несколько предположений:
(1) Во-первых, как показывает NCPA, для достоверной реконструкции истории группы “A2” недостаточно данных: здесь исследовано лишь три гаплотипа, что, несомненно, слишком мало. Однако степень дивергенции между филогенетическими линиями “A1” и “A2” слишком велика, и навряд ли удастся найти сколь-либо полный ряд переходных гаплотипов.
(2) Во-вторых, геоклиматические изменения, а именно большое плейстоценовое оледенение, могло привести к эффекту “бутылочного горлышка” для популяций Северной Америки. Вероятно, подобные процессы протекали и на территории Европы, что и наблюдается в значительно меньшем генетическом разнообразии “европейской” клады “A1” по сравнению с популяциями клады “B”.
(3) Наконец, нельзя однозначно исключить наличие неких исторически закрепившихся механизмов поддержания низкого уровня генетического разнообразия у представителей D. magna филогенетической линии “А1”.
Хотя эти возможные объяснения нельзя сразу отвергнуть, их реализация в природных условиях представляется довольно сомнительной. Таким образом, можно констатировать, что на территории Северной Америки в настоящее время существуют популяции из двух филогенетических линий (рис. 2), относящихся к кладам “A1” (тождественные современным европейским дафниям) и “A2” (вероятно, древняя группа, происходящая от европейских дафний, но генетически значительно отличающаяся от них в настоящее время). Такой сложный характер генетических отношений для ареала D. magna объясняет провал теста Мантеля (p < 0.05), показывающего отсутствие значимой связи между генетическими различиями и географическим положением выборок. Результат теста Мантеля по корреляции между генетическими и географическими дистанциями позволяет выделить лишь одну группу, дифференцированную от других (см. дополнительные материалы https://osf.io/8ec3m/). Вероятно, в данном случае отражается как раз базальное положение североамериканской клады “A2”. Как правило, тест Мантеля хорошо описывает географические паттерны генетической дивергенции в природных популяциях (Diniz-Filho et al., 2013) (Diniz-Filho et al., 2013). В случае изучения генетической структуры для D. magna провал теста может свидетельствовать об искусственном объединении разных гаплотипов в одной локации. Итак, для объяснения наблюдаемых фактов следует использовать данные не только о современной ситуации, но и знания об экологических и эволюционных процессах, лежащих в основе генетических изменений у этого вида, а также учитывать вероятность участия человека как вектора при расселении животных.
Дальнейший анализ примечаний к последовательностям в GenBank (NCBI) и запрос к авторам, депонировавшим их, позволил установить, что особи, генетически идентичные европейским, но собранные на американском континенте, в действительности прямо происходят из лабораторных и коммерческих культур, либо из популяций, образовавшихся в результате их попадания в естественные водоемы. Вероятно, эти чужеродные популяции изначально происходили из Европы и в настоящее время успешно акклиматизировались на американском континенте. Таким образом, мы смогли выделить в общем пуле последовательности, однозначно принадлежащие чужеродным особям, и провести биогеографический анализ исходя уже из точных представлений о происхождении конкретных последовательностей, что позволяет совместить наблюдаемые эффекты истории расселения и географии совместно распространенных групп (Gutierrez-Garcia, Vazquez-Dominguez, 2011).
В случае кладоцер с их покоящимися стадиями, устойчивыми к внешнему воздействию и легко распространяющимися разнообразными векторами (Смирнов, 1975; Котов, 2013; Incagnone et al., 2015), во всех выборках для филогеографических построений высока вероятность того, что часть популяций появилась в результате неучтенной нами антропогенной интродукции в недавнее время. В ряде случаев на основании анализа стандартного набора митохондриальных и ядерных генов можно довольно четко установить факт инвазии и регион-донор, как это было сделано для D. galeata, недавно появившейся в Австралии (Karabanov et al., 2018). В случае же анализа таких таксонов как D. magna требуется провести большую аналитическую работу, при этом не во всех случаях удается получить однозначное заключение об имевшем место случае биологической инвазии.
Можно предложить следующий алгоритм выявления чужеродного компонента при проведении филогеографических исследований:
(1) При проведении теста Мантеля отсутствие корреляций при сравнении матриц генетических и географических дистанций может свидетельствовать о несоответствии имеющихся данных модели последовательного расселения.
(2) Анализ сетей гаплотипов с упором на географическое положение клад, в случае провала теста χ2 вплоть до клад высшего порядка, может помочь вычленить линии, вносящие дисбаланс в результаты анализа вложенных клад (NCPA).
(3) Наконец, выделив вероятных контаминантов, можно провести биогеографическую реконструкцию. Вероятно, для адвентивных линий биогеографическая модель будет колебаться между событием дисперсии и викарирования, с большей вероятностью расселения.
Однако даже выполнение всех этих условий во многих случаях может быть лишь поводом для более пристального изучения массива исходных генетических данных на предмет выявления в них последовательностей, принадлежащих неаборигенным особям. Наша небольшая работа показывает, что на сегодняшний день влияние биологических инвазий на результаты филогеографических построений сильно недооценено. Требуются специальные усилия по разработке как понятийного аппарата, прежде всего – математических алгоритмов выявления и оценки вклада биологических инвазий в популяционно-генетическую структуру вида, так и специализированного программного обеспечения для выявления инвазийных процессов.
Список литературы
Горбачев В.В., 2011. Новое ограничение микросателлитных маркеров для их применения в популяционных исследованиях (на примере панмиктических популяций) // Вавиловский журнал генетики и селекции. Т. 15. С. 746–749.
Дгебуадзе Ю.Ю., Петросян В.Г., Хляп Л.А. (Ред.), 2018. Самые опасные инвазионные виды России (ТОП-100). М.: Товарищество научных изданий КМК. 688 с.
Котов А.А., 2013. Морфология и филогения Anomopoda (Crustacea: Cladocera). М.: Товарищество научных изданий КМК. 638 с.
Смирнов Н.Н., 1975. Биология ветвистоусых ракообразных // Итоги науки и техники. Вып. 3. Зоология беспозвоночных. М.: ВИНИТИ АН СССР. 116 с.
Элтон Ч., 1960. Экология нашествий животных и растений. М.: Издательство иностранной литературы. 232 с.
Avise J.C., 2000. Phylogeography: The history and formation of species. Cambridge (MA), London: Harvard University Press. 464 p.
Avise J.C., Bowen B.W., Ayala F.J., 2016. In the light of evolution X: Comparative phylogeography // Proceedings of the National Academy of Sciences of the United States of America. V. 113. P. 7957–7961.
Bekker E.I., Karabanov D.P., Galimov Y.R., Haag C.R., Neretina T.V., Kotov A.A., 2018. Phylogeography of Daphnia magna Straus (Crustacea: Cladocera) in Northern Eurasia: Evidence for a deep longitudinal split between mitochondrial lineages // PLoS One. V. 13. e0194045. https://doi.org/10.1371/journal.pone.0194045
Berg D.J., Garton D.W., 1994. Genetic differentiation in North American and European populations of the cladoceran Bythotrephes // Limnology and Oceanography. V. 39. P. 1503–1516.
Bouckaert R., 2016. Phylogeography by diffusion on a sphere: whole world phylogeography // PeerJ. V. 4. e2406. https://doi.org/10.7717/peerj.2406
Bouckaert R., Vaughan T.G., Barido-Sottani J., Duchene S., Fourment M., et al., 2019. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis // PLoS Computational Biology. V. 15. e1006650. https://doi.org/10.1371/journal.pcbi.1006650
Clark J.R., Ree R.H., Alfaro M.E., King M.G., Wagner W.L., Roalson E.H., 2008. A comparative study in ancestral range reconstruction methods: retracing the uncertain histories of insular lineages // Systematic Biology. V. 57. P. 693–707.
Clement M., Posada D., Crandall K.A., 2000. TCS: a computer program to estimate gene genealogies // Molecular Ecology. V. 9. P. 1657–1659.
De Gelas K., De Meester L., 2005. Phylogeography of Daphnia magna in Europe // Molecular Ecology. V. 14. P. 753–764.
Diniz-Filho J.A.F., Soares T.N., Lima J.S., Dobrovolski R., Landeiro V.L., Campos Telles M.P., et al., 2013. Mantel test in population genetics // Genetics and Molecular Biology. V. 36. P. 475–485.
Dormontt E.E., Gardner M.G., Breed M.F., Rodger J.G., Prentis P.J., Lowe A.J., 2014. Genetic bottlenecks in time and space: reconstructing invasions from contemporary and historical collections // PLoS One. V. 9. e106874. https://doi.org/10.1371/journal.pone.0106874
Drummond A.J., Suchard M.A., Xie D., Rambaut A., 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7 // Molecular Biology and Evolution. V. 29. P. 1969–1973.
Fields P.D., Obbard D.J., McTaggart S.J., Galimov Y., Little T.J., Ebert D., 2018. Mitogenome phylogeographic analysis of a planktonic crustacean // Molecular Phylogenetics and Evolution. V. 129. P. 138–148.
Forster P., Torroni A., Renfrew C., Rohl A., 2001. Phylogenetic star contraction applied to Asian and Papuan mtDNA evolution // Molecular Biology and Evolution. V. 18. P. 1864–1881.
Frisch D., Havel J.E., Weider L.J., 2013. The invasion history of the exotic freshwater zooplankter Daphnia lumholtzi (Cladocera, Crustacea) in North America: a genetic analysis // Biological Invasions. V. 15. P. 817–828.
Ho S.Y.W., Tong K.J., Foster C.S.P., Ritchie A.M., Lo N., Crisp M.D., 2015. Biogeographic calibrations for the molecular clock // Biology Letters. V. 11. 20150194. https://doi.org/10.1098/rsbl.2015.0194
Huson D.H., Bryant D., 2006. Application of phylogenetic networks in evolutionary studies // Molecular Biology and Evolution. V. 23. P. 254–267.
Huson D.H., Rupp R., Scornavacca C., 2011. Phylogenetic networks: Concepts, algorithms and applications. Cambridge: Cambridge University Press. 362 p.
Incagnone G., Marrone F., Barone R., Robba L., Naselli-Flores L., 2015. How do freshwater organisms cross the “dry ocean”? A review on passive dispersal and colonization processes with a special focus on temporary ponds // Hydrobiologia. V. 750. P. 103–123.
Ishida S., Taylor D.J., 2007. Quaternary diversification in a sexual Holarctic zooplankter, Daphnia galeata // Molecular Ecology. V. 16. P. 569–582.
Jost L., 2008. G(ST) and its relatives do not measure differentiation // Molecular Ecology. V. 17. P. 4015–4026.
Karabanov D.P., Bekker E.I., Shiel R.J., Kotov A.A., 2018. Invasion of a Holarctic planktonic cladoceran Daphnia galeata Sars (Crustacea: Cladocera) in the Lower Lakes of South Australia // Zootaxa. V. 4402. P. 136–148.
Kotov A.A, Garibian P.G., Bekker E.I., Taylor D.J., Karabanov D.P., 2020. A new species group from the Daphnia curvirostris species complex (Cladocera: Anomopoda) from the eastern Palaearctic: taxonomy, phylogeny and phylogeography // Zoological Journal of the Linnean Society. zlaa046. https://doi.org/10.1093/zoolinnean/zlaa046
Kotov A.A., Korovchinsky N.M., 2006. First record of fossil Mesozoic Ctenopoda (Crustacea, Cladocera) // Zoological Journal of the Linnean Society. V. 146. P. 269–274.
Kotov A.A., Taylor D.J., 2011. Mesozoic fossils (145 Mya) suggest the antiquity of the subgenera of Daphnia and their coevolution with chaoborid predators // BMC Evolutionary Biology. V. 11. P. 129. https://doi.org/10.1186/1471-2148-11-129
Kumar S., Stecher G., Li M., Knyaz C., Tamura K., 2018. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms // Molecular Biology and Evolution. V. 35. P. 1547–1549.
Lampert W., 2011. Daphnia: Development of a model organism in ecology and evolution. Oldendorf, Luhe, Germany: International Ecology Institute. 250 p.
Landis M.J., Matzke N.J., Moore B.R., Huelsenbeck J.P., 2013. Bayesian analysis of biogeography when the number of areas is large // Systematic Biology. V. 62. P. 789–804.
Matzke N.J., 2013. Probabilistic historical biogeography: New models for founder-event speciation, imperfect detection, and fossils allow improved accuracy and model-testing // Frontiers of Biogeography. V. 5. P. 242–248.
Matzke N.J., 2014. Model selection in historical biogeography reveals that founder-event speciation is a crucial process in Island Clades // Systematic Biology. V. 63. P. 951–970.
Mergeay J., Verschuren D., de Meester L., 2005. Cryptic invasion and dispersal of an American Daphnia in East Africa // Limnology and Oceanography. V. 50. P. 1278–1283.
Panchal M., Beaumont M.A., 2010. Evaluating nested clade phylogeographic analysis under models of restricted gene flow // Systematic Biology. V. 59. P. 415–432.
Posada D., Crandall K.A., Templeton A.R., 2000. GeoDis: a program for the cladistic nested analysis of the geographical distribution of genetic haplotypes // Molecular Ecology. V. 9. P. 487–488.
Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A., 2018. Posterior summarization in Bayesian phylogenetics using Tracer 1.7 // Systematic Biology. V. 67. P. 901–904.
Ree R.H., Smith S.A., 2008. Maximum likelihood inference of geographic range evolution by dispersal, local extinction, and cladogenesis // Systematic Biology. V. 57. P. 4–14.
Rosenberg M.S., Anderson C.D., 2011. PASSaGE: Pattern Analysis, Spatial Statistics and Geographic Exegesis. Version 2 // Methods in Ecology and Evolution. V. 2. P. 229–232.
Sharma P., Kotov A.A., 2015. Establishment of Chydorus sphaericus (O.F. Muller, 1785) (Crustacea: Cladocera) in Australia: consequences of mass fish stocking from Northern Europe? // Journal of Limnology. V. 74. P. 225–233.
Smirnov N.N., 1992. Mesozoic Anomopoda (Crustacea) from Mongolia // Zoological Journal of the Linnean Society. V. 104. P. 97–116.
Smirnov N.N., 2017. Physiology of the Cladocera, Second edition. Amsterdam: Academic Press. 418 p.
Taylor D.J., Connelly S.J., Kotov A.A., 2020. The Intercontinental phylogeography of neustonic daphniids // Scientific Reports. V. 10. 1818. https://doi.org/10.1038/s41598-020-58743-8
Templeton A.R., 1998. Nested clade analyses of phylogeographic data: testing hypotheses about gene flow and population history // Molecular Ecology. V. 7. P. 381–397.
Templeton A.R., 2004. Statistical phylogeography: Methods of evaluating and minimizing inference errors // Molecular Ecology. V. 13. P. 789–809.
Templeton A.R., 2009. Statistical hypothesis testing in intraspecific phylogeography: Nested clade phylogeographical analysis vs. approximate Bayesian computation // Molecular Ecology. V. 18. P. 319–331.
Williford D., Deyoung R.W., Honeycutt R.L., Brennan L.A., Hernandez F., 2016. Phylogeography of the bobwhite (Colinus) quails // Wildlife Monographs. V. 193. P. 1–49.
Xiang Q.-Y., Soltis D.E., 2001. Dispersal-vicariance analyses of intercontinental disjuncts: historical biogeographical implications for Angiosperms in the Northern Hemisphere // International Journal of Plant Sciences. V. 162. S29–S39.
Xu S., Hebert P.D.N., Kotov A.A., Cristescu M.E., 2009. The noncosmopolitanism paradigm of freshwater zooplankton: Insights from the global phylogeography of the predatory cladoceran Polyphemus pediculus (Linnaeus, 1761) (Crustacea, Onychopoda) // Molecular Ecology. V. 18. P. 5161–5179.
Yu Y., Harris A.J., Blair C., He X., 2015. RASP (Reconstruct Ancestral State in Phylogenies): A tool for historical biogeography // Molecular Phylogenetics and Evolution. V. 87. P. 46–49.
Zink R.M., Blackwell-Rago R.C., Ronquist F., 2000. The shifting roles of dispersal and vicariance in biogeography // Proceedings of the Royal Society of London. Series B, Biological sciences. V. 267. P. 497–503.
Дополнительные материалы отсутствуют.
Инструменты
Зоологический журнал