

Биоорганическая химия, 2020, T. 46, № 4, стр. 425-434
Исследование структурной организации активного центра немодифицированной рекомбинантной сульфатазы из мицелиального гриба Fusarium proliferatum LE1
Н. В. Колчина 1, 2, Г. Н. Рычков 1, 2, А. А. Кульминская 1, 2, Ф. М. Ибатуллин 1, М. Г. Петухов 1, 3, К. С. Бобров 1
1 ФГБУ “Петербургский институт ядерной физики им. Б.П. Константинова научно-исследовательского центра “Курчатовский институт”
188300 Гатчина, Ленинградская область, Орлова роща, 1, Россия
2 ФГАОУ ВО “Санкт-Петербургский политехнический университет Петра Великого”
195251 Санкт-Петербург, ул. Политехническая, д. 29, Россия
3 ФГБУ “Российский научный центр радиологии и хирургических технологий имени академика А.М. Гранова” Министерства здравоохранения Российской Федерации
197758 Санкт-Петербург, поселок Песочный, ул. Ленинградская, д. 70, Россия
Поступила в редакцию 03.02.2020
После доработки 27.02.2020
Принята к публикации 28.02.2020
Аннотация
Сульфатазы катализируют реакцию гидролиза эфиров серной кислоты и играют ключевую роль в ряде биологических процессов как высших эукариот, так и прокариот. По литературным данным для осуществления катализа некоторым представителям этой группы ферментов требуется пост-трансляционная модификация остатков серина или цистеина в активном центре в уникальную аминокислоту Cα-формилглицин. Тем не менее, подтверждено, что даже в отсутствии данной модификации некоторые сульфатазы способны катализировать реакцию гидролиза сульфоэфиров. В настоящей работе были проведены исследования структурно-функциональных особенностей активной рекомбинантной сульфатазы из мицелиального гриба Fusarium proliferatum LE1, содержащей в активном центре каталитический остаток цистеина, не подвергшийся модификации. Впервые была построена теоретическая атомарная модель фермента, определена структурная организация его активного центра и выявлены ключевые аминокислотные остатки, принимающие участие в связывании п-нитрофенил сульфата и катализирующие его гидролиз. Точечные аминокислотные замены выявленных остатков привели к инактивации фермента. В частности, мутантные формы фермента с заменой каталитического цистеина на серин и треонин, содержащие гидроксильную группу в боковой цепи, полностью утрачивали активность, что свидетельствует о непосредственном участии меркаптогруппы цистеина в реакции гидролиза.
ВВЕДЕНИЕ
Сульфатазы (КФ 3.1.6.-) представляют собой широко распространенный в природе класс гидролаз, катализирующих реакцию гидролиза различных эфиров серной кислоты, таких как стероиды, гормоны, а также различные гликоконъюгаты. Данная группа ферментов обнаружена во множестве микроорганизмов, в растениях, а также в большинстве животных и человеческих тканей [1]. У человека было идентифицировано большое количество сульфатаз, играющих ключевые роли в таких биологических процессах как синтез гормонов, деградацию гликозаминогликанов и гликолипидов, репродуктивные процессы, развитие костей и хрящей и др. [2]. Нарушение их функций связано с рядом заболеваний, включающих множественную сульфатазную недостаточность, Х-связанный ихтиозис и др. [3, 4]. Микробные сульфатазы изучены менее подробно [5]. Известно, что сульфатазы микроорганизмов участвуют в процессах утилизации альтернативных источников серы и экспрессируются в условиях недостаточности неорганического сульфата [6]. Сульфатазы на поверхности клеток некоторых патогенных микроорганизмов участвуют в процессе взаимодействия “хозяин-патоген” [7]. Помимо этого, данные ферменты играют важную роль в росте и морфологии грибов рода Fusarium, что делает их потенциальной мишенью для разработки новых фунгицидов [8].
Современная классификация сульфатаз основана на сравнении аминокислотных последовательностей и разделяет ферменты на четыре семейства [9]. При этом почти 90% известных сульфатаз являются представителями семейства S1 [10]. Ферменты, входящие в это семейство, содержат в активном центре уникальный аминокислотный остаток Cα-формилглицин (FGly), образующийся в результате пост-трансляционной модификации серина или цистеина в консервативном мотиве (C/S)XPXR на N-конце белка [11, 12]. Считается, что наличие FGly строго необходимо для проявления сульфатазной активности, а ферменты с немодифицированным каталитическим остатком образуют с субстратом ковалентный комплекс, в дальнейшем негидролизуемый по традиционной схеме [13–16]. Далее под модифицированным ферментом дикого типа мы будем подразумевать фермент без аминокислотных замен, содержащий каталитический остаток FGly, соответственно немодифицированным будем называть фермент, содержащий каталитический остаток Cys или Ser.
Тем не менее, в последнее время стали появляться работы, доказывающие, что некоторые ферменты, не подвергшиеся пост-трансляционной модификации каталитического остатка, все-таки способны катализировать гидролиз сульфатированных соединений. Так, отсутствие FGly в рекомбинантной холинсульфатазе из Ensifer (Sinorhizobium) meliloti было подтверждено методом масс-спектрометрии, но при этом фермент проявлял детектируемую активность в гидролизе холин-, п-нитрофенил- и метилумбеллиферил-сульфатов [17]. Аналогично, ранее охарактеризованная нами рекомбинантная немодифицированная сульфатаза из мицелиального гриба Fusarium proliferatum LE1 также катализировала реакцию гидролиза п-нитрофенил сульфата (pNPS) [18].
Опубликованные результаты указывают на необходимость дальнейшего тщательного изучения структурных особенностей, обеспечивающих каталитическую активность как модифицированных, так и немодифицированных сульфатаз. В настоящей работе мы провели исследование структурной организации активного центра рекомбинантной немодифицированной сульфатазы из мицелиального гриба F. proliferatum LE1 для выявления каталитических остатков, участвующих в катализе гидролиза модельного субстрата pNPS, методами молекулярного моделирования и белковой инженерии.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Анализ первичной структуры и субстратная специфичность
В работе использовали созданную нами ранее систему экспрессии рекомбинантной сульфатазы из мицелиального гриба F. proliferatum LE1 (F.p.Sulf-6His) [18]. Так, было установлено, что данный фермент проявляет гидролитическую активность в отношении таких субстратов, как pNPS (удельная активность 2.3 × 10–2 ед. акт./мг белка) и п-нитрофенил фосфат (удельная активность 3.8 × 10–4 ед. акт./мг белка). При этом отсутствие пост-трансляционной модификации каталитического цистеина в FGly было подтверждено методом масс-спектрометрии [18]. Анализ аминокислотной последовательности F.p.Sulf-6His показал, что изучаемый белок относится к семейству S1 сульфатаз, подсемейству S1_S12 (холинсульфатазы) [10]. В настоящей работе было проведено исследование гидролитической активности F.p.Sulf-6His в отношении его предполагаемого субстрата холинсульфата.
Анализ реакционной смеси показал отсутствие в ней холина, являющегося непосредственным продуктом реакции гидролиза холинсульфата. Следовательно, можно заключить, что F.p.Sulf-6His не катализирует реакцию гидролиза холинсульфата.
Молекулярное моделирование
С помощью методов молекулярного моделирования построили атомарные модели F.p.Sulf-6His, взяв за основу пространственные структуры сульфатаз с гомологичными аминокислотными последовательностями из трех организмов: Sinorhizobium meliloti (SmCS, идентичность последовательности 44%, PDB код: 4UG4, рис. 1а), Bacteroides thetaiotaomicron (BtS, идентичность последовательности 32%, PDB код: 5G2V, рис. 1б) и Homo sapiens (HsS, идентичность последовательности 33%, PDB код: 4MHX, рис. 1в).
Рис. 1.
Пространственная структура сульфатаз из S. melliloti (SmCS, идентичность последовательности 44%, PDB код: 4UG4) (а), B. thetaiotaomicron (BtS, идентичность последовательности 32%, PDB код: 5G2V) (б) и H. sapiens (HsS, идентичность последовательности 33%, PDB код: 4MHX) (в). Салатовым цветом окрашена модель F.p.Sulf-6His, построенная по соответствующему структурному шаблону, темно-зеленым – фермент SmCS, синим – BtS, бирюзовым – HsS. Желтой сферой обозначен ион металла в активном центре. Красным цветом выделены участки третичной структуры ферментов относящиеся к активному центру, и отличающиеся в модели и структурном шаблоне.
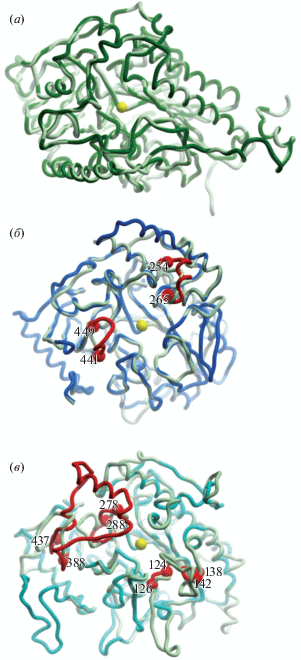
Наиболее полное совпадение модели и шаблона по аминокислотному составу, ходу полипептидной цепи и пространственному расположению аминокислот активного центра наблюдается только для структуры F.p.Sulf-6His, построенной на основе сульфатазы SmCS (рис. 1а). Поэтому именно эту модель использовали для исследования фермент-субстратного комплекса F.p.Sulf-6His с pNPS. В свою очередь аминокислотный состав активных центров модели и шаблона существенным образом отличается для модели, построенной, как на основе BtS, так и на основе HsS. Из-за разницы в количестве аминокислотных остатков между моделью и шаблоном отличается также ход полипептидной цепи на участках активного центра, затрагивающих остатки 254–265 и 441–449 BtS (рис. 1б) и в еще большей степени на участках 124–126, 138–142, 278–288, 388–437 HsS (рис. 1в).
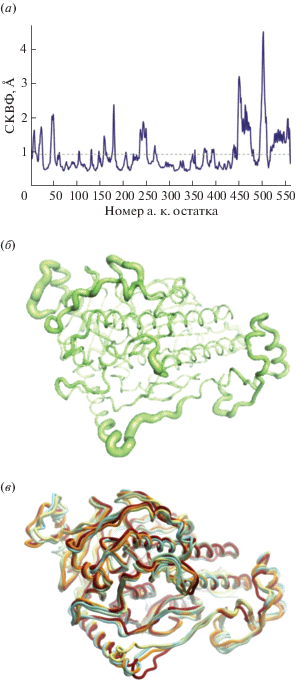
Релаксация сконструированной модели белка F.p.Sulf-6His методом молекулярной динамики (МД) в водном окружении в течение 150 нс не выявила нестабильности третичной структуры глобулы белка (рис. 2). Усредненная величина среднеквадратичных флуктуаций атомов C, Cα, N основной цепи составляет 0.89 ± 0.55Å. Подвижностью выше средней обладали лишь участки на N-конце (5–13, 18–26) и на C-конце (515–557) белка, информация о которых не содержалась в структурном шаблоне SmCS а также участки петель и концевые участки альфа-спиралей, не принимающие участия в формировании активного центра (43–51, 157–161, 175–181, 234–249, 265–269, 445–475, 492–507) (рис. 2а и 2б). Среди наиболее заселенных конформаций белка, найденных методами кластеризации (рис. 2в), была выбрана конформация с открытым входом в активный центр, объем которого был достаточен для размещения субстрата pNPS. N- и С-концы фермента не загораживают вход в активный центр, располагаясь на расстоянии более 25 Å от него.
Рис. 2.
Подвижность третичной структуры модели белка F.p.Sulf-6His в ходе МД. (а) Среднеквадратичные флуктуации (СКВФ) основной цепи аминокислотных остатков. Зеленой пунктирной линией обозначена средняя величина СКВФ. (б) Утолщения полипептидной цепи пропорциональны величине СКВФ этих участков цепи. (в) Пространственное совмещение медианных структур из пяти кластеров, полученных по результатам анализа траектории МД.
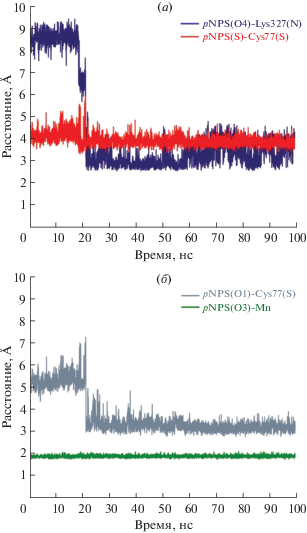
Возможные варианты связывания pNPS в активном центре белка F.p.Sulf-6His установили с помощью методов докинга с последующей проверкой стабильности комплекса методом МД в водном окружении. Ранее масс-спектрометрический анализ показал, что в отличие от SmCS на месте каталитического FGly в ферменте F.p.Sulf-6His находится остаток цистеина в положении 77 [18]. Ближайшими аминокислотными остатками в радиусе 4 Å от pNPS являются: D36, С77, A78, R81, K125, M126, H127, Y145, D314, H315 и K327 (рис. 3). Аминокислотные остатки D36, D314, H315 координируют ион марганца, а остаток R81 для этого правильным образом располагает в пространстве остаток D314, образуя с ним водородную связь. Рисунок 4 демонстрирует наблюдаемое в ходе МД изменение расстояний между атомами сульфатной группы pNPS и взаимодействующими с ней ключевыми атомами в структуре F.p.Sulf-6His: S–SγC77, O1–SγC77, О3–NζK327, О4–Mn2+. Начиная с 21-ой нс, когда субстрат pNPS изменил исходное положение в активном центре фермента, структура комплекса F.p.Sulf-6His–pNPS оставалась стабильной до конца траектории; среднеквадратичные флуктуации атомов pNPS составили 0.82 Å. Средние рассчитанные расстояния равны 3.90 ± 0.19, 3.31 ± 0.24, 1.96 ± 0.05 и 3.30 ± 0.50 Å для пар атомов S–SγC77, O1–SγC77, О3–NζK327, О4–Mn2+ соответственно. Атом кислорода О4 сульфогруппы pNPS прочно удерживается ионом марганца на одном расстоянии. Атом О3 образует водородную связь с аминогруппой лизина 327, но расстояние между донором и акцептором водородной связи меняется в ходе МД из-за подвижности длинной боковой цепи лизина. В непосредственной близости от SγC77 находится атом кислорода О1, соединяющий сульфатную и нитрофенольную группы pNPS. 4% времени атом SγC77 находится в плотном ван-дер-ваальсовом контакте с атомом серы сульфогруппы pNPS, когда расстояния между ними не превышает расстояния атаки 3.6 Å. При этом минимальное наблюдаемое расстояние S–SγC77 равняется 3.35 Å.
Рис. 3.
(а) Расположение субстрата pNPS в активном центре F.p.Sulf-6His, полученное методами молекулярного моделирования. (б) Диаграмма взаимодействия аминокислотных остатков F.p.Sulf-6His с pNPS. Зеленым цветом показана гидрофобная область, голубым затенением показаны акцепторы водородной связи, серыми дугами показана поверхность, доступная растворителю.
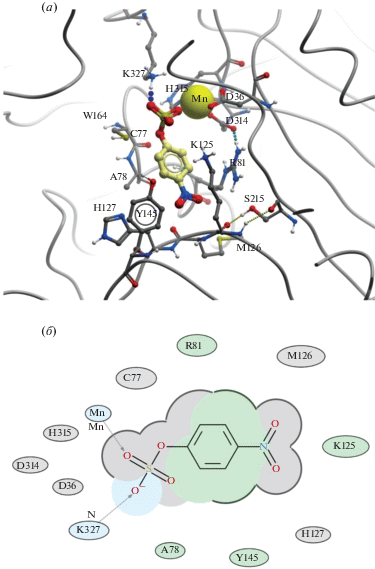
Рис. 4.
Изменение расстояния между атомами сульфатной группы pNPS и взаимодействующими с ними атомами белка.
Построенная атомарная модель комплекса F.p.Sulf-6His–pNPS позволила сделать предположения о роли отдельных аминокислотных остатков активного центра фермента и предложить одиночные аминокислотные замены для проверки этих предположений. Необходимо было выяснить, действительно ли остаток С77 участвует в реакции гидролиза pNPS и важно ли для катализа наличие его меркаптогруппы, или же ее роль может выполнять гидроксильная группа. Для проверки первого предположения решили сделать аминокислотную замену C77A; для проверки второго – рекомендовали провести замены C77S и C77T. Остатки серина и треонина по химическому составу боковых цепей напоминают цистеин с той лишь разницей, что тиольная группа заменена на гидроксильную.
Для определения других ключевых остатков в активном центре фермента F.p.Sulf-6His, способных участвовать в связывании субстрата и оказывать влияние на ход каталитической реакции, предложили аминокислотные замены K327A, K125A, H127A, R81A. По аналогии с описанным механизмом действия сульфатазы SmCS [19] аминокислотный остаток K327 может отвечать за протонирование уходящей группы при гидролизе сульфатных эфиров, однако в построенном нами комплексе F.p.Sulf-6His–pNPS остатки K125 и H127 располагаются на удалении от сульфогруппы (расстояние от атома S pNPS до атома N остатка K125 составляет 5.8 Å, от атома S pNPS до атома N остатка H127 – 8.9 Å), ближе ко входу в активный центр и взаимодействуют с уходящей группой субстрата.
Анализ гидролитической активности мутантных форм F.p.Sulf-6His
Нами были получены, выделены и очищены мутантные формы белка F.p.Sulf-6His, содержащие предложенные аминокислотные замены. Уровень экспрессии мутантных форм соответствовал уровню экспрессии белка дикого типа (данные не показаны). При этом ни один из выделенных ферментов не продемонстрировал детектируемой сульфатазной активности.
ОБСУЖДЕНИЕ
Полученные результаты указывают на то, что катализ реакции гидролиза pNPS ферментами с каталитическим FGly и немодифицированной сульфатазой F.p.Sulf-6His происходит в том же активном центре. Так, замена обсуждаемых аминокислот в SmCS с модифицированным каталитическим остатком приводит к снижению активности фермента на 3–4 порядка [19]. Вероятно, данное снижение происходит и в случае немодифицированной F.p.Sulf-6His, но, поскольку исходная активность фермента невысока, то гидролитическую активность мутантных форм оказалось невозможно детектировать используемыми в работе методами, так как снижение данной величины на три порядка приведет к тому, что измеряемые величины оптической плотности реакционной смеси станут соизмеримы с погрешностью спектрофотометра. Увеличение времени реакции в нашем случае также не способствовало детекции гидролитической активности по причине низкой стабильности исследуемого фермента. При этом инактивация мутантной формы F.p.Sulf-6His C77A указывает на участие немодифицированного цистеина в реакции гидролиза сульфоэфиров.
С учетом высокой гомологии активных центров белков F.p.Sulf-6His и SmCS и результатов молекулярного моделирования, мы предположили, что арилсульфатаза F.p.Sulf-6His катализирует реакцию гидролиза pNPS путем атаки SH-группы цистеина С77 на атом серы pNPS. Такой атаке способствует электрон-акцепторное влияние нитро-группы, увеличивающей дробный отрицательный заряд на кислородном атоме нитрофенильного остатка, что облегчает разрыв связи между ним и атомом серы сульфата. Дробный отрицательный заряд на нитрофенильном атоме кислорода может также способствовать частичному депротонированию SH-группы цистеина С77, что также должно увеличивать нуклеофильность меркаптогруппы, а следовательно, ускорять реакцию замещения остатка нитрофенола на остаток цистеина. В результате остаток цистеина, предположительно, превращается в тиосульфатное производное, в котором остаток сульфата присоединен к сере посредством S–S связи. Активирующее влияние нитрофенильной группы не вызывает сомнения, так как мы показали, что фермент не способен гидролизовать алкилсульфаты, в частности, холинсульфат. Однако остается вопрос, что происходит с ферментом после того, как SH-группа цистеина С77 превращается в остаток тиосульфата. S-Алкилтиосульфаты или, так называемые, соли Бунтэ хорошо известны в органической химии [20]. Кислотный гидролиз таких производных приводит к отщеплению сульфата и высвобождению алкил-тиолов, что является одним из препаративных способов получения последних. Однако обычно такой гидролиз протекает в достаточно жестких условиях, к примеру, путем кипячения в кислой среде в течение нескольких часов. Основания также способны гидролизовать алкилтиосульфаты, но известно лишь, что реакция протекает по сложному механизму. Тем не менее, поскольку в результате гидролиза pNPS не происходит инактивации фермента, это, вероятно, означает, что тиосульфатное производное цистеина 77 каким-то образом способно гидролизоваться в физиологических условиях, высвобождая исходную SH-группу и переводя, таким образом, фермент в активную форму.
Необходимо отметить, что замена каталитического цистеина на серин инактивировала F.p.Sulf-6His. Известно, что оба этих аминокислотных остатка в сульфатазах могут модифицироваться в FGly [2, 21]. Однако гидролитическую активность проявлял лишь F.p.Sulf-6His с цистеином в активном центре. Мутантная форма фермента, в которой каталитический цистеин был заменен на треонин, также не проявила детектируемой гидролитической активности, указывая на необходимость участия меркаптогруппы цистеина в реакции гидролиза, катализируемой F.p.Sulf-6His.
В литературе описываются случаи наличия сульфатазной активности у немодифицированных ферментов, содержащих в активном центре как цистеин, так и серин. Так, гидролиз сульфоэфиров, помимо F.p.Sulf-6His, были способны осуществлять немодифицированная сульфатаза из S. meliloti [17], в активном центре которой находится цистеин, и сульфатаза из Pseudomonas aeruginosa, в активный центр которой методом сайт-направленного мутагенеза был введен серин [13]. Однако в работе Williams с соавт. было показано, что реакция распада фермент-сульфатного комплекса (переходного состояния) предположительно идет по механизму элиминирования E2, а не нуклеофильного замещения SN2 [16]. Для протекания данной реакции необходимо наличие геминальной гидроксигруппы у каталитической аминокислоты. Именно ее отсутствием авторы объясняют невозможность распада ковалентного комплекса немодифицированных сульфатаз с серином в активном центре и сульфатной группы. Наличие же гидролитической активности у сульфатазы из P. aeruginosa Williams с соавт. объяснили загрязнением мутантной формы белка диким типом фермента. Аналогично было объяснено наличие сульфатазной активности у C69S мутантной формы сульфатазы из H. sapiens [14]. Также было показано, что две сульфатазы из B. thetaiotaomicron, содержащие серин в активном центре, являются неактивными [22].
Известно, что как серин, так и цистеин могут непосредственно участвовать в катализе, осуществляемыми гидролазами [23]. Типичными представителями таких ферментов являются сериновые и цистеиновые протеазы. Такие ферменты имеют дополнительные аминокислоты, необходимые для активации и депротонирования гидроксильной (серин) или тиольной (цистеин) группы. В целом, считается, что снижение pKa остатка цистеина вызывают положительно заряженные группы соседних аминокислот и/или усиление электростатических взаимодействий между этой группой и атомом серы в результате увеличения электронной плотности на атоме серы цистеина [24]. При этом тиольная группа является более сильным нуклеофилом, чем гидроксильная группа, и для ее активации может использоваться более короткая цепь переноса протонов [23].
Таким образом, можно сделать следующие предположения. Субстрат pNPS связывается как с модифицированной, так и с немодифицированной сульфатазой в одном и том же активном центре. В результате данного связывания в обоих случаях происходит образование ковалентного фермент-сульфатного комплекса. Однако, в то время как комплекс субстрата с модифицированной сульфатазой расщепляется по энергетически выгодному механизму элиминации E2, комплекс субстрата с немодифицированным ферментом по данному механизму расщепляться не может ввиду отсутствия геминальной гидроксильной группы у каталитической аминокислоты. Поэтому данная стадия может протекать по альтернативному механизму нуклеофильного замещения SN2, что требует депротонирования каталитической аминокислоты. Цепь переноса протонов, имеющаяся в активном центре F.p.Sulf-6His, вероятно, недостаточно длинная, чтобы депротонировать гидроксильную группу серина или треонина, поэтому мутантные формы фермента C77S и C77T не проявляют гидролитической активности. При этом депротонирование тиольной группы цистеина по этой цепи происходит, что позволяет протекать реакции гидролиза фермент-субстратного комплекса с регенерацией фермента. Стоит отметить, что заряды боковых групп аминокислотных остатков, окружающих каталитический цистеин и влияющих на его свойства, зависят от кислотности среды [24]. Наши предыдущие исследования показали, что F.p.Sulf-6His обладает активностью в достаточно узком диапазоне pH [18]. Вероятно, при значениях pH около 6 зарядовое состояние аминокислотных остатков вблизи цистеина наиболее благоприятно для увеличения его реакционноспособности.
ЗАКЛЮЧЕНИЕ
В представленной работе мы впервые построили теоретическую атомарную модель немодифицированного фермента F.p.Sulf-6His. Методами молекулярного моделирования и белковой инженерии показали, что связывание субстрата происходит в активном центре, как и в случае модифицированного белка, и выявили ключевые аминокислотные остатки активного центра, принимающие участие в связывании и гидролизе субстрата pNPS. При этом тиольная группа С77, не подвергшаяся пост-трансляционной модификации в FGly, принимает непосредственное участие в катализе гидролиза сульфоэфиров.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы
В работе использовали реактивы производства компании Sigma-Aldrich (США), если не указано иное.
Субстрат pNPS был синтезирован согласно опубликованному протоколу [25]; субстрат холинсульфат был синтезирован из холинбромида (Chemapol, Чехословакия) аналогично синтезу β-аминоэтилсерной кислоты [26].
ПЦР проводили с использованием высокоточной Q5 полимеразы (NEB). Очистка ДНК из реакционных смесей проводилась с использованием набора реактивов Cleanup Standard (Евроген, Россия). Ферменты, используемые для работ по молекулярной биологии, были производства Thermo Scientific (Германия) или СибЭнзим (Россия). Электропоратор MicroPulser и 0.2 см кюветы были производства компании Bio-Rad (США).
Молекулярное моделирование
В базах данных открытого доступа информация о пространственном строении F.p.Sulf-6His отсутствует. Поиск ферментов, гомологичных F.p.Sulf-6His, провели в Базе данных структур белков (PDB), используя интернет-сервер SWISS-MODEL [27]. Атомарные модели F.p.Sulf-6His построили в программе Molsoft ICM-Pro [28], взяв в качестве структурных шаблонов сульфатазы, обладающие наибольшей схожестью пептидной последовательности с исследуемым ферментом (коды PDB: 4UG4, 4MHX, 5G2V). Координаты атомов главной цепи F.p.Sulf-6His задавались положением Cα атомов гомологичного белка, для всех неидентичных остатков проводился поиск низкоэнергетических конформаций методом глобальной оптимизации с учетом их аминокислотного окружения.
Построенную атомарную модель F.p.Sulf-6His релаксировали методом МД в явно заданном водном растворителе с периодическими граничными условиями, по протоколу, описанному ниже. Кластеризацию структур F.p.Sulf-6His, наблюдаемых в ходе МД, проводили с помощью иерархического агломеративного алгоритма, задав ограничение на расстояние между кластерами равным 2. Для построения комплекса F.p.Sulf-6His–pNPS выбрали медианную структуру фермента с открытым входом в активный центр, чей объем был достаточен для размещения pNPS.
Атомарную модель pNPS построили в программе Molsoft ICM-Pro и оптимизировали ее геометрию с использованием силового поля MMFF [29]. Процедуру “гибкого” докинга проводили по протоколу, подробно описанному авторами программы [28, 30, 31]. Докинг pNPS запускали трижды из различных начальных положений в активном центре и конформаций. Так как в процессе докинга для поиска возможных конформаций лиганда используются метод псевдо-броуновского перемещения лиганда совместно со смещено-вероятностным методом Монте-Карло [31], то трех запусков вполне достаточно для относительно полного исследования конформационного пространства субстрата в изучаемом сайте связывания.
Стабильность обнаруженного в результате докинга комплекса F.p.Sulf-6His–pNPS, обладавшего наименьшей величиной оценочной функции, исследовали методом МД по протоколу, описанному ниже.
Протокол молекулярной динамики
Расчеты МД и обработку траекторий проводили в программном пакете AMBER 16 [32], используя силовые поля ff14 для белка и GAFF2 для pNPS. Исследуемые модели F.p.Sulf-6His и его комплекса с pNPS помещали в октаэдрический водный бокс, размеры которого выбрали таким образом, чтобы толщина водного слоя вокруг белка была не менее 12 Å. Молекулы воды были представлены моделью TIP3P; параметры для одновалентных ионов – Li, Song и Merz [33], для двухвалентых ионов – Li и Merz [34]. В систему добавляли 18 ионов Na+, для нейтрализации заряда. МД проводили по стандартному протоколу, включающему: 1) двухстадийную минимизацию, затрагивающую сначала только молекулы растворителя, а затем и всю моделируемую систему (белок, субстрат и растворитель); 2) постепенный нагрев системы в течение 100 пс с наложением ограничений на подвижность атомов белка; 3) уравновешивание системы в течение 1 нс; 4) запуск продуктивной молекулярной динамики системы продолжительностью 150 нс для релаксации модели F.p.Sulf-6His и 100 нс для исследования комплекса F.p.Sulf-6His–pNPS.
Шаг интегрирования МД равнялся 2 фс. Для удержания величин длин ковалентных связей и величин валентных углов использовали алгоритм SHAKE. Температура системы поддерживалась на уровне 300 К с помощью динамики Ланжевена. Для поддержания давления в системе равного 1 атм использовали баростат Берендсена с константой времени взаимодействия равной 2 пс. Дальнодействующие взаимодействия учитывали методом PME (particle mesh Ewald – суммирование Эвальда по пространственной сетке частиц). Расстояние отсечки невалентных взаимодействий атомов установили равным 10 Å.
Определение гидролитической активности белков
Гидролитическая активность ферментных препаратов с использованием в качестве субстрата pNPS определялась по ранее опубликованному стандартному протоколу [18]. Единицу активности определяли как количество фермента, гидролизующее 1 мкмоль субстрата pNPS в минуту.
Исследование реакции гидролиза холин сульфата ферментом дикого типа в стандартных условиях проводилось спектрофотометрически. Для определения гидролитической активности фермента к реакционной смеси добавляли 1 мл реактива, содержащего 0.1 г/л холиноксидазы, 0.1 г/л пероксидазы хрена, 0.3 М трис-фосфатный буфер (рН 7), 40% глицерина и 0.1 г/л о-дианизидина. После инкубации смеси при 37°С в течение 30 минут к ней добавляли 2 мл 5 М HCl и определяли оптическую плотность раствора при длине волны 525 нм. Количество холина в реакционной смеси определяли по калибровочному графику, полученному для растворов холин бромида с разной концентрацией. Единицу активности определяли как количество фермента, гидролизующее 1 мкмоль субстрата холин сульфата в минуту.
Сайт-направленный мутагенез и выделение белков
Внесение предложенных мутаций в ген белка F.p.Sulf-6His проводили с использованием плазмиды pPIC3-F.p.Sulf-6His [18]. Дизайн праймеров осуществляли при помощи он-лайн сервиса NEBaseChanger® [35]. Продукты ПЦР, полученные для каждой пары праймеров, последовательно обрабатывали ферментами DpnI, Т4 полинуклеотидкиназой и Т4-лигазой, согласно протоколам производителей. Конечные реакционные смеси трансформировали в бактериальные клетки Escherichia coli DH5α методом электропорации. Наличие точечных мутаций в гене белка F.p.Sulf-6His подтверждали секвенированием. Плазмидную ДНК, несущую гены белка F.p.Sulf-6His с требуемыми мутациями, выделяли, обрабатывали рестриктазой Eco147I и трансформировали в клетки Pichia pastoris GS115 методом электропорации согласно протоколу компании Invitrogen. Экспрессию, выделение и очистку белков F.p.Sulf-6His, содержащих точечные мутации, осуществляли по протоколу, опубликованному ранее [18]. Гомогенность полученных белковых препаратов проверяли методом ПААГ-электрофореза.
Сульфатазную активность полученных белковых препаратов проверяли с использованием pNPS в качестве субстрата.
Список литературы
Stressler T., Seitl I., Kuhn A., Fischer L. // Appl. Microbiol. Biotechnol. 2016. V. 100. № 21. P. 9053–9067.
Hanson S.R., Best M.D., Wong C.-H. // Angew. Chem. Int. Ed. 2004. V. 43. № 43. P. 5736–5763.
Diez-Roux G., Ballabio A. // Annu. Rev. Genomics Hum. Genet. 2005. V. 6. № 1. P. 355–379.
Rižner T.L. // Front. Pharmacol. 2016. V. 7. P. 30.
Швецова С.В., Кульминская А.А. // Вестник Московского Университета. 2018. Т. 59. № 4. С. 243–256.
Toesch M., Schober M., Faber K. // Appl. Microbiol. Biotechnol. 2014. V. 98. № 4. P. 1485–1496.
Hossain Md.M., Kawarabayasi Y., Kimura M., Kakuta Y. // J. Biochem. (Tokyo). 2009. V. 146. № 6. P. 767–769.
Markham P., Robson G.D., Bainbridge B.W., Trinci A.P. // FEMS Microbiol. Rev. 1993. V. 10. № 3–4. P. 287–300.
Barbeyron T., Brillet-Guéguen L., Carré W., Carrière C., Caron C., Czjzek M., Hoebeke M., Michel G. // PLoS One. 2016. V. 11. № 10. P. e0164846.
http://abims.sb-roscoff.fr/sulfatlas/index.html.
Miech C., Dierks T., Selmer T., von Figura K., Schmidt B. // J. Biol. Chem. 1998. V. 273. № 9. P. 4835–4837.
Schmidt B., Selmer T., Ingendoh A., von Figura K. // Cell. 1995. V. 82. № 2. P. 271–278.
Olguin L.F., Askew S.E., O’Donoghue A.C., Hollfelder F. // J. Am. Chem. Soc. 2008. V. 130. № 49. P. 16 547–16 555.
Recksiek M., Selmer T., Dierks T., Schmidt B., von Figura K. // J. Biol. Chem. 1998. V. 273. № 11. P. 6096–6103.
Stressler T., Reichenberger K., Glück C., Leptihn S., Pfannstiel J., Swietalski P., Kuhn A., Seitl I., Fischer L. // Appl. Microbiol. Biotechnol. 2018. V. 102. № 6. P. 2709–2721.
Williams S.J., Denehy E., Krenske E.H. // J. Org. Chem. 2014. V. 79. № 5. P. 1995–2005.
Sánchez-Romero J.J., Olguin L.F. // Biochem. Biophys. Rep. 2015. V. 3. P. 161–168.
Korban S.A., Bobrov K.S., Maynskova M.A., Naryzhny S.N., Vlasova O.L., Eneyskaya E.V., Kulminskaya A.A. // Protein Eng. Des. Sel. 2017. V. 30. № 7. P. 477–488.
van Loo B., Schober M., Valkov E., Heberlein M., Bornberg-Bauer E., Faber K., Hyvönen M., Hollfelder F. // J. Mol. Biol. 2018. V. 430. № 7. P. 1004–1023.
Distler H. // Angew. Chem. Int. Ed. Engl. 1967. V. 6. № 6. P. 544–553.
Dierks T., Miech C., Hummerjohann J., Schmidt B., Kertesz M.A., von Figura K. // J. Biol. Chem. 1998. V. 273. № 40. P. 25560–25564.
Cartmell A., Lowe E.C., Baslé A., Firbank S.J., Ndeh D.A., Murray H., Terrapon N., Lombard V., Henrissat B., Turnbull J.E. // Proc. Natl. Acad. Sci. 2017. V. 114. № 27. P. 7037–7042.
Варфоломеев С.Д., Гариев И.А., Упоров И.В. // Успехи химии. 2005. Т. 74. № 1. С. 67–83.
Nagahara N. // Journal of Amino Acids. 2010. V. 2011. P. 709 404.
Huggins C., Smith D.R. // J. Biol. Chem. 1947. V. 170. № 1. P. 391–398.
Лазурьевский Г.В., Терентьева И.В., Шамшурин А.А. Практические работы по химии природных соединений. Москва. Высшая школа, 2-е изд. 1966. 335 с.
Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Gallo Cassarino T., Bertoni M., Bordoli L. // Nucleic Acids Res. 2014. V. 42. Web Server issue. P. W252–W258.
Abagyan R., Totrov M., Kuznetsov D. // J. Comput. Chem. 1994. V. 15. № 5. P. 488–506.
Halgren T.A. // J. Comput. Chem. 1996. V. 17. № 5–6. P. 490–519.
Fernández-Recio J., Totrov M., Abagyan R. // Proteins. 2003. V. 52. № 1. P. 113–117.
Totrov M., Abagyan R. // Proteins. 1997. V. Suppl. 1. P. 215–220.
Salomon-Ferrer R., Case D.A., Walker R.C. // WIREs Comput. Mol. Sci. 2013. V. 3. № 2. P. 198–210.
Li P., Song L.F., Merz K.M. // J. Chem. Theory Comput. 2015. V. 11. № 4. P. 1645–1657.
Li P., Song L.F., Merz K.M. // J. Phys. Chem. B. 2015. V. 119. № 3. P. 883–895.
http://nebasechanger.neb.com/.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия