

Биоорганическая химия, 2020, T. 46, № 4, стр. 340-359
Экспериментальные методы исследования механизма взаимодействия липидных мембран с низкомолекулярными лекарствами
И. М. Ле-Дейген 1, А. А. Скуредина 1, Е. В. Кудряшова 1
1 ФГБОУ ВО МГУ имени М.В. Ломоносова, химический факультет
119991 Москва, Ленинские горы, д. 1, стр. 3, Россия
Поступила в редакцию 08.02.2020
После доработки 20.02.2020
Принята к публикации 22.02.2020
Аннотация
Обзор посвящен методам исследования механизма взаимодействия липидных мембран с низкомолекулярными лекарствами. Представлены методы, позволяющие выяснить влияние включения активной молекулы на состояние липидного бислоя: ИК- и ЭПР-спектроскопия, флуоресцентный анализ, дифференциальная сканирующая калориметрия, методы микроскопии. Рассмотрены методы определения размера и заряда везикул: динамическое светорассеяние и анализ траектории наночастиц. Методы разделены на требующие введения дополнительных меток и не требующие таковых. Важной задачей обзора, помимо анализа последних достижений инструментальных методов анализа, является поиск оптимальной стратегии исследования путем подбора информативных, современных подходов к изучению взаимодействия лекарств с липидными мембранами.
ВВЕДЕНИЕ
Разработка новых биологически активных веществ и их лекарственных форм ставит перед современной наукой существенные вопросы, связанные с безопасностью и эффективностью терапии. Включение активных молекул в липидные системы доставки позволяет бороться с выраженными побочными эффектами лекарств, повышает биодоступность и биосовместимость препаратов. Таким образом, выявление механизма взаимодействия лекарство – мембрана и влияния активной молекулы на физико-химические свойства бислоя является одними из ключевых задач при создании новых лекарственных форм.
Обширен арсенал физико-химических методов, используемых в данных целях. Однако лавинообразно увеличивающееся количество публикаций по данному вопросу обусловливает потребность в глубоком анализе накопленных данных для обнаружения новых комплексных подходов к изучению взаимодействия липидных мембран с низкомолекулярными лекарствами.
Данный обзор посвящен анализу последних достижений инструментальных методов исследования липидных систем. Отдельно рассматриваются методы, требующие и не требующие введения дополнительных меток. Среди методов, не требующих введения дополнительной метки, рассмотрены как классические подходы, такие как дифференциальная сканирующая калориметрия (ДСК), атомно-силовая микроскопия (АСМ), сканирующая электронная микроскопия (СЭМ), криоэлектронная микроскопия, так и относительно новые, например, метод малоуглового рассеяния рентгеновского излучения, ИК-спектроскопия Фурье, ЭСЭМ-микроскопия (Environmental Scanning Electron Microscopy), метод дифракции излучения лазера на свободных электронах.
В ряду методов, требующих введения дополнительной метки, рассмотрены флуоресцентные методы анализа, в том числе методы анализа поляризации флуоресценции, разрешено-временная флуоресцентная анизотропия (РВФА), флуоресцентная корреляционная спектроскопия (ФКС), ЭПР-спектроскопия.
Использование нескольких из вышеуказанных методов позволяет оценить расположение и состояние лекарства в липосоме, определить сайты связывания, влияние активной молекулы на фазовый переход и на морфологию везикул.
Отдельно рассмотрены методы “контроля качества” получаемых липидных везикул, оценки эффективности включения активной молекулы, определения размера и заряда везикул: динамическое светорассеяние (ДСР) и анализ траектории наночастиц (НТА), хроматографические методы анализа, УФ-спектроскопия и флуоресцентная спектроскопия. При рассмотрении данного аспекта необходимо принимать во внимание факт существенного влияния способа получения везикул на их физико-химические свойства.
Важной задачей обзора, помимо анализа последних достижений инструментальных методов анализа, является поиск оптимальной стратегии исследования путем подбора информативных, современных подходов к изучению взаимодействия лекарств с липидными мембранами.
1. МЕТОДЫ “КОНТРОЛЯ КАЧЕСТВА” ВЕЗИКУЛ, ЗАГРУЖЕННЫХ ЛЕКАРСТВЕННЫМ ПРЕПАРАТОМ
Получение липидных везикул, загруженных лекарственным препаратом, – хорошо отработанная процедура [1]. Необходимы методы контроля получаемых липосомальных формуляций; обычно следует подтвердить гомогенность везикул, определить их гидродинамический радиус и дзета-потенциал, а также оценить эффективность включения лекарственного препарата. Рассмотренные в данном разделе методы обычно предваряют анализ полученных липосомальных систем иными методами для установления механизма взаимодействия лекарство – мембрана.
1.1.1. Динамическое светорассеяние
Классическим методом определения размера липосом является метод динамического светорассеяния [2]. Данный метод позволяет получить данные о распределении по размерам нано- и субмикронных частиц в разбавленных растворах. Луч лазера, проходя через раствор и достигая частицы, отражается и рассеивается, т.к. ее размер сравним с длиной волны. Непрерывное броуновское движение частиц приводит к постоянному перемещению источникa рассеивания. Флуктуация локальной концентрации частиц соответствует флуктуации интенсивности рассеянного света [3]. На основе данных временных флуктуаций интенсивности рассеянного света (рис. 1а) строится автокорреляционная кривая G(τ) (рис. 1б), которая описывается уравнением (1):
(1)
$G\left( \tau \right) = b\left[ {1 + \varepsilon ~{\text{exp}}\left( {{{ - \tau } \mathord{\left/ {\vphantom {{ - \tau } {{{\tau }_{{\text{c}}}}}}} \right. \kern-0em} {{{\tau }_{{\text{c}}}}}}} \right)} \right]~,$Рис. 1.
Схематическое представление (а) флуктуаций интенсивности светорассеяния, регистрируемых в малом объеме в микросекундном временном интервале и соответствующей автокорреляционной функции G(τ) (б).
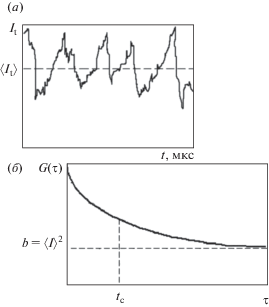
Коэффициент диффузии D частиц обратно пропорционален времени затухания τc рассматриваемой корреляционной функции:
где k – константа (модуль волнового вектора рассеяния).Расчет гидродинамического радиуса наночастиц осуществляется по уравнению Стокса–Эйнштейна через коэффициент диффузии (3),
где D — коэффициент диффузии, kB — постоянная Больцмана, T — абсолютная температура, η — вязкость жидкости, r — радиус частицы.Метод вошел в рутинный арсенал исследователей липосомальных систем [4], и без представления данных о гидродинамическом радиусе и коэффициенте полидисперсности (PdI) трудно себе представить достоверное исследование по взаимодействию мембран с лекарствами [5].
ДСР позволяет изучать суспензии частиц с размером от нескольких нанометров до микрона. У метода, однако, есть ряд существенных ограничений. Во-первых, наличие в суспензии крупных частиц микронного размера (пылинки, агрегаты) может исказить результаты определения. Для реальных систем предельная точность измерения позволяет определить обычно лишь средний размер частиц, ширину и асимметрию распределения частиц по размерам. “Разрешить” пики, соответствующие двум популяциям везикул различных размеров, удается, если диаметры различаются в 2–3 раза. Например, с помощью ДСР удалось доказать образование популяции малых везикул с диаметром около 100 нм при инкубации гигантских моноламеллярных липосом (диаметр около 2000 нм) с АТФ и ГТФ в течение 15 минут [6]. Если же в системе присутствуют популяции, например, липосом, покрытых полимерной оболочкой, и непокрытых, чаще всего двух независимых пиков зарегистрировать не удается, однако наблюдается смещение основного максимума в сторону больших размеров [7]. Так, для анионных липосом 60 ± 2 нм покрытие поликатионом ПЭГ-хитозаном приводило к увеличению гидродинамического радиуса до 85 ± 4 нм. Если же стоит задача разделить две популяции везикул с близкими размерами, можно предложить использовать метод анализа траектории наночастиц.
1.1.2. Анализ траектории наночастиц
Данный подход часто используется для определения концентрации наночастиц в растворе и их распределения по размерам. Раствор облучают лазером, при этом частицы размером менее длины волны ведут себя как точечные рассеиватели света. Наблюдение производится через ультрамикроскоп, расположенный сверху, под прямым углом к лазеру. Высокочувствительная камера записывает видео броуновского движения частиц. Далее компьютер, исходя из известного объема области наблюдения и количества зарегистрированных частиц, рассчитывает концентрацию частиц в растворе (рис. 2) [8]. Например, по данным метода НТА в суспензии анионных липосом, состоящих из 80% массовых дипальмитоилфосфатидилхолина (ДПФХ) и 20% кардиолипина (КЛ), при общей концентрации липидов 3 мг/мл содержится 1 × 1016 везикул/литр [9], что согласуется с теоретическими расчетами. Это позволило определить число полимерных молекул, приходящееся на каждую липосомальную частицу в составе комплекса липосом с ПЭГ-хитозаном.
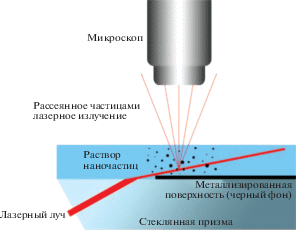
Как и в методе динамического светорассеяния, гидродинамический радиус наночастицы вычисляется по уравнению Стокса–Эйнштейна. К достоинствам НТА следует отнести возможность различать популяции везикул с небольшим различием в размерах (20–30%), возможность работать с очень низкими концентрациями образца (обычно 106–109 частиц/мл), возможность исключить фактор крупных пылинок или агрегатов. С другой стороны, пробоподготовка в методе НТА требует использования высокочистой воды (обычно используется система Milli-Q, в которой вода проходит через ионообменные смолы и картриджи с активированным углем, а затем через фильтры с размером пор 0.22 микрона, так, чтобы значение удельного сопротивления составляло 18.2 МОм см). Однако, работа при больших разбавлениях (в случае суспензии липосом 3 мг/мл необходимо разбавлять образец минимум в 10 000 раз) может приводить к диссоциации нековалентных липосомальных комплексов, которые, как правило, имеют константы диссоциации порядка 10–4 М.
Сравнение методов ДСР и НТА приведено в работе [10]. Рассматривалась суспензия катионных липосом состава: яичный фосфатидилхолин : : 1,2-диолеил-sn-глицеро-3-фосфоэтаноламин: 1,2-диолеил-3-триметил аммонийпропан в мольном соотношении 4 : 2 : 1. По данным ДСР средний гидродинамический диаметр составил 117 нм с индексом полидисперсности 0.248, что указывает на гетерогенность популяции везикул. Однако, по данным НТА липосомы оказались крупнее (154 нм) и гомогеннее.
Несовпадение результатов определения размеров липосом по данным ДСР и НТА часто встречается на практике [11], что связано с принципиальной разницей в физических основах методов. При этом однозначно отклонить или рекомендовать тот или иной метод для решения рутинных задач не представляется возможным, поскольку у обоих подходов есть ряд ограничений. В случае ДСР обычно учитывают гидродинамический радиус, причем для получения дополнительной информации рассматривают распределение по размерам, взвешенное по интенсивности и по числу частиц, что позволяет выявить влияние крупных агрегатов. Для метода НТА наиболее информативен комплексный анализ траектории движения каждой частицы по видеозаписи, включающий анализ полной длины траектории везикулы (finite track length adjustment FTLA) для наиболее корректного отбора траекторий в расчет гидродинамического радиуса.
Наилучшим решением может быть комбинация обоих методов, как, например в работе [12]. Дополнительные сведения о размере липосом можно получать с помощью методов электронной микроскопии, о которых речь пойдет в следующих разделах обзора.
1.1.3 Определение ζ-потенциала липосом
Часто приборы ДСР оснащены возможностью измерения ζ-потенциала везикул (иногда его называют электрокинетическим потенциалом). Это понятие вводится при рассмотрении двойного электрического слоя движущейся коллоидной частицы (рис. 3) и его удобно использовать как для оценки заряда липосом, так и предположений об их стабильности. Известно, что заряженные везикулы медленнее агрегируют, также их поверхность легче функционализировать полимерами для использования в системах адресной доставки [7, 13, 14]. Следует отметить, что ζ-потенциал очень чувствителен к изменениям условий: на его величину влияет рН, ионная сила, температура и т.д.
Рис. 3.
Строение двойного электрического слоя: потенциал в плоскости скольжения соответствует электрокинетическому ζ-потенциалу везикулы. На рисунке φ0 – потенциал стенки, φd – Штерновский потенциал.
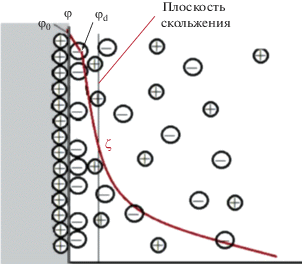
Измерение ζ-потенциала проводится в специальной кювете, оснащенной двумя электродами. Наложение электрического поля приводит в движение заряженные везикулы со скоростью, пропорциональной ζ-потенциалу. Скорость (электрофоретическая подвижность Ue) же определяют по рассеянию света. Расчет ζ-потенциала производят по уравнению Генри (4):
где ε – диэлектрическая проницаемость, η – вязкость среды, f(κa) – функция Генри, κ – дебаевская длина частицы, a – дебаевский радиус частицы. В аппроксимации Смолуховского для водных сред функцию Генри принимают равной 1.5, а в неводных средах – 1.В работе [15] проведен сравнительный анализ ζ-потенциала для липосом различного липидного состава и заряда. Варьируя содержание анионного или катионного липида в нейтральной матрице, авторы обнаружили линейную зависимость ζ-потенциала от мольного содержания заряженного липида. В работах [9, 16] продемонстрирована плавная нейтрализация ζ-потенциала анионных липосом с выходом на насыщение при добавлении поликатионов – производных хитозана. Встраивание же лекарственных молекул в бислой или включение во внутреннюю полость липосом редко может значительно повлиять на суммарный заряд везикулы: в литературе описаны небольшие изменения, чаще всего в пределах доверительного интервала [17, 18].
Следует отметить, что помимо ζ-потенциала для оценки поверхностного заряда частиц часто используют близкий параметр – электрофоретическую подвижность [14]. Данную величину измеряют методом лазерного микроэлектрофореза. Например, при исследовании взаимодействия поликатионов c липосомами на основе яичного лецитина и кардиолипина с исходным отрицательным значением электрофоретической подвижности сорбция полимера приводила к полной нейтрализации заряда, а дальнейшая сорцбия приводила к перезарядке поверхности везикулы.
Рассмотренная выше группа методов определения размера, заряда и гомогенности везикул служит, таким образом, не только для контроля качества получаемых везикул, но также предоставляет ценную информацию о популяциях частиц. Изменение размера частиц или их заряда при включении лекарственного препарата может предоставить доказательства в пользу того или иного механизма взаимодействия активной молекулы с бислоем.
1.2. Методы определения эффективности включения лекарственного препарата
Ключевой характеристикой липосом как систем доставки лекарственных средств является эффективность включения (ЭВ) активной молекулы. Различают два параметра: собственно ЭВ, рассчитываемую как отношение массы (или моль) включившегося лекарства к общей добавленной массе (или моль) к липидной суспензии, и массовое соотношение (МС) лекарство : липиды в полученной липосомальной форме. Оба параметра важны для характеристики системы доставки: ЭВ подходит для сравнения разных методик загрузки одного и того же лекарства, в то время как МС важнее при сравнении липосомальных систем доставки с другими носителями в вопросе оптимизации соотношения лекарственного вещества к общей массе системы доставки. Таким образом, оба эти параметра зачастую необходимо рассчитывать. Обычно известно изначально добавленное количество лекарственного препарата, таким образом, необходимо либо определять содержание лекарства в самих липосомах, либо определять содержание невключившегося препарата. Общая схема определения представлена на рис. 4.
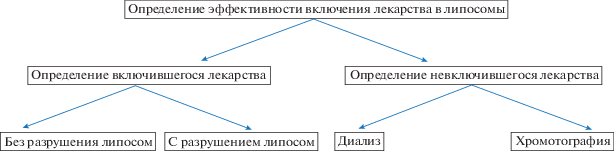
Более распространены методики определения количества невключившегося лекарства. Для отделения липосомальной фракции от фракции со свободным лекарством обычно используют диализ или гель-проникающую хроматографию. Достоинства и недостатки данных методов отражены в табл. 1. Оба метода заслуженно активно применяются в исследовательской практике.
Таблица 1.
Методы разделения липосомальной фракции и фракции невключившегося лекарства
| Метод | Достоинства | Недостатки |
|---|---|---|
| Диализ | Методическая простота Доступность (подходят диализные мешки с большой массой отсечения) |
Необходим контроль сохранности липосом Сильное разбавление фракции свободного лекарства Длительность: от нескольких часов до суток |
| Гель-проникающая хроматография | Методическая простота Быстрота Возможна работа с малыми объемами (менее 1 мл) |
Необходим подбор носителя для колонки Возможное разбавление липосомальной фракции |
Определяют содержание лекарства в промывных водах (в случае диализа) или во фракциях, свободных от липосом. Выбор метода в основном обусловлен природой активной молекулы: зачастую походит УФ-спектроскопия, высокоэффективная жидкостная хроматография, часто – флуоресцентные методы. Для некоторых лекарственных препаратов, например, рапамицина, высокоэффективная жидкостная хроматография является практически единственным возможным решением из-за отсутствия аналитически значимых спектров УФ и флуоресценции [19]. Использование ИК-спектроскопии допустимо, если содержание активной молекулы в изучаемой фракции не менее 0.2–0.5 мг/мл в виду предела обнаружения метода.
При разрушении липосом детергентом для определения содержания лекарства используют те же методы с поправкой на возможное изменение сигнала от активной молекулы. Например, при разрушении липосом, загруженных доксорубицином, поверхностно-активным веществом Triton-X необходимо определять содержание лекарства по калибровочным кривым, полученным для лекарства в присутствии соответствующего содержания детергента, поскольку потенциальное мицеллообразование [20] может значительно изменить коэффициент молярного поглощения препарата и исказить определение ЭВ.
Направление, связанное с определением ЭВ в липосомах без предварительного разрушения, разработано гораздо слабее по сравнению с остальными. Это обусловлено рядом причин: во-первых, липосомальные системы часто оптически непрозрачны, что затрудняет применение многих спектральных методов. Во-вторых, присутствие липидов как таковых и их коллоидных везикул существенно может искажать сигнал от активной молекулы.
2. МЕТОДЫ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ ВЗАИМОДЕЙСТВИЯ ЛИПИДНОГО БИСЛОЯ И ЛЕКАРСТВЕННОГО ПРЕПАРАТА
2.1. Методы исследования липидных систем, не требующие введения дополнительных меток
Рассматриваемые выше методы служат в первую очередь для контроля качества получаемых липосомальных форм активных молекул, но не предоставляют информации о механизме взаимодействия лекарство – бислой. Далее представлены методы, которые могут быть полезны для изучения основных закономерностей данного процесса. Важным их достоинством является отсутствие необходимости вводить дополнительные метки в бислой, что упрощает интерпретацию данных и ускоряет анализ.
2.1.1. Дифференциальная сканирующая калориметрия
Рассмотрим подробнее метод дифференциальной сканирующей калориметрии, который изначально нашел свое широкое применение в изучении фазовых переходов в липосомах. В работе [21] приведен исчерпывающий обзор, представляющий собой описание возможностей ДСК для анализа структуры и свойств липосом, однако метод хорошо подходит и для установления особенностей взаимодействия низко- и высокомолекулярных веществ с бислоем на основе различий в калориметрических кривых.
В работе [22] методом ДСК установили, что при взаимодействии липосомальной мембраны (100% дистеароилфосфатидилхолин) с тамоксифеном фазовый переход смещается в область меньших температур, при этом максимум на кривой ДСК уширяется, что указывает на дестабилизацию мембраны, по-видимому, за счет встраивания лекарства в бислой.
Более точная регистрация кривых ДСК позволяет получать информацию о состоянии лекарства в липосомах и определении локализации лекарства. Фоггиа с коллегами изучали взаимодействие нестероидного противовоспалительного препарата ибупрофена с липосомами из димистроилфосфатидилхолина [23]. Регистрация кривых ДСК при различных значениях рН позволила установить, что протонированная форма ибупрофена при рН 3 способна проникать в неполярную часть бислоя, в то время как при рН 7 взаимодействие депротонированного ибупрофена с холиновым группами фосфолипида препятствует дальнейшему проникновению препарата.
Иехезкель Баренхольц [24] с коллегами при разработке нового липофильного пролекарства митомицина С и изучении его влияния на мембрану ПЭГилированных липосом обнаружили, что наличие двух остатков стеариновой кислоты в структуре пролекарства обеспечивают стабильное встраивание в липосомный бислой, о чем свидетельствует изменение максимума на кривой ДСК: пик становится высокоинтенсивным и узким с максимумом при 56°С и плечом при 53°С, причем для контрольных липосом пик был широким с максимумом при 53°С. Авторы отмечают, что такой тип фазового перехода характерен для мембраны, содержащей холестерин, и объясняют наблюдаемое явление формированием богатых пролекарством доменов в мембранах, в которых гидрофобные цепи плотно упакованы в высокоупорядоченные кристаллоподобные структуры.
Внимание исследователей привлекает изучение нанокристаллов активных молекул, которые образуются при включении в липосомы [25]. Ли с коллегами в работе фокусировались на методах получения подобных нанокристаллических систем в липосомах, упоминая ДСК как возможный метод обнаружения кристаллов в везикулах. Ранее уже было показана кристалличность сульфата доксорубицина и некоторых других препаратов с помощью калориметрии высокого разрешения, однако о их природе и свойствах ведутся споры. В работе 2012 года [26] не обнаруживали существенного влияния на фазовый переход дипильмитоилфосфатидилхолиновых липосом встраивания доксорубицина. Однако в 2017 году усовершенствованная техника позволила Перинелли с коллегами обнаружить упорядоченное состояние нанокристаллов доксорубицина внутри ПЭГилированных липосом (формуляция Доксила), причем кристаллы оставались стабильны даже после нагревания до 80°C [27].
Применение ДСК позволяет следить за микрофазовым разделением в мембране липосом смешанного состава. Так, для смешанных анионных липосом ДПФХ/КЛ (80/20%) комбинацией методов ДСК и ИК-спектроскопии обнаружено расслоение в липидном бислое с образованием двух доменов с различными температурами плавления [18]. Калориметрическая кривая смешанных липосом характеризовалась широким фазовым переходом с пиком при 39°С и плечом при 35°С, которые отвечали плавлению двух микрофаз ДПФХ/КЛ с различным соотношением компонентов. Для таких липосом загруженных моксифлоксацином, удалось доказать структурирование и стабилизацию гелеобразной фазы за счет электростатического взаимодействия аминогруппы активной молекулы с отрицательно заряженным кардиолипином. Температура основного фазового перехода липосом, загруженных моксифлоксацином сдвинулась на 3–5 градусов.
Таким образом, метод ДСК позволяет изучать детали процесса фазового перехода в липосомах и влияния на него лекарств, косвенно обнаружить образование нанокристаллов или интеграцию активной молекулы в бислой.
В качестве альтернативы ДСК в работе [28] предложено использовать метод ДСР, причем аналитическим сигналом будет служить величина average count rate (средняя скорость счета). Включение в димистроилфосфатидилхолиновые липосомы препарата целекоксиба приводило к достоверному смещению кривой средняя скорость роста – температура в область больших температур. Сравнение кривых для загруженных и свободных липосом при рН 5.0 и 7.4 выявило, что при обоих значениях рН кривые смещаются единообразно, но в нейтральной среде эффект более выражен (расчетные температуры плавления составили 21.7 и 20.7°C против контрольного значения для свободных липосом 22.0°C). Авторы предполагают, что в кислой среде встраивание целексосиба влияет на кооперативность фазового перехода, а не на значение температуры фазового перехода, в то время как при рН 7.4 целекоксиб разупорядочивает мембрану, что проявляется в потере кооперативности и уменьшении температуры фазового перехода.
2.1.2. Метод малоуглового рассеяния рентгеновского излучения
В методе малоуглового рассеяния рентгеновского излучения (ММРР) измеряют интенсивность рентгеновских лучей, рассеянных от образца в зависимости от угла рассеяния (0.1°–5°C). Ключевым преимуществом данного метода относительно классических рентгеновских методов является его универсальность для многих типов наноматериалов и способность регистрировать спектры от образцов, как в порошковой, так и в гелеообразной и жидкой дисперсной форме. Установки ММРР позволяют исследовать усредненные структурные особенности от образца большого объема, что может быть полезно при работе с неоднородными коллоидными системами. Данный метод хорошо подходит для изучения взаимодействия мембран с активными молекулами, кристаллизующимися во внутренней полости везикул.
Важным достоинством ММРР является возможность определять форму везикул. Известно, что методы динамического светорассеяния и анализа траектории наночастиц предоставляют информацию о гидродинамическом радиусе частиц из предположения об их сферической форме, в то время как липосомы, содержащие нанокристаллы вытянутой формы, могут принимать и эллиптическую форму. Аналитически ценным параметром из спектра ММРР является вектор рассеяния q; обработка зависимости интенсивности рассеяния от q методом Фурье-преобразований позволяет построить график функции распределения парных расстояний.
Значительных успехов в изучении липосомальных форм нанокристаллов антибиотиков достигли Чиполла и Чен, Университет Сиднея. Их работы посвящены в первую очередь изучению нанокристаллов ципрофлоксацина – антибиотика широкого спектра действия фторхинолонового ряда [25, 29, 30]. Например, комбинацией методов ММРР, криоэлектронной микроскопии и деполяризованного динамического светорассеяния авторам удалось доказать [30] циллиндрическую форму липосом, загруженных ципрофлоксацином.
К недостаткам метода ММРР можно отнести слабый сигнал, накапливаемый от липосомальных суспензий, возможное перекрывание спектров везикул и нанокристаллов и потенциальное разрушение образца в процессе исследования.
Рентгеновские методы активно применяют для исследования липосомальных форм неорганических наночастиц – фотосенсибилизаторов, например наночастиц иридия [31], с целью подтвердить неизменность кристалличности неорганической фракции после включения в везикулы. Однако взаимодействие липосом с неорганическими наночастицами представляет собой отдельную тему, достойную объемного обзора, и несколько выходит за рамки обсуждаемой в настоящей работе тематики.
2.1.3. ИК-спектроскопия Фурье
ИК-спектроскопия Фурье заслуженно вошла в арсенал исследователей липидных систем как один из наиболее информативных и точных методов, позволяющий получать уникальную информацию о микроокружении функциональных групп липидов, их взаимодействии с различными лигандами и фазовых переходах. Использование ИК‑спектроскопии для анализа биосистем стало возможным с развитием аппаратуры и методов математической обработки (Фурье-преобразования) исходного сигнала прибора – интерферограммы.
Рассмотрим три оптических эффекта, обеспечивающих высокую точность метода ИК-спектроскопии Фурье. Первый из них получил название выигрыш Жакино. Его суть состоит в том, что в схеме Фурье-спектрометра, в отличие от классического дисперсионного, отсутствует щель, так что все излучение достигает детектора. При фиксированной разрешающей силе светосила Фурье-спектрометра в 200 раз превышает светосилы дисперсионного спектрометра.
Следующий эффект получил название выигрыш Фельжета или фактор мультиплексности. Он связан с одновременной регистрацией в спектре всех частот. Например, при одновременной регистрации и обработке спектрального интервала 4000 см–1 при разрешении 1 см–1 соотношение сигнал-шум для Фурье-спектрометра будет в 63 раза выше, а скорость регистрации спектра в 4000 раз выше. В настоящее время регистрация одного спектра в высоком разрешении, пригодного для тонкого анализа спектральных полос, занимает не более двух минут, что, несомненно, является важным методическим достоинством метода.
Третий эффект получил название выигрыш Конна. Использование в ИК-спектрометрах гелий-неоновых лазеров как внутренних стандартов длин волн и для контроля интерферограммы в зависимости от положения подвижного зеркала позволило значительно повысить точность определения волновых чисел. В настоящее время некоторые приборы характеризуются разрешением 0.01 см–1. Подобные машины требуют тонкой наладки перед каждым запуском, в то время как рутинные измерения можно надежно проводить при разрешении 1–2 см–1.
На сегодняшний день в исследовании сложных биологических систем наиболее широко используется режим нарушенного полного внутреннего отражения (НПВО) [32]. Явление НПВО основано на проникновении излучения из оптически более плотной среды (кристалла n1) в менее плотную среду (образец n2) на некоторую длину (т.н. penetration depth dp), примерно равную длине волны, при падении света под углом, большим критического, при этом часть падающего излучения проникает в образец на dp и там поглощается. Критический угол определяют как угол падения, при котором угол преломления составляет 90°, его величина зависит от коэффициентов n1 и n2.
Следует отметить, что поскольку dp прямо пропорциональна длине волны излучения, в длинноволновой области спектра НПВО следует ожидать большую интенсивность полос поглощения. Если n2/n1 постоянно, то dp линейно зависит от длины волны. Большинство современных программ для анализа ИК-спектров предлагают функцию ATR-correction, которая линейно корректирует интенсивности полос, приводя спектр к виду близкому к спектру пропускания.
Пионерские работы по использованию ИК-спектроскопии Фурье для анализа белковых и липидных систем принадлежат Эрику Гурматигху и его научной группе в Свободном Университете Брюсселя, Бельгия. В его лаборатории разработаны многочисленные протоколы регистрации спектров с подбором оптимальных условий: типом кристалла, количеством сканирований, способом атмосферной компенсации и т.д. и методы их анализа.
ИК-спектр фосфолипидов имеет следующие характеристические пики функциональных групп: две полосы симметричных и ассиметричных колебаний углеводородных связей, колебания карбонильной группы C=O, колебания фосфатной и холиновой групп (рис. 5). Полное соотнесение полос поглощения и соответствующих колебаний дипальмитоилфосфатидилхолина представлено в табл. 2. Положение полос и их форма чувствительны к связыванию бислоя с лигандами, к образованию водородных связей, агрегации и окислению и т.д. [7, 11, 18, 26, 33–37].
Рис. 5.
Нормализованный ИК-спектр нарушенного полного внутреннего отражения дипальмитоилфосфатидилхолина.
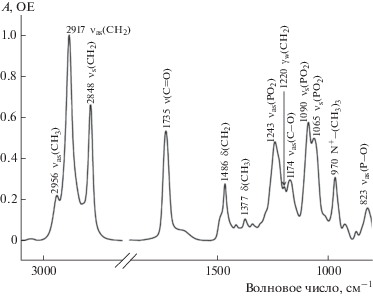
Таблица 2.
Характерные полосы в ИК-спектре липидов по данным. Даны в сокращении названия колебаний: вал. – валентное, деф. – деформационное, ножн. – ножничное, маят. – маятниковое, асим. – асимметричное, сим. – симметричное
| Волновое число, см–1 | Колебание | Волновое число, см–1 | Колебание |
|---|---|---|---|
| 3010 | =C–H (вал.) | 1378 | CH3 (сим. деф.) |
| 2956 | CH3 (асим. вал.) | 1400–1200 | CH2 (веерные) |
| 2920 | CH2 (асим. вал.) | 1228 | ${\text{PO}}_{{\text{2}}}^{ - }$ (асим. вал.) |
| 2870 | CH3 (сим. вал.) | 1170 | CO–O–C (асим. вал.) |
| 2850 | CH2 (сим. вал.) | 1085 | ${\text{PO}}_{{\text{2}}}^{ - }$ (сим. вал.) |
| 1730 | C=O (вал.) | 1070 | CO–O–C (сим. вал.) |
| 1485 | (CH3)3N+ (асим. деф.) | 1047 | C–O–P (вал.) |
| 1473, 1472, 1468, 1463 | CH2 (ножн.) | 972 | (CH3)3N+ (асим. вал.) |
| 1460 | CH3 (асим. деф.) | 820 | P–O (асим. вал.) |
| 1405 | (CH3)3N+ (сим. деф.) | 730, 720, 718 | CH2 (маят.) |
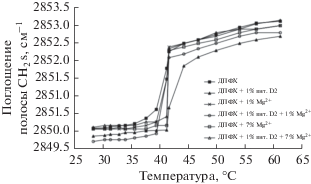
Симметричным и асимметричным валентным колебаниям СН2 группы соответствуют полосы в регионе 2853 ± 5 и 2926 ± 5 см–1, деформационным колебаниям соответствует полоса при 1465 ± ± 5 см–1 (так называемая частота метиленовой деформации). Симметричные деформационные колебания СН3 группы характеризуются полосой поглощения при 1375 ± 5 см–1. Изменение конформации липидных цепей сопровождается частотным сдвигом полос поглощения групп СН2, причем эти изменения коррелируют с числом цис-изомеров ненасыщенных остатков гидрофобных хвостов. Цис-изомеры упакованы менее плотно по сравнению с транс-изомерами, ацильные цепи могут менять свое направление и размер. Увеличение подвижности сопровождается высокочастотным сдвигом на ИК-спектре [7]. Влияние на фазовый переход различных лекарственных препаратов можно оценивать как по смещению полос поглощения симметричных и асимметричных валентных колебаний CH2 групп, так и с использованием метода главных компонент (principal component analysis PCA). Классическим подходом является регистрация зависимости положения полос поглощения CH2 as и CH2 s в зависимости от температуры. Например, в работе [33] исследовали влияние витамина D2 в присутствии ионов Mg2+: установлено, что фазовый переход существенно замедляется, однако, ход кривых остается единообразным, что указывает на упорядочивающий эффект активной молекулы на бислой, сопровождающийся уменьшением доли числа транс-конформеров (рис. 6). В качестве вспомогательного источника информации авторы использовали ширину данной полосы поглощения (на уровне 0.75 высоты пика), которая уменьшалась при введении в липосомы витамина D2, что подтверждает данные термограммы.
Рис. 6.
Зависимость положения полосы поглощения валентных колебаний CH2 в липосомах ДПФХ в присутствии витамина D2 и ионов Mg2+.
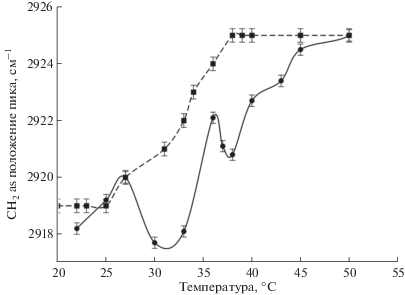
Однако, для мультикомпонентных липосом, склонных к образованию микрофаз, например, содержащих 80% ДПФХ и 20% КЛ, ход термограмм может значительно изменяться в присутствии активных молекул. Для контрольных незагруженных везикул ход кривой плавный, S-образный (рис. 7), отдельные микрофазы не различаются. При этом, в работе [18] был обнаружен эффект независимого взаимодействия моксифлоксацина с микрофазами с разным содержанием КЛ, что отражается принципиальным изменением хода кривой термограммы положение CH2 as – температура, а именно минимумами в диапазоне температур 28–35°С, указывающими на локальное упорядочивание бислоя (рис. 7).
Рис. 7.
Зависимость положения полосы поглощения валентных колебаний CH2 в липосомах ДПФХ 80%, кардиолипин 20% (пунктир) и липосомах того же состава, загруженных моксифлоксацином.
Распространен подход, связанный с математической обработкой спектров по методу главных компонент, который также может предоставить дополнительную информацию о фазовом переходе липосомальных форм активных молекул, особенно в случае накопления большого объема данных. В работе [38] рассматривалось взаимодействие флуфеназина с липосомами из яичного сфингомиелина. Тонкий анализ спектральных изменений позволил авторам сделать вывод о том, что включение лекарства в бислой приводит к снижению температуры фазового перехода пропорционально содержанию активной молекулы, что в свою очередь приводит к быстрому высвобождению флуфеназина в водную фазу.
Метод ИК-спектроскопии чувствителен к изменениям в плотности упаковки ацильных цепей и в случае внешнего воздействия на мембрану, например, под действием колебательно-вращательного движения наностержней магнетита в низкочастнотном негреющем переменном магнитном поле [11]. В данном случае метод деконволюции, то есть разложения наблюдаемой полосы поглощения на несколько компонент, позволил количественно оценить эффект разрыхления бислоя.
Полосы поглощения карбонильной и фосфатной группы в первую очередь чувствительны к изменению степени гидратации данных функциональных групп, в том числе за счет образования ионных связей и агрегации.
Гидратированная карбонильная С=О группа в липидах и липосомах поглощает в регионе 1730 см–1. На кислороде находится частично отрицательный заряд, который в свободных липосомах компенсируется за счет образования водородных связей с молекулами воды. При снижении степени гидратации, то есть разрыве части водородных связей, характеристическое волновое число возрастает. Так происходит, например, при взаимодействии липосом с поликатионами или рядом биологически-активных полярных веществ [7, 34]. Изменения в области поглощения карбонильной группы чрезвычайно информативны при анализе изменений на границе раздела фаз липид-вода, в том числе для контроля за сохранностью липосом при хранении [7].
Для фосфатной группы определены две основных полосы поглощения, соответствующих симметричным 1088 см–1 и асимметричным 1250–1230 см–1 валентным колебаниям. Наибольший интерес представляет полоса асимметричных колебаний, как более чувствительная к степени гидратации. Низкочастотный сдвиг положения данной полосы поглощения связан с повышением степени гидратации [34], и наоборот. Вид и форма данной полосы существенным образом может измениться при добавлении активной молекулы, как показано в работе [35] на примере липосом с включенными в них нестероидными противовоспалительными средствами.
Помимо анализа смещения полос поглощения карбонильной и фосфатной групп исключительно полезной может быть процедура деконволюции, поскольку становится возможным выявить компоненты, соответствующие функциональным группам, связанным с активной молекулой. Например, при встраивании в анионные липосомы доксорубицина, модифицированного фрагментом спермина и стеариновой кислоты, происходит двукратное уменьшение доли высокогидратированных фосфатных групп, которым соответствует компонента 1219 см–1, и двукратное увеличение доли низкогидратированных карбонильных групп (компонента 1745 см–1) [34].
Таким образом, метод ИК-спектроскопии информативен при изучении многих параметров липосомальных систем: процесса фазового перехода, электростатического взаимодействия с лекарствами, изменениям микроокружения на поверхности раздела фаз липид-вода за счет связывания с лигандами или при агрегации. В табл. 3 приведены основные способы обработки полос поглощения липосом и информация, которую можно получить.
Таблица 3.
Информация от основных полос поглощения в ИК-спектре липосом
| Полоса поглощения | Получаемая информация | Возможные методы анализа спектров | Ссылки |
|---|---|---|---|
| CH2 as, CH2 s | Влияние активной молекулы на плотность упаковки липидов | Анализ положения полос поглощения (смещения в область больших волновых чисел указывают на разрыхление, в область меньших – на упорядочивание) | [11, 33] |
| Влияние активной молекулы на фазовый переход | Анализ термограмм CH2 as (T), CH2 s (T), метод главных компонент | [11, 18, 33] | |
| CO | Влияние активной молекулы на поверхность раздела фаз, образование водородных связей с катионными лигандами | Анализ положения полосы поглощения (смещения в область больших волновых чисел указывают на увеличение степени гидратации, в область меньших – на образование электростатических связей) | [7] |
| Деконволюция полосы поглощения для количественной оценки изменения степени гидратации карбонильной группы | [7, 34, 36, 37] | ||
| PO2 as | Влияние активной молекулы на гидрофильную поверхность липосом | Анализ положения полосы поглощения (смещения в область больших волновых чисел указывают на образование электростатических связей) | [26, 35] |
| Деконволюция полосы поглощения для количественной оценки изменения степени гидратации фосфатной группы | [34] |
2.1.4. Методы на чипе
Методы на чипе [39] лишь набирают популярность для изучения взаимодействия поверхности липосом с лекарством. Для этого хорошо подходит метод гигантского плазмонного резонанса. Авторы предложили сначала на модифицированную алканами декстрановую матрицу на чипе сорбировать липосомы с разным содержанием холестерина, а далее во внешний раствор вводить лекарственный препарат. Метод предполагает использование отклика системы на связывание на чипе лекарства с липосомальной мембраной. При этом важную роль играет кинетика сорбции, по которой можно сделать выводы о силе взаимодействия лекарства с мембраной. Так, удалось установить, что анионные лекарства быстро диссоциируют с поверхности липосом, в то время как катионные лекарства в зависимости от своей природы более прочно связываются с бислоем.
2.1.5. Методы микроскопии
Как известно, размер моноламеллярных липосом (30–150 нм) значительно меньше дифракционного предела, поэтому для изучения взаимодействия везикул с лекарственными молекулами могут быть применены методы электронной микроскопии и атомной силовой микроскопии [40]. Специфика данных методов позволяет исследовать топологию поверхности везикул. Обширный обзор по методам визуализации липосом представлен в работе [41], в настоящем обзоре хотелось бы выделить особенности применения методов микроскопии для изучения влияния загрузки лекарственных препаратов на морфологию липосом.
К преимуществам АСМ можно отнести возможность работы, как в контактном, так и в бесконтактном режиме, и получение информации о морфологии липосом без дополнительной пробоподготовки (окрашивания, фиксации или введения меток). Однако, движения кантилевера могут вызывать деформацию бислоя [42], а сами везикулы должны быть нанесены на подложку, что может влиять на их морфологию [41].
В работе [43] показана применимость метода АСМ для 3D анализа популяции липосом, загруженных доксорубицином: везикулы с наибольшими кристаллами доксорубицина приобретали вытянутую форму; данные подтверждали методом криоэлектронной микроскопии.
Применимость сканирующей электронной микроскопии для липосомальных систем ограничена в виду структурных изменений в условиях глубокого вакуума. Однако именно методы электронной микроскопии позволяют определять не только размеры везикул, но и оценивать их ламеллярность. Негативное контрастирование, например, с помощью UO2(CH3COO)2, может быть полезно для оценки. Более того, необходимость высушивания, фиксации и окраски образца также затрудняет получение достоверных результатов. Решение данной проблемы было предложено Данилатосом, автором метода ЭСЭМ (Environmental Scanning Electron Microscopy), в котором допустима работа с образцами в газовой среде, включая воздух.
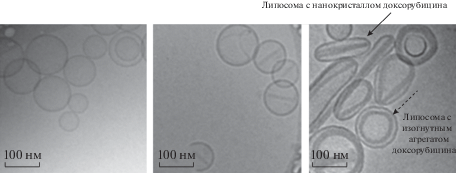
Заслуженно значимую роль в визуализации и характеризации липосомальных форм лекарственных препаратов играет криоэлектронная микроскопия (рис. 8). Метод позволяет изучать липосомы в их нативном состоянии, без применения дополнительных процедур фиксации или окрашивания. В зависимости от толщины образца можно добиться разрешения до 5 нм [41]. Данный метод позволяет отследить влияние мольного соотношения активной молекулы и липидов на морфологию везикул, так в работе [44] при мольном соотношении 0.05 липосомы ожидаемо напоминают кофейные зерна с нанокристаллом доксорубицина внутри, но при больших мольных соотношениях (0.37) неожиданно обнаруживались даже треугольные структуры, в которых три нанокристалла доксорубицина образуют правильный треугольник в пределах одной липосомы.
Рис. 8.
Криоэлектронная микроскопия для (слева-направо) контрольных незагруженных липосом, содержащих 20% мольных доксорубицина, 40% мольных доксорубицина.
В случае, если загруженный препарат не образует кристаллическую форму в липосоме, метод криоэлектронной микроскопии также может быть полезен. Так, в работе [45] по увеличенной электронной плотности внутренней полости липосом по сравнению с изображениями контрольных незагруженных везикул судили о включении винкристина, винбластина и винорелбина во внутреннюю полость липосом.
В лаборатории проф. Баренхольца недавно был предложен подход к реконструкции 3D структуры липосом по 2D криоэлектронным микрофотографиям [46], который позволяет получать дополнительную информацию о распределении препарата в везикулах. Данный подход может быть чрезвычайно полезным при исследовании взаимодействия липосом с, например, липофильно-модифицированными лекарствами, которые могут искажать строение мембраны.
2.1.6. Метод дифракции излучения лазера на свободных электронах
Исключительных результатов удалось добиться при визуализации в растворе липосом, загруженных доксорубицином, методом дифракции излучения лазера на свободных электронах (Free-electron coherent diffraction imaging) [47]. По дифракционной картине от одиночных загруженных препаратом липосом авторам удалось реконструировать реальную форму кристалла доксорубицина в водной полости липосом. Данный метод пока ограниченно применяется, поскольку кристаллические формы в липосомах образуют не все активные молекулы, однако может быть чрезвычайно полезными при исследовании встраивания нанокристаллов в везикулы.
2.2.1. Методы, основанные на введении флуоресцентных меток
Важную роль в исследовании структуры липосом и их комплексов играет флуоресцентная спектроскопия. Поскольку природные липиды собственных флуорофорофоров в структуре не имеют, необходимо введение в везикулы меток-репортеров, в зависимости от свойств которых можно получать информацию о состоянии гидрофобной или гидрофильной части липосом [48]. Можно сформулировать ряд ключевых свойств флуоресцентых меток, которые следует учитывать при выборе:
1. Основные характеристики флуорофоров, а именно длины волн возбуждения и испускания, квантовый выход и флуоресцентное время жизни;
2. Часто для изучения липидных систем предпочтение отдают меткам, флуоресцентные свойства которых резко меняются при изменении полярности среды;
3. Желательна простота методики встраивания флуоресцентной метки в липосомы, при этом следует отдельно удостовериться в локализации метки в везикуле.
Надежной и удобной меткой-репортером для липосом является пирен, по изменению спектра флуоресценции которого можно судить об изменении подвижности гидрофобных цепей липидов (рис. 9).
Рис. 9.
Флуоресцентные метки-репортеры для изучения состояния гидрофобной части бислоя. (а) Спектр флуоресценции пирена, приведены формулы мономера и эксимера. (б) Структура и спектры поглощения (1) и флуоресценции (2) N-[7-(4,4-дифтор-1,3,5,7-тетраметил-4-бор-3a,4a-диазо-s-индекан-8)гептил]сфингозин-1-фосфотидилхолина.
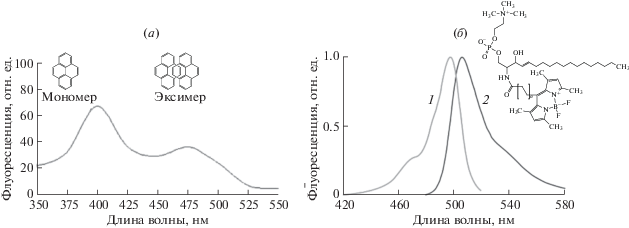
Одной из первых работ, в которой предложено использовать пирен для изучения состояния гидрофобной части бислоя, является [49]. Авторы сравнили ряд органических молекул: пирен, нафталин и 1,12-бензперилен – с точки зрения их поведения в бислое, в том числе в процессе фазового перехода. Установлено, что завершение фазового перехода приводит к заглублению пирена в бислой. Аналогичный эффект был обнаружен для 1,12-бензперилена, в то время как нафталин оставался на неизменной позиции.
Известно, что в спектре флуоресценции пирена присутствуют два максимума: 394 нм (мономера) и 475 нм (эксимера-димера, состоящего из возбужденной и невозбужденной молекулы пирена). Дальнейшие исследования [50] показали, что аналитически значимой величиной является отношение интенсивностей данных максимумов. При разрыхлении мембраны вероятность образования эксимера увеличивается, что приводит к пропорциональному увеличению интенсивности соответствующего максимума. В работе [51] показано, например, что связывание липосомальных мембран с некоторыми противоартимическими средствами (этмозин, ксантиверин) сопровождается снижением микровязкости бислоя. Однако в той же работе отмечается и недостаток пирена как метки – зачастую сигналы от мономера или эксимера перекрываются с сигналами от самих лекарств, что делает расчет отношения недостоверным.
Перспективным является использование интенсивно флуоресцирующих меток на основе борфторидных комплексов дипирролилметена BODIPY (рис. 9). Для более эффективного встраивания подобных меток в липосомы часто получают липидоподобные молекулы, в которых присутствует фрагмент BODIPY. Значительных успехов в синтезе подобных молекул достигла лаборатория проф. Е.Л. Водовозовой. Так, была получена липидоподобная метка [52], обладающая высоким ε = 90 000 и встраивающаяся в гидрофобную часть бислоя. Применение подобных меток значительно упрощает проведение анализа, например, при изучении белок-мембранных взаимодействий [52], однако следует учесть ряд особенностей такого подхода. Метка предоставляет информацию о своем ближайшем микроокружении, в то время как для комплексов липосом с полиэлектролитами часто наблюдается микрофазовое разделение; сведения в таком случае могут быть сложными для анализа. Более того, для надежного встраивания метки в бислой часть требуется ее липофильная модификация; без этого возможно перемещение метки по бислою в процессе, например, фазового перехода.
2.2.2. Флуоресцентная корреляционная спектроскопия
Метод ФКС основан на том, что диффузия флуоресцентных молекул вызывает флуктуации в интенсивности флуоресценции в малом элементе объема (0.1–0.5 фемталитр), который создается в конфокальном микроскопе (рис. 10). Данный метод позволяет получать информацию о локальной концентрации биомолекул, а также следить за агрегацией, за взаимодействием с лигандами, за трансляционной диффузией и внутренней подвижностью флуоресцентно меченных биополимеров. Измеряемой величиной является интенсивность излучения F(t) от нескольких флуоресцентных объектов в освещенном лазерном объеме. Флуктуация интенсивности обусловлена броуновским движением флуоресцентных объектов в освещенном лазерном объеме. Время, за которое данный объект пересекает освещенный объем связано с коэффициентом диффузии D [53]. Для расчета D, а в дальнейшем и гидродинамического радиуса анализируют автокорреляционную функцию G(t) (5):
(5)
$G\left( t \right) = ~\frac{{{{{\left\langle I \right\rangle }}^{2}} + \left\langle {\delta I\left( t \right)\delta I\left( {t + \tau } \right)} \right\rangle }}{{{{{\left\langle I \right\rangle }}^{2}}}},$Рис. 10.
Принцип метода флуоресцентной корреляционной спектроскопии. (а) Сфокусированный лазерный луч освещает фиксированный малый объем измеряемой системы (1 фемтолитр). (б) Измерение флуктуации интенсивности флуоресценции в объеме детекции (0.1–0.5 фемтолитр) как функция от времени. (в) Автокорелляционный анализ интенсивности позволяет определять среднее число частиц в объеме детекциии время и трансляционной диффузии исследуемых флуоресцентных молекул. (г) Статистический анализ распределения частиц по молекулярной яркости (число детектируемых флуоресцентных фотонов в пересчете на молекулу в секунду).
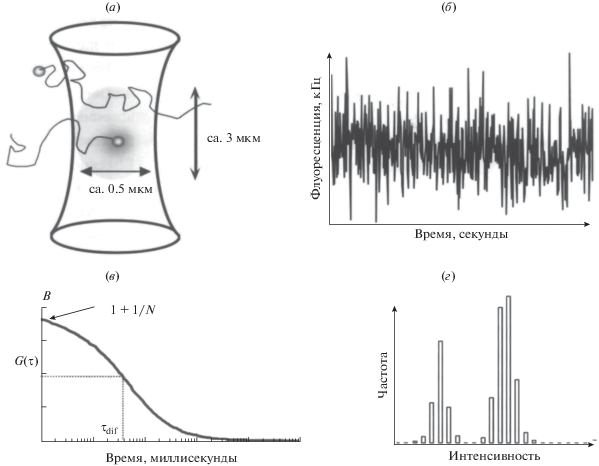
Из анализа автокорреляционной функции G(t) определяют время трансляционной диффузии, τd, которое характеризует время пребывания молекулы в рассматриваемом элементе объема (6):
(6)
$G\left( \tau \right) = \frac{1}{{N\left( {1 + {\tau \mathord{\left/ {\vphantom {\tau {{{\tau }_{d}}}}} \right. \kern-0em} {{{\tau }_{d}}}}} \right){{{\left( {1 + \left( {{\tau \mathord{\left/ {\vphantom {\tau {{{a}^{2}}{{\tau }_{d}}}}} \right. \kern-0em} {{{a}^{2}}{{\tau }_{d}}}}} \right)} \right)}}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}.$Метод относят к методам детекции одиночных молекул, поскольку он позволяет работать с минимальными концентрациями флуорофора (единичные молекулы в освещенном объеме).
Особенно информативен метод ФКС, если само лекарство является флуорофором. Например, в работе [54] рассмотрено взаимодействие хорошо изученного противогрибкового препарата сангвинарина, для которого недавно обнаружили противоопухолевую активность, с липидным бислоем. Коэффициент диффузии сангвинарина, включенного в липосомы (63.8 ± 3 мкм2 с–1), в 7 раз ниже оного для лекарства в буферном растворе (9.07 ± 0.42 мкм2 с–1). Напротив, введение бета-циклодекстрина, олигосахарида, способного взаимодействовать с бислоем, приводило к увеличению D почти в 5 раз (25.76 ± 1.21 мкм2 с–1). Повреждение липосомальной мембраны под действием бета-циклодекстрина приводит к существенно большей подвижности лекарственной молекулы, что отражается на сигнале ФКС.
Если лекарство не является флуорофором, можно ввести соответствующую метку, причем ее концентрация может быть минимальной в виду того, что метод ФКС может быть отнесен к методам детекции единичных молекул. По изменению D судят о вязкости липосом; существенное изменение указывает на размягчение или наоборот затвердевание бислоя.
Большое количество работ в последние годы посвящено методу ФКС для анализа взаимодействия липосомальных лекарственных форм с белками плазмы крови. Например, в работе [55] рассмотрено поведение липосомальной формы флуоресцентно меченого ди-β-D-галактопиранозида в присутствии сывороточного альбумина, трансферрина, аполипопротеи на А1 и фибриногена. Авторы использовали ФКС для отслеживания высвобождения содержимого липосом (для этого во внешний раствор при проведении экспериментов по высвобождению вводили галактозидазу, которая расщепляла связь между флуоресцеином и галактопиранозидом, после чего содержание свободного флуоресцеина определяли по ФКС) и расчета констант диссоциации комплекса с белками. Для этого необходимо было использовать белки, меченные красителем Алекса 488, которые титруют липосомальной суспензией. Связывание с белками приводит к изменению сигнала ФКС; математическая обработка данных может позволить даже рассчитать константы диссоциации комплексов.
2.2.3. Метод разрешенно-временной анизотропии
Метод разрешенно-временной анизотропии – относительно новый микроспектроскопический метод, широко используемый для исследования структуры биомолекул и их комплексов в растворах. Он позволяет следить за вращательной динамикой как комплекса биомолекул, так и каждого из фрагментов, обладающих независимым вращением, и в итоге получать детальную информацию о пространственной структуре липосомальных систем [53, 56, 58].
В РВФА эксперименте определяется величина флуоресцентной анизотропии как функция от времени [57]. Анизотропия сферических молекул, вращение которых симметрично (изотропно), затухает согласно моноэкспоненциальному закону, в общем случае “затухание” анизотропии описывается мультиэкспоненциальной зависимостью (7):
(7)
$r\left( t \right) = \sum {{{\beta }_{j}}} {\text{exp}}\left( { - {t \mathord{\left/ {\vphantom {t {{{\varphi }_{j}}}}} \right. \kern-0em} {{{\varphi }_{j}}}}} \right),$Так как анизотропия r(t) вычисляется по разности между наблюдаемыми компонентами эмиссии флуоресценции I||(t) и I⊥(t), надежные значения анизотропии r(t) могут быть вычислены только для времен, при которых наблюдается значительное испускание флуоресценции. По этой причине обычно в качестве метки выбирают флуорофоры со временем затухания, сравнимым с предполагаемым корреляционным временем вращения. Если флуоресцентное время жизни τ много меньше φ, то интенсивность флуоресценции затухает раньше, чем произойдет заметная потеря анизотропии.
В биосистемах существует несколько причин потери анизотропии (или деполяризации): быстрое движение флуоресцентной метки относительно места ее прикрепления, более медленное внутреннее движение сегментов биополимера, медленное вращение всей молекулы биополимера, очень медленное вращение надмолекулярного комплекса, а также, перенос энергии между хромофорами. При взаимодействии липосомальных форм лекарственных средств с белками, например, белками крови, спад анизотропии принимает сложный вид: появляются дополнительные источники деполяризации, связанные с вращением всего комплекса и/или его отдельных сегментов, что отражается в появлении соответствующих корреляционных времен вращения.
Метод РВФА применим для исследования как гомогенных, так и микрогетерогенных и гетерогенных систем [53, 58]. Рассмотрим несколько примеров исследования надмолекулярных структур на основе липосом методом РВФА.
В работе [59] на примере липосом, меченых 1,6-дифенил-1,3,5-гексатриеном, изучали поведение противогрибкового препарата итраконазола. Комбинацией флуоресцентных методов и метода молекулярной динамики авторы показали, что активная молекула способна вызывать упорядочивание бислоя, уменьшая подвижность гидрофобных цепей.
Методом РВФА было исследовано взаимодействие поверхностного белка из бактериофага М13 с малыми моноламеллярными везикулами димиристоилфосфатидилхолин/димиристоилфосфатидовая кислота (80/20 по массе). За флуоресценцией следили по триптофановому остатку белка [60]. Спад анизотропии характеризуется двумя корреляционными временами вращения. Короткое корреляционное время ϕ1 (0.5 нс) отражает быстрый деполяризационный процесс внутри фрагмента белка, и долгое ϕ2 (более 20 нс) соответствует вращению всей липосомальной системы, содержащей белок (ϕбелок + липосома). Корреляционное время вращения, соответствующее свободному вращению белка в системе отсутствует, что указывают на полное связывание белка с липосомой.
В работе [61] с применением методов РВФА и ФКС исследовали изменение состояния липидного бислоя при окислении жирных кислот. С использованием флуоресцентного аналога фосфолипида, который способен включаться в липидный бислой BP-C11 определяли изменение плотности упаковки липидов (по молекулярной подвижности метки в бислое – через корреляционные времена вращения) в процессе окисления ненасыщенных жирных кислот (арахидоновой кислоты) в модельном фосфолипиде. Обнаружено, что при окислении арахидоновой кислоты липидный бислой становится существенно менее подвижным. Аналогичный вывод был получен из анализа данных ФКС с использованием флуоресцентной метки монопиренил-фосфатидилхолин, где наблюдалось замедление латеральной диффузии при окислении липидов в той же липосомальной системе. В итоге определен молекулярный механизм изменения структуры бислоя при перекисном окислении липидов биомембран.
2.2.4. ЭПР-спектроскопия
Метод электронного парамагнитного резонанса дает ценную информацию о природе, основном состоянии парамагнитных центров и об их ближайшем окружении. К парамагнитным центрам можно отнести атомы и молекулы с нечетным числом электронов, ионы, имеющие частично заполненные внутренние электронные оболочки, свободные радикалы и точечные дефекты в твердых телах.
Использование ЭПР-спектроскопии для изучения биосистем ограничено, поскольку естественных парамагнитных центров в подобных объектах мало. Говоря о диагностике взаимодействия липосомальных мембран с лекарствами, целесообразно рассмотреть виды спин-меток, предназначенных для введения в толщу бислоя. Тогда информация, полученная из ЭПР-спектра метки, может быть использована для анализа текучести мембраны и обнаружения в ней дефектов.
Структура ЭПР-спектра сложна, однако современные методы математического моделирования позволяют рассчитывать предполагаемые спектры. Взаимодействие электрона только с внешним магнитным полем приводит к одиночной линии в спектре ЭПР, следует учитывать взаимодействие магнитного момента электрона с магнитным моментом расположенных поблизости ядер, так называемое сверхтонкое взаимодействие (СТВ), приводящее к дополнительному расщеплению спектральных линий парамагнитных систем в магнитном поле (так называемое сверхтонкое расщепление – СТР).
Время корреляции вращательной диффузии и константы СТВ являются основными количественными параметрами в спектрах меток-репортеров для липосом.
Неочевидным ограничением применимости метода ЭПР-спектроскопии для анализа липосомальных систем является следующий технический факт. Наиболее распространены спектрометры ЭПР, регистрирующие сигнал от порошковых образцов, однако корректная трактовка подобных результатов для липосомальных систем затруднена; необходимо искать способы регистрации спектров от жидких образцов. Существуют специальные ячейки на основе капилляров, например, система AquaX компании Bruker, требующая небольшой объем пробы (18–30 мкл на см капиллярной системы), однако подобные установки встречаются крайне редко. Вероятно, этот факт объясняет относительно редкое использование такого ценного метода как ЭПР-спектроскопия для анализа липосомальных систем.
Метки чаще всего содержат нитроксильный радикал (рис. 11). Хорошо изучено поведение иминонитроксильных радикалов в липидном бислое: считается, что они вращаются по типу волчков с аксиальной симметрией [62].
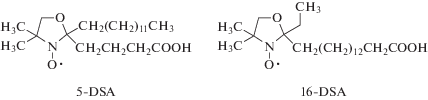
“Излюбленными” спин-метками в изучении липосомальных систем стали 5-доксил-стеариновая кислота (5-ДСК) и 16-доксил-стеариновая кислота (16-ДСК), поскольку в совокупности информация от этих двух меток хорошо описывает приполярный (5-ДСК) и гидрофобный участки бислоя (16-ДСК). Изменение вязкости мембраны можно оценить по изменению величины СТР 2Amax (рис. 12) в спектрах липосом, меченных 5-ДСК, поскольку оно отражает вращательную подвижность молекул липидов. Известно, что 2Amax увеличивается при понижении вязкости мембраны [63]. Из спектров 16-ДСК меченных липосом высчитывают время вращательной корреляции τ из параметров спектра W0, h0 и h–1 (рис. 12) по уравнению (8):
(8)
$\tau = \left( {6.5 \times {{{10}}^{{ - 10}}}} \right){{W}_{0}}\left[ {{{{\left( {\frac{{{{h}_{0}}}}{{{{h}_{{ - 1}}}}}} \right)}}^{{0.5}}} - 1} \right].$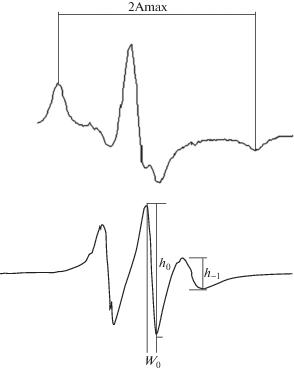
Увеличение времени вращательной корреляции также указывает на уменьшение вязкости бислоя.
Анализ обоих этих параметров позволил доказать, что встраивание паклитаксела в ДПФХ липосомы вызывает увеличение вязкости бислоя [63], что может быть использовано не только для создания новых лекарственных форм, но и для более глубокого понимания механизма взаимодействия активных молекул с мембраной.
Меткой могут быть ионы металлов, например, меди [64], причем ионы металлов вводятся на стадии диспергирования тонкой липидной пленки. Изменение интенсивности сигнала ЭПР спектра и расчетные значения спин-гамильтониана позволили точнее установить структуру липосомального препарата CPX-351, содержащего цитарабин и даунорубицин: ионы меди в параллельной плоскости связываются с первой активной молекулой, а в перпендикулярной – со второй. При этом обнаружено, что даунорубицин образует комплексы с медью в стехиометрии 1 : 1 или 2 : 1.
ЗАКЛЮЧЕНИЕ
Механизм взаимодействия лекарств с биомембраной – важнейший вопрос, который стоит при разработке новых биосовместимых форм лекарственных препаратов с улучшенными биофармацевтическими характеристиками. В первую очередь это обусловлено тем, что значительная доля активных молекул, в частности, противоспалительные нестероидные средства и большинство антибиотиков, имеют значительные побочные эффекты, включая фототоксичность, нарушения гемодинамики, тромбозы, гепатотоксичность и нейротоксичность. В ряде случаев механизм развития побочных эффектов обусловлен природой самого лекарства, которое, помимо воздействия на мишень в организме, вызывает неспецифические изменения в клеточной мембране. Такой эффект обнаружен, например, для нестероидных противовоспалительных средств [35]: взаимодействие активных молекул с ацильными цепями вызывает разрыхление мембраны и образование в ней дефектов. Для антибактериальных препаратов класса фторхинолонов на примере анионных липосом смешанного состава показано, что моксифлоксацин вызывает перераспределение липидов в бислое и образование гексагональных мезафаз. Подобные изменения могут приводить к нарушению целостности клеточной мембраны, увеличение тромбогенности и прочим побочным эффектам.
Описанные в представленном обзоре методы позволяют глубоко исследовать механизмы взаимодействия лекарственных веществ с липидным бислоем и выявлять тонкую структуру липосомальных формуляций активных молекул в зависимости от их химической природы и состава липидной матрицы. Однако каждый метод в отдельности зачастую не предоставляют полной картины происходящего процесса. Поэтому актуальным является вопрос комбинации методов. В табл. 4 приведено краткое резюме по информации, которую можно получить от того или иного метода. Обращают на себя внимание наиболее информативные методы: ИК-спектроскопия и группа флуоресцентных методов. Данные методы обеспечивают широкий спектр возможностей при исследовании состояния биомембраны при фазовых переходах, при взаимодействии липидного бислоя с лекарственными субстанциями. При изучении надмолекулярных ансамблей на основе липосом исключительно информативны методы ФКС и РВФА, которые позволяют получать детальную информацию о пространственной структуре и молекулярной подвижности как всего комплекса на основе липосом, так и его отдельных компонентов. Комбинация их с методами, предоставляющими информацию о морфологии везикул, позволяет получить полную картину взаимодействия активной молекулы с бислоем. Можно ожидать, что в ближайшем будущем область применения рассматриваемых в обзоре методов в первую очередь ИК-спектроскопии и флуоресцентных для изучения биологических объектов будет расширяться, поскольку с усовершенствованием технического оснащения станут доступными принципиально новые приложения этих методов, например, возможность слежения за динамикой отдельных везикул в растворе.
Таблица 4.
Сравнение методов исследования механизма взаимодействия липосом с лекарственными препаратами
| Методы | Подвижность цепей | Температура фазового перехода | Морфология | Электростатическое взаимодействие | Взаимодействие с белками плазмы крови | Морфология кристаллов в липосоме |
|---|---|---|---|---|---|---|
| ИК-спектроскопия | + | + | + | + | ||
| Флуоресцентные методы | + | + | + | |||
| ЭПР-спектроскопия | + | |||||
| ДСК | + | + | ||||
| ММРР | + | |||||
| Метод дифракции излучения лазера на свободных электронах | + | |||||
| Методы на чипе | + | + | ||||
| Методы микроскопии | + | + |
Список литературы
Manaia E.B. et al. // Int. J. Nanomedicine. 2017. V. 12. P. 4991–5011.
Kinuta M., Takei K. // Cell Struct. Funct. 2002. V. 27. № 2. P. 63–69.
Куликов К.Г., Кошлан Т.В. // Журнал технической физики. 2015. V. 85. № 12. P. 26–32.
Yaroslavov A.A. et al. // Colloid J. 2011. V. 73. № 3. P. 430–435.
Sánchez-Purrà M. et al. // Int. J. Pharm. 2016. V. 511. № 2. P. 946–956.
Kinuta M. et al. // Proc. Natl. Acad. Sci. U. S. A. 2002. V. 99. № 5. P. 2842–2847.
Deygen I.M., Kudryashova E.V. // Russ. J. Bioorganic Chem. 2014. V. 40. № 5. P. 547–557.
Eremenko A.V. et al. // Electroanalysis. 2012. V. 24. № 3. P. 573–580.
Deygen I.M., Kudryashova E.V. // Colloids Surfaces B Biointerfaces. 2016. V. 141. P. 36–43.
Filipe V., Hawe A., Jiskoot W. // Pharm. Res. 2010. V. 27. № 5. P. 796–810.
Le-Deygen I.M. et al. // Nanomedicine Nanotechnology, Biol. Med. Elsevier Inc., 2019. V. 21. P. 102 065.
Shin M. et al. // Mol. Pharm. 2018. V. 15. № 3. P. 721–728.
González-Rodríguez M.L., Rabasco A.M. // Expert Opin. Drug Deliv. 2011. V. 8. № 7. P. 857–871.
Yaroslavov A. A. et al. // Adv. Colloid Interface Sci. 2008. V. 142. № 1–2. P. 43–52.
Smith M.C. et al. // Anal. Bioanal. Chem. Analytical and Bioanalytical Chemistry. 2017. V. 409. № 24. P. 5779–5787.
Kim M.W., Niidome T., Lee R. // Mar. Drugs. 2019. V. 17. № 10. P. 581–593.
Singh S. // Int. J. Nanomedicine. 2018. V. 13. P. 11–13.
Le-Deygen I.M. et al. // Chem. Phys. Lipids. 2020. V. 228. P. 104 891.
Rouf M.A. et al. // J. Liposome Res. 2009. V. 19. № 4. P. 322–331.
Robson R.J., Dennis E.A. // J. Phys. Chem. 1977. V. 81. № 11. P. 1075–1078.
Biltonen R.L., Lichrenberg D. // Chem. Physicis Lipids. 1993. V. 64. P. 129–142.
Bilge D. et al. // Spectrochim. Acta. A. Mol. Biomol. Spectrosc. Elsevier B.V., 2014. V. 130. P. 250–256.
Di Foggia M. et al. // J. Therm. Anal. Calorim. 2017. V. 127. № 2. P. 1407–1417.
Wei X. et al. // Mol. Pharm. 2017. V. 14. № 12. P. 4339–4345.
Li T. et al. // J. Control. Release. 2018. V. 288. № July. P. 96–110.
Mady M.M. et al. // Cell Biochem. Biophys. 2012. V. 62. № 3. P. 481–486.
Perinelli D.R. et al. // Int. J. Pharm. Elsevier. 2017. V. 534. № 1–2. P. 81–88.
Pereira-Leite C. et al. // J. Phys. Chem. B. 2012. V. 116. № 46. P. 13 608–13 617.
Cipolla D. et al. // Pharm. Res. Pharmaceutical Research. 2016. V. 33. № 11. P. 2748–2762.
Cipolla D. et al. // RSC Adv. 2016. V. 6. № 8. P. 6223–6233.
Feng L. et al. // Biomaterials. 2018. V. 181. P. 81–91.
Kleinschmidt J.H. // Lipid-Protein Interactions: Methods and Protocols / New York: Humana Press, 2013. 464 p.
Toyran N., Severcan F. // J. Mol. Struct. 2007. V. 839. № 1–3. P. 19–27.
Deygen I.M. et al. // Langmuir. 2016. V. 32. № 42. P. 10 861–10 869.
Manrique-Moreno M. et al. // Biochim. Biophys. Acta. 2009. V. 1788. № 6. P. 1296–1303.
Lewis R.N. et al. // Biophys. J. 1994. V. 67. № 6. P. 2367–2375.
Manrique-Moreno M. et al. // Lett. Drug Des. Discov. 2009. V. 7. № 1. P. 50–56.
Kuć M. et al. // Chem. Phys. 2015. V. 458. P. 9–17.
Baird C.L., Courtenay E.S., Myszka D.G. // Anal. Biochem. 2002. V. 310. № 1. P. 93–99.
Ruozi B. et al. // Int. J. Nanomedicine. 2011. V. 6. P. 557–563.
Robson A.L. et al. // Front. Pharmacol. 2018. V. 9. P. 1–8.
Ruozi B. et al. // Eur. J. Pharm. Sci. 2005. V. 25. № 1. P. 81–89.
Takahashi N. et al. // J. Pharm. Sci. American Pharmacists Association. 2018. V. 107. № 2. P. 717–726.
Johnston M.J.W. et al. // J. Liposome Res. 2008. V. 18. № 2. P. 145–157.
Zhigaltsev I.V. et al. // J. Control. Release. 2005. V. 104. № 1. P. 103–111.
Shamrakov D. et al. // Int. J. Pharm. 2018. V. 547. № 1–2. P. 648–655.
Chang W.-H. et al. // Nanoscale. 2018. V. 10. № 6. P. 2820–2824.
Alves A.C. et al. // Sci. Rep. 2017. V. 7. № 1. P. 1–11.
Lianos P., Mukhopadhyay A.K., Georghiou S. // Photochem. Photobiol. 1980. V. 32. № 3. P. 415–419.
Macdonald A.G. et al. // Biochim. Biophys. Acta. 1988. V. 938. P. 231–242.
Береговая Е.Г. и др. // Биополимеры и клетка. 1993. Т. 9. № 5. С. 19–26.
Boldyrev I. et al. New BODIPY lipid probes for fluorescence studies of membranes // J. Lipid Res. 2007. V. 48. № 7. P. 1518–1532.
Кудряшова Е.В. // Функционирование и структура белков на поверхностях раздела фаз. Новые методы исследования / Palmarium Academic Publishing AV Akademikerverlag GmbH & Co., 2013. 146 p.
Paul B.K., Ghosh N., Mukherjee S. // Colloids Surfaces B Biointerfaces. 2018. V. 170. P. 36–44.
Mittag J.J. et al. // Eur. J. Pharm. Biopharm. 2017. V. 119. P. 215–223.
Cundall R., Dale R.E. // Time-Resolved Fluorescence Spectroscopy in Biochemistry and Biology / Eds. Cundall R.B., Dale R.E. New York: Plenum Press, 1983. P. 555–605.
Krishnamoorthy G. // J. Biosci. Springer India. 2018. V. 43. № 3. P. 555–567.
Кудряшова Е.В., Гладилин А.К., Левашов А.В. // Успехи биологической химии. 2002. Т. 42. С. 257–294.
Poojari C. et al. // Chem. Phys. Lipids. 2019. V. 223. P. 104 784.
Van Slooten M.L. et al. // J. Pharm. Sci. 2000. V. 89. № 12. P. 1605–1619.
Borst J.W. et al. // Biochim. Biophys. Acta – Mol. Cell Biol. Lipids. 2000. V. 1487. № 1. P. 61–73.
Иванов Л.В., Картель Н.Т. // Reports Natl. Acad. Sci. Ukr. 2012. V. 5. P. 139–145.
Zhao L. et al. // Int. J. Pharm. 2007. V. 338. № 1–2. P. 258–266.
Dicko A. et al. // Int. J. Pharm. 2010. V. 391. № 1–2. P. 248–259.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия