

Биоорганическая химия, 2020, T. 46, № 4, стр. 385-395
Оптимизация твердофазного синтеза инграмона – пептидного антагониста моноцитарного хемотаксического белка 1 (МСР-1) человека
М. В. Сидорова 1, У. С. Дудкина 1, Д. В. Авдеев 1, М. Е. Палькеева 1, А. С. Молокоедов 1, М. В. Овчинников 1, А. А. Азьмуко 1, С. Б. Гречишников 2, Е. В. Кудрявцева 2, В. Н. Бушуев 1, Т. И. Арефьева 1
1 ФГБУ “Национальный медицинский исследовательский центр кардиологии” Минздрава РФ
121552 Москва, 3-я Черепковская ул., 15а, Россия
2 ЗАО “Синтез пептидов”
121552 Москва, 3-я Черепковская ул., 15а, Россия
Поступила в редакцию 20.01.2020
После доработки 29.01.2020
Принята к публикации 31.01.2020
Аннотация
Оптимизирован способ твердофазного синтеза (ТФС) инграмона H-Asp-His-Leu-Asp-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH – пептидного антагониста моноцитарного хемотаксического белка 1 (МСР-1) человека, обладающего противовоспалительным действием. Изучены побочные продукты ТФС этого пептида с использованием Fmoc-методологии, их структура установлена спектроскопией 1Н-ЯМР и масс-спектрометрией и подтверждена встречным синтезом. Найдены методические приемы, позволяющие минимизировать образование примесей в ходе ТФС инграмона. Разработана воспроизводимая методика получения инграмона, позволяющая свести к минимуму образование побочных продуктов – соответствующих [Asi4]-, [D-Asp4]- и [βAsp4]пептидов – в ходе его твердофазного синтеза.
ВВЕДЕНИЕ
Ранее нами было показано, что 12-членный пептидный фрагмент (65–76) моноцитарного хемотаксического белка 1 (МСР-1) человека H-Asp-His-Leu-Asp-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH является его антагонистом [1, 2]. Этот пептид получил название инграмон11 (ингибитор гранулоцитов и моноцитов). В серии экспериментальных работ, выполненных на грызунах и приматах, доказана способность этого пептида подавлять миграцию моноцитов и оказывать противовоспалительное действие [3–5], установлено, что инграмон, меченный технецием (99mTc), накапливается в очаге воспаления [4].
В модели экспериментального рестеноза артерий после ангиопластики ежедневные инъекции инграмона замедляли формирование неоинтимы [6]. Показано, что противовоспалительное действие инграмона реализуется благодаря конкурированию с МСР-1 за связывание с гликозаминогликанами на поверхности клеток и в составе внеклеточного матрикса; т. о. препарат препятствует созданию концентрационного градиента хемокина, необходимого для привлечения моноцитов в очаг воспаления [7, 8]. Результаты доклинического токсикологического исследования показали, что препарат нетоксичен, не обладает мутагенными, тератогенными свойствами, не влияет на репродуктивную функцию, не проявляет аллергизирующих и иммунотоксических свойств. Безопасность инграмона была подтверждена у здоровых добровольцев и в клинических исследованиях, в том числе у пациентов с сердечно-сосудистыми заболеваниями. Противовоспалительная активность инграмона была доказана у пациентов с ИБС, стентированием коронарных артерий: дополнительное к стандартной терапии ежедневное внутривенное введение препарата до и после процедуры ангиопластики препятствовало подъему концентрации в крови белков острой фазы [8, 9].
Данная работа является продолжением наших исследований инграмона и посвящена изучению состава образующихся в ходе его ТФС примесей, поискам путей минимизации побочных процессов и созданию оптимизированной методики его синтеза.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Изучение структуры примесей, образующихся при ТФС инграмона
Для разработки схемы ТФС инграмона за основу была принята его Fmoc-методология. Для блокирования функциональных групп боковых цепей аминокислот были выбраны следующие защиты: Boc – для ε-аминогрупп лизина; But – для карбоксильных групп аспарагиновой кислоты и гидроксильных групп треонина и Trt – для карбоксамидных функций глутамина и имидазольного кольца гистидина. Автоматический ТФС инграмона был проведен по стандартной программе автоматического пептидного синтезатора “Tribute UV” с иcпользованием раствора 2% 4-MePip/2% DBU/ DMF для отщепления Fmoc-защит и метода урониевых солей (TBTU/NMM) для создания амидных связей. На рис. 1а представлен профиль аналитической ВЭЖХ неочищенного продукта ТФС. Наиболее существенные примеси – соединения (II) и (III) на рис. 1а, были выделены и проанализированы с помощью масс-спектрометрии и 1Н-ЯМР-спектроскопии. Было установлено, что оба примесных соединения являются продуктами побочных реакций по остатку Asp4, вещество, соответствующее пику II, имело корректную для целевого продукта молекулярную массу (1411.8 Да) и, следовательно, являлось либо оптическим, либо стерическим его изомером. На основании данных спектроскопии 1Н-ЯМР, представленных в табл. 1, можно предполагать, что это соединение является диастереомерным [D-Asp4]инграмоном, т.к. наиболее заметные изменения наблюдаются у сигнала протона при хиральном α-углеродном атоме остатка Asp4: в инграмоне – α-СН (4.58 м.д.), в побочном продукте – при 4.68 м.д. и амидного протона остатка Lys5: в инграмоне – NH (7.79 м.д.), в побочном продукте – 8.08 м.д.
Рис. 1.
Аналитическая ВЭЖХ (условия 1, см. эксперим. ч.) продуктов синтеза инграмона (I): а – неочищенный продукт ТФС, полученный с использованием для отщепления Fmoc-защит смеси 2% DBU/ 2% 4-MePip/DMF; б – неочищенный продукт, полученный с использованием деблокирующей смеси 25% 4-МеPip/DMF/0.1 M HOBt; в – искусственная смесь образцов целевого (I) и побочных продуктов (II)–(IV); г – пептид (I), очищенный с помощью препаративной ВЭЖХ (см. эксперимент. ч.) (условия 1). Детекция при 220 нм.
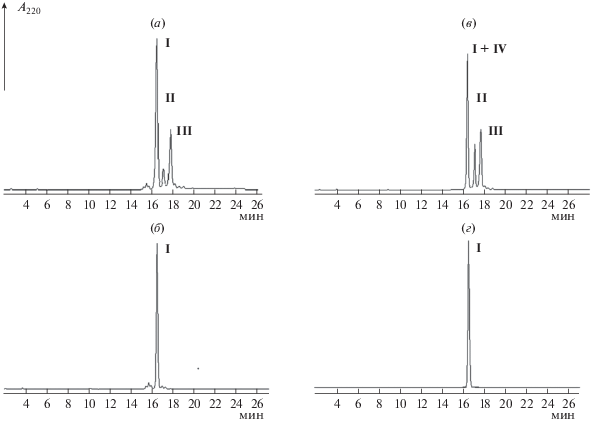
Таблица 1.
Химические сдвиги (δ, м.д.) сигналов протонов инграмона и побочных продуктов (II)–(IV), образующихся в ходе его ТФС, в DMSO-d6 при 300 K
| Остаток | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Инграмон (I) | [D-Asp4]инграмон (II) | [Asi4]инграмон (III) | [βAsp4]инграмон (IV) | |||||||||||||||||
| HN | Hα | Hβ | Hγ | Hδ | HN | Hα | Hβ | Hγ | Hδ | HN | Hα | Hβ | Hγ | Hδ | HN | Hα | Hβ | Hγ | Hδ | |
| Asp1 | 8.16 | 4.13 | 2.80; 2.68 |
8.16 | 4.13 | 2.80; 2.68 |
8.18 | 4.13 | 2.80; 2.70 |
8.16 | 4.13 | 2.80; 2.68 |
||||||||
| His2 | 8.73 | 4.66 | 3.10; 3.02 |
8.73 | 4.66 | 3.08; 2.99 |
8.75 | 4.68 | 3.12; 1.98 |
8.74 | 4.65 | 3.09; 3.00 |
||||||||
| Leu3 | 8.18 | 4.31 | 1.45 | 1.58 | 0.87; 0.84 |
8.15 | 4.32 | 1.44 | 1.56 | 0.86; 0.83 |
8.22 | 4.28 | 1.48 | 1.56 | 0.87; 0.84 |
8.15 | 4.31 | 1.49 | 1.63 | 0.88; 0.84 |
| Asp4/ D-Asp4/Asi4/ βAsp4 | 8.43 | 4.58 | 2.72; 2.52 |
8.46 | 4.63 | 2.68; 2.52 |
8.80 | 4.49 | 3.05; 2.68 |
8.365 | 4.54 | 2.66; 2.57 |
||||||||
| Lys5 | 7.79 | 4.25 | 1.635; | 1.28; | 1.52 | 8.08 | 4.30 | 1.68; | 1.29 | 1.52 | – | 4.49 | 2.04; | 1.29 | 1.49 | 8.17 | 4.266 | 1.65; | 1.29 | |
| 1.50 | 1.52 | 1.53 | 1.83 | 1.53 | ||||||||||||||||
| Gln6 | 8.14 | 4.31 | 1.90; 1.78 |
2.14 | 8.27 | 4.29 | 1.92; 1.79 |
2.15 | 8.05 | 4.27 | 1.86; 1.74 | 2.10 | 8.17 | 4.31 | 1.73; 1.42 |
2.14 | ||||
| Thr7 | 7.81 | 4.22 | 4.01 | 1.02 | 7.78 | 4.22 | 4.01 | 1.02 | 7.87 | 4.20 | 4.01 | 1.02 | 7.78 | 4.22 | 4.09; 4.03 |
1.02 | ||||
| Gln8 | 7.87 | 4.38 | 1.87; 1.72 |
2.10 | 7.87 | 4.375 | 1.84; 1.78 |
2.10 | 7.87 | 4.38 | 1.84; 1.72 | 2.10 | 7.86 | 4.37 | 1.88; 1.70 |
2.11 | ||||
| Thr9 | 8.05 | 4.37 | 3.84 | 1.13 | 8.08 | 4.36 | 3.83 | 1.13 | 8.04 | 4.37 | 3.84 | 1.12 | 8.03 | 4.36 | 3.83 | 1.13 | ||||
| Pro10 | – | 4.37 | 2.03 | 1.92; 1.84 |
3.72; 3.62 |
– | 4.37 | 2.03 | 1.90; 1.84 |
3.72; 3.64 |
– | 4.37 | 2.03; 1.85 | 1.93; 1.84 |
3.72; 3.64 |
– | 4.37 | 2.02 | 1.90; 1.84 |
3.77; 3.62 |
| Lys11 | 8.06 | 4.32 | 1.70; 1.54 |
1.36 | 1.52 | 8.05 | 4.32 | 1.71; 1.54 |
1.36 | 1.52 | 8.07 | 4.32 | 1.70; 1.51 |
1.36 | 8.06 | 4.32 | 1.70 | 1.36 | 1.52 | |
| Thr12 | 7.65 | 4.18 | 4.14 | 1.04 | 7.65 | 4.18 | 4.14 | 1.04 | 7.65 | 4.18 | 4.14 | 1.04 | 7.65 | 4.18 | 4.14 | 1.04 | ||||
Соединение, соответствующее пику III (рис. 1а), имело молекулярную массу (1393.5 Да) на 18 Да меньше, чем у целевого инграмона. По всей вероятности, пептид (III) является продуктом дегидратации. В спектре 1Н-ЯМР этого вещества (см. табл. 1) присутствуют сигналы протонов всех аминокислотных остатков, входящих в состав инграмона. Однако химические сдвиги сигналов ряда остатков побочного продукта отличаются от их значений в спектре целевого вещества (I). В первую очередь это относится ко всем сигналам остатков Asp4 и Lys5. Так, сигналы Asp4 в спектре инграмона – α-СН (4.58 м.д.), β-СН2 (2.52; 2.72 м.д.) и NH (8.43 м.д.), в побочном продукте наблюдаются соответственно при 4.49 для α-СН, 2.68; 3.05 для β‑СН2 и 8.80 м.д. для NH. Столь значительные различия (особенно сигналов протонов при β-углеродном атоме) можно объяснить модификацией боковой цепи остатка аспарагиновой кислоты. Сигналы протонов остатка Lys5 также сильно отличаются: в инграмоне – α-СН (4.25 м.д.), β-СН2 (1.50; 1.635 м.д.) и NH (7.79 м.д.), в побочном продукте наблюдаются – α-СН (4.49 м.д.), β-СН2 (1.83; 2.04 м.д.), сигнал амидного протона в спектре вещества (III) отсутствует. Отсутствие сигнала этого протона свидетельствует об образовании циклического продукта с участием α-аминогруппы остатка Lys5. На основании данных масс-спектрометрии и 1Н-ЯМР-спектроскопии было сделано предположение, что соединение (III) представляет собой аспартимид – промежуточный циклический продукт α → β-перегруппировки амидной связи, образуемой карбоксильной группой остатка Asp4. В результате щелочного гидролиза полученного вещества (III) (1% раствором гидроокиси аммония при рН 9.0 в течение 16 ч) получаются 2 продукта (рис. 2а), один из которых по ВЭЖХ совпадает с инграмоном, а второй, вероятно, представляет собой побочный изоаспартилпептид ([βAsp4]инграмон) (IV). Известно, что при синтезе аспартилпептидов на стадиях отщепления Fmoc-защит действием вторичных аминов происходит образование циклического аспартимида, результатом чего является α → β-перегруппировка амидной связи с образованием изомерного побочного продукта [10]. Образование аспартимида, катализируемое кислотой или основанием, является одной из наиболее серьезных побочных реакций в ТФС пептидов, содержащих аспарагиновую кислоту, и до сих пор не разработана экономически эффективная стратегия ее подавления [11, 12]. На рис. 3 схематически представлена α → β-перегруппировка амидной связи, образуемой карбоксильной группой остатка Asp4 в последовательности инграмона. Степень протекания транспептидации главным образом определяется природой соседнего с аспарагиновой аминокислотой остатка, но зависит также и от конкретной аминокислотной последовательности пептида. Надо отметить, что последовательность Asp-Lys, судя по литературным данным [12], не является “критической” с точки зрения возможности протекания этой побочной реакции. В обзоре [12] показано, что при использовании Fmoc-методологии наиболее подвержены транспептидации последовательности Asp-Gly и Asp-Asn [12]. Однако мы применяли для деблокирования α-аминогрупп сильное основание – DBU, которое может ускорить побочный процесс образования аспартимида [12, 13].
Рис. 2.
Аналитическая ВЭЖХ (условия 2, см. эксперимент. ч.): а – продуктов щелочного гидролиза [Asi4]инграмона (1% NH4OH, pH 9.0, 16 ч); б – искусственной смеси образцов инграмона (I) и [βAsp4]инграмона (IV); в – пептид (I), очищенный с помощью препаративной ВЭЖХ (см. эксперимент. часть).
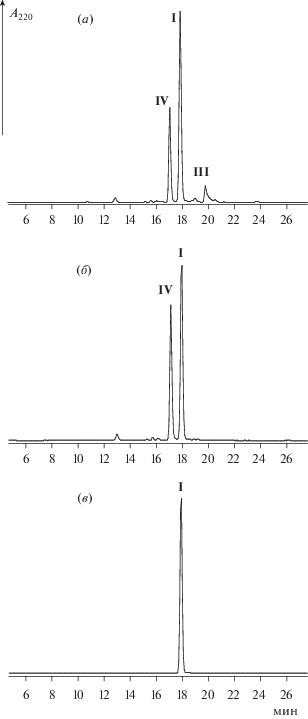
Рис. 3.
α → β-Перегруппировка амидной связи, образуемой карбоксильной группой остатка Asp4 в последовательности инграмона H-Asp-His-Leu-Asp-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH.
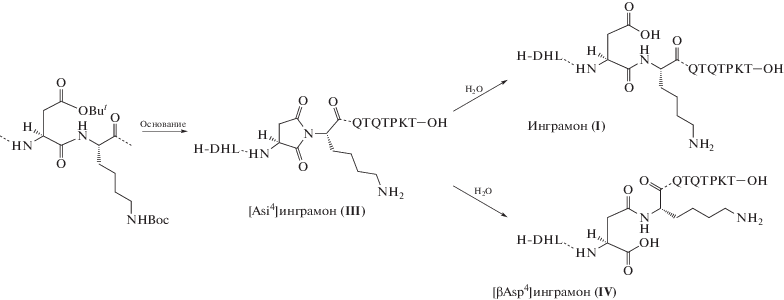
Следующим этапом работы было подтверждение структуры примесей, образующихся при ТФС инграмона. Для этого был проведен направленный ТФС [D-Asp4]инграмона (II) с использованием производного Fmoc-D-Asp(OBut)-OH. Также был синтезирован [βAsp4]инграмон (IV) c использованием соответствующего производного с незамещенной β-карбоксильной группой Fmoc-Asp-OBut. Изоаспартилпептид (IV) был необходим для идентификации продуктов гидролиза побочного [Asi4]инграмона (III) и ВЭЖХ-анализа продуктов ТФС. Полученные пептиды (II) и (IV) были очищены с использованием ВЭЖХ, их характеристики приведены в табл. 2. Сравнение соединения (II) с примесью, выделенной из пика II реакционной смеси синтеза инграмона, показало их идентичность (данные аналитической ВЭЖХ, 1H-ЯМР и MС). На рис. 1в приведен профиль ВЭЖХ искусственной смеси инграмона (I) и примесей: [D-Asp4]- (II), [Asi4]- (III) и [βAsp4]инграмона (IV). Видно, что инграмон (I) и [βAsp4]инграмон (IV) имеют одинаковое время удерживания при рН 3.0, а диастереомеры по остатку Asp4 – соединения (I) и (II) – удается разделить.
Таблица 2.
Характеристики пептидов
| Пептид | Последовательность (Мол. масса) | Чистота по ВЭЖХ, % | ВЭЖХ*, Rt (мин) | МC (MALDI), m/z | |
|---|---|---|---|---|---|
| рН 3.0 | рН 5.95 | ||||
| (I) | H-Asp-His-Leu-Asp-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH Инграмон (М 1411.52) |
99 | 16.82 | 17.90 | 1411.826 [M + H]+ 1433.858 [M + Na]+ 1449.854 [M + K]+ |
| (II) | H-Asp-His-Leu-D-Asp-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH [D-Asp4]Инграмон (М 1411.52) |
95 | 17.35 | 17 .90 | 1411.787 [M + H]+ 1433.814 [M + Na]+ 1449.807 [M + K]+ |
| (III) | H-Asp-His-Leu-Asi-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH [Asi4]Инграмон (М 1393.52) |
95 | 18.04 | 19.72 | 1393.371 [M + H]+ 1455.334 [M + Na + K]+ |
| (IV) | H-Asp-His-Leu-Asp(Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH)-OH [βAsp4]Инграмон (М 1411.52) |
96 | 16.82 | 17.07 | 1411.845 [M + H]+ 1433.808 [M + Na]+ 1449.800 [M + K]+ |
Для анализа качества целевого пептида было очень важно подобрать условия ВЭЖХ, в которых изомерные пептиды с α- и β-амидной связью, образуемой карбоксильной группой остатка Asp4, имеют разное время удерживания (делятся). Оказалось, что при повышении значения рН подвижной фазы в ВЭЖХ до 5.95 соединения (I) и (IV) удается разделить (см. рис. 2б). Следует отметить, что при рН 5.95 не происходит разделения диастереоизомерных пептидов (I) и (II). Поэтому для анализа качества инграмона необходимо проводить ВЭЖХ при разных значениях рН подвижной фазы. Сравнение продуктов гидролиза [Asi4]инграмона с продуктами встречного синтеза пептидов (I) и (IV) подтвердило наше предположение о том, что в ходе ТФС инграмона при деблокировании α-аминогрупп раствором 2%DBU/2% 4-MePip/DMF происходит α → β-перегруппировка амидной связи в последовательности Asp4-Lys5 с образованием циклического аспартимида (III).
Таким образом, были идентифицированы наиболее существенные примеси, образующиеся при ТФС инграмона с использованием Fmoc-методологии, и их структура была подтверждена независимыми физико-химическими методами и встречным синтезом. Следующей задачей была оптимизация методики получения инграмона.
Оптимизация методики синтеза инграмона
Для разработки оптимальной методики было изучено влияние различных факторов на состав неочищенного продукта ТФС. Нами был получен октапептидилполимер H-Lys(Boc)-Gln(Trt)-Thr(But)-Gln(Trt)-Thr(But)-Pro-Lys(Boc)-Thr(But)-полимер (V), образец которого был полностью деблокирован для оценки его чистоты c помощью ВЭЖХ. Содержание основного вещества в этом продукте составляло не менее 90%. В дальнейшем аликвоты октапептидилполимера (V) были использованы в качестве исходных соединений в серии тестовых синтезов инграмона, в которых меняли реагенты для отщепления Fmoc-группы и способ создания амидной связи.
Из литературы известны различные методические приемы подавления транспептидации: используют объемные, стерически затрудненные защитные группы для β-карбоксильной функции аспарагиновой кислоты; полимерные носители со стерически затрудненными якорными группами; модифицированные реагенты для отщепления Fmoc-защит, сокращают суммарное время воздействия основных реагентов [12, 14]. В первую очередь было изучено влияние состава деблокирующего реагента на качество неочищенного продукта ТФС, результаты этих опытов приведены в табл. 3.
Таблица 3.
Влияние реагента для отщепления Fmoc-защит на состав неочищенного продукта ТФС инграмона на полимере Ванга
| Деблокирующий реагент | Время деблокиро-вания, мин (Σ*, мин) | Конденсирующий реагент | Содержание инграмона (I) в неочищенном продукте по ВЭЖХ**, % | Содержание примесей по ВЭЖХ, % | |
|---|---|---|---|---|---|
| [D-Asp4]инграмон (II) | [Asi4]инграмон (III) | ||||
| 2% 4-МеPip/2% DBU/ DMF | 10 (40) | TBTU/NMM | 46.80 | 5.90 | 21.70 |
| 25% 4-МеPip/DMF | 15 (60) | DCC/HOBt | 77.78 | 0.68 | 2.67 |
| 25% 4-МеPip/DMF/ 0.1 M HOBt | 15 (60) | DCC/HOBt | 84.20 | 0.52 | 0.60 |
Представленные в табл. 3 данные показывают, что использование сильного основания – DBU – в составе смеси для отщепления Fmoc-защит приводит к образованию более 20% побочного аспартимида, отсутствие DBU в деблокирующем реагенте практически на порядок снижает содержание аспартимида (до 2.67%), а добавка 1-гидроксибензотриазола к 25% раствору 4-метилпиперидина [14] в DMF минимизирует протекание этого побочного процесса (содержание аспартимида (III) в этом случае колеблется от 0.6 до 0.9%). Вариация суммарного времени деблокирования в пределах 40–60 мин в наших экспериментах не отражалось на результатах синтеза.
В серии экспериментов, проводившихся с использованием оптимального деблокирующего реагента, было изучено влияние способа создания амидных связей и используемого полимерного носителя на качество неочищенного инграмона. Результаты этих опытов приведены в табл. 4 и 5.
Таблица 4.
Влияние способа создания амидной связи на состав неочищенного продукта ТФС инграмона при использовании 25% 4-МеPip/DMF/0.1 M HOBt для отщепления Fmoc-защит
| Масштаб ТФС, ммоль стартовой аминокислоты |
Конденсирующий агент | Содержание инграмона (I) в неочищенном продукте по ВЭЖХ, % | Содержание примесей по ВЭЖХ, % | |
|---|---|---|---|---|
| [D-Asp4]инграмон (II) | [Asi4]инграмон (III) | |||
| 0.5 | TBTU/NMM | 74.5 | 0.89 | 0.72 |
| 0.5 | DIC/HOBt | 84.2 | 0.52 | 0.60 |
| 9.0 | DIC/HOBt | 83.7 | 1.16 | 0.87 |
Таблица 5.
Влияние природы полимерного носителя на состав неочищенного продукта ТФС инграмона в масштабе 0.5 ммоль при использовании смеси 25% 4-МеPip/DMF/0.1 M HOBt для отщепления Fmoc-защит и DIC/HOBt-метода для создания амидных связей
| Полимерный носитель | Содержание стартовой аминокислоты, моль/г | Содержание инграмона (I) в неочищенном продукте по ВЭЖХ, % | Содержание примесей по ВЭЖХ, % | Выход*, % | |
|---|---|---|---|---|---|
| [D-Asp4]инграмон (II) | [Asi4]инграмон (III) | ||||
| Полимер Ванга | 0.50 | 84.20 | 0.52 | 0.60 | 48.7 |
| 2-СТС-полимер | 0.46 | 85.51 | 0.30 | 0.32 | 54.2 |
Из данных, представленных в табл. 4, следует, что при ступенчатом наращивании пептидной цепи лучшие результаты получаются при использовании карбодиимидного метода, применение солей урония в реакциях пептидообразования дает более высокий выход диастереомерного продукта – [D-Asp4]инграмона (II). Однако при 20-кратном увеличении масштаба синтеза даже при использовании смеси DCC/HOBt содержание примеси диастереомерного пептида (II) растет. Видимо, это связано с увеличением времени, необходимого для проведения отдельных процедур ТФС при увеличении масштаба синтеза.
Данные, приведенные в табл. 5, показывают, что при использовании оптимальных деблокирующего и конденсирующего реагентов результаты ТФС инграмона на стандартном полимере Ванга с 4-гидроксиметилфеноксиметильной якорной группой и полимере с объемной 2-хлортритилхлоридной (2-CTC) якорной группой сравнимы с точки зрения содержания целевого пептида (I), однако содержание примесей (II) и (III) в последнем случае почти вдвое меньше. Выход пептида (I) при синтезе на 2-СТС-полимере также несколько выше – 54.2%. При тест-синтезе на полимере Ванга выход пептида (I) составлял 48.7%. Этот результат согласуется с литературными данными [12] о том, что в некоторых синтезах использование 2-СТС-полимера обеспечивало снижение образования побочного аспартимида.
В результате проведенных исследований
‒ разработана методика ТФС инграмона, согласно которой использование модифицированного реагента для отщепления Fmoc-защит (раствора 25% 4-МеPip/DMF с добавкой 0.1 M HOBt), карбодиимидного способа создания пептидных связей (DIC/HOBt) и полимерного носителя с 2‑хлортритилхлоридной якорной группой обеспечивают получение целевого пептида с хорошей чистотой и низким содержанием побочных продуктов транспептидации и рацемизации по остатку Asp4 (рис. 1г и 2в);
‒ идентифицированы примеси родственных инграмону пептидов, образование которых возможно в процессе его ТФС;
‒ разработана методика контроля качества продуктов ТФС инграмона, предполагающая обязательное использование при его ВЭЖХ буферов с различными значениями рН.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали производные аминокислот (Novabiochem, ФРГ), DMF, NMM, HOBt, TBTU, TIS, дихлорметан и трифторуксусную кислоту (Fluka, Швейцария). Для ВЭЖХ применяли ацетонитрил фирмы “Carl Roth GmbH” (ФРГ).
Аналитическую ВЭЖХ проводили на хроматографе Knauer (ФРГ) на колонке (4.6 × 250 мм) Kromasil 100-5 C18 (Швеция), размер частиц сорбента 5 мкм, размер пор порядка 100А. В качестве элюентов использовали: буфер А – 0.05 М КН2РО4, pH 3.0 (условия 1) или 0.05 М КН2РО4, pH 5.95 (условия 2); в условиях 1 и 2 буфер Б – 70% ацетонитрил в буфере А, элюцию проводили со скоростью 1 мл/мин в градиенте концентрации буфера Б в буфере А от 0 до 40% за 40 мин. Детекция – при длине волны 220 нм. Препаративную ВЭЖХ пептидов (II)–(IV) осуществляли на колонке Kromasil 100-10-С18 (30 × 250 мм) с размером частиц сорбента 10 мкм. В качестве элюентов использовали буфер А – 0.1% водный раствор TFA, содержащий 3% ацетонитрила, буфер Б – 80% ацетонитрила в буфере А. Элюцию проводили от 100% буфера А в градиенте концентрации 0.5%/мин буфера Б со скоростью 20 мл/мин. ВЭЖХ пептида (I) выполняли на колонках Kromasil 100-10-С18 (30 × 250 мм) или (50 × 250 мм) с размером частиц сорбента 10 мкм. В качестве элюентов использовали буфер А – 0.01 М водный раствор ацетата аммония с рН 4.5, содержащий 3% ацетонитрила, буфер Б – 80% ацетонитрила в буфере А. Элюцию проводили от 100% буфера А в градиенте концентрации 0.5% мин буфера Б со скоростью 50 мл/мин. Пептиды детектировали при длине волны 220 нм. Фракции, соответствующие целевому веществу, объединяли, концентрировали в вакууме и лиофилизовали.
1Н-ЯМР-спектры регистрировали на спектрометре WH-500 Bruker 500 МГц (ФРГ) в DMSO-d6 при 300 K, концентрация пептидов составляла 2–3 мг/мл, химические сдвиги (δ, м.д.) измеряли относительно тетраметилсилана. Отнесение сигналов к определенным группам протонов аминокислотных остатков проводили с помощью метода дифференциального двойного резонанса. Масс-спектры регистрировали на масс-спектрометре Bruker Autoflex speed (Bruker Daltonics Inc., ФРГ), оснащенном твердотельным УФ-лазером с λ 355 нм и рефлектроном, в режиме регистрации положительно заряженных ионов. Для регистрации масс-спектров MALDI использовали стальную мишень MTP 384 ground steel. Частота облучения 50 Гц, при регистрации масс-спектра суммировались данные, полученные при 10 последовательных облучениях.
Твердофазный синтез пептидов
Автоматический синтез пептидов H-Asp-His-Leu-D-Asp-Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH (II) и H-Asp-His-Leu-Asp(Lys-Gln-Thr-Gln-Thr-Pro-Lys-Thr-OH)-OH (IV) проводили на синтезаторе Tribute-UV (Protein Technologies, Inc., США) в масштабе 0.5 ммоль в соответствии с программой однократной конденсации Fmoc-аминокислот. Пептидную цепь наращивали с С-конца по одной аминокислоте, исходя из Fmoc-Thr(But)-полимера Ванга (Novabiochem, ФРГ), содержащего 0.50 ммоль/г стартовой аминокислоты. Цикл ТФС включал следующие стадии: 1) промывки DMF Fmoc-аминоацил- или пептидилполимера; 2) деблокирование α-аминогрупп 25% 4-МеPip/ DMF/0.1 M HOBt (2 × 5 мин), 3) конденсация 0.4 М раствора 4-кратного избытка ацилирующего агента (2.0 ммоль Fmoc-аминокислоты + 2.0 ммоль HOBt + + 2.0 ммоль DIC) в DMF в течение 1 ч; 4) промывки DMF. Пептиды отщепляли от полимерного носителя действием смеси TFA/TIS/H2O (90 : 5 : 5 v/v) в течение 1.5 ч при комнатной температуре. Полимер отфильтровывали, промывали на фильтре деблокирующей смесью, фильтрат упаривали, продукт осаждали сухим диэтиловым эфиром. Осадок отфильтровывали, промывали эфиром, DCM, эфиром и высушивали. Пептиды очищали с помощью ВЭЖХ, как описано выше. Выходы пептидов (II) и (IV) составили 48.2 и 45.0% соответственно, в расчете на стартовую аминокислоту. Их характеристики приведены в табл. 1 и 2.
ТФС на полимере Ванга
Синтез пептида (I) проводили в ручном режиме, исходя из 22 г (11.0 ммоль) Fmoc-Thr(But)-полимера Ванга (содержание стартовой аминокислоты – 0.5 ммоль/г).
Получение октапептидилполимера Fmoc-Lys(Boc)- Gln(Trt)-Thr(But)-Gln(Trt)-Thr(But)-Pro-Lys(Boc)-Thr(But)-полимера (V). Цикл ТФС включал следующие стадии: 1) промывки Fmoc-аминоацил-/пептидилполимера DMF; 2) деблокирование α‑аминогрупп 25% 4-МеPip/DMF (5 + + 10 мин), 3) конденсация c 0.4 М раствором 4-кратного избытка ацилирующего агента (44.0 ммоль Fmoc-аминокислоты + 44.0 ммоль HOBt + 44.0 ммоль DIC) в DMF в течение 1.5 ч; 4) промывки DMF, 5) тест с нингидрином на остаточные аминогруппы [15]. Для оценки качества промежуточного октапептида образец Nα –свободного пептидил-полимера (V) был обработан деблокирующей смесью TFA/TIS/H2O (90 : 5 : 5 v/v) в течение 1 ч и после осаждения диэтиловым эфиром продукт анализировали ВЭЖХ в условиях 1. Содержание основного вещества в образце составило 91%. Аликвоты пептидилполимера (V), содержание пептида в которых составляло ≈0.5 ммоль, использовали для тест-синтезов инграмона (I), условия и результаты которых представлены в табл. 3–5. Основную часть октапептидилполимера (V) с содержанием пептида ≈9.0 ммоль использовали для получения целевого продукта (I). Цикл ТФС включал следующие стадии: 1) промывки Fmoc-аминоацил-/пептидилполимера DMF; 2) деблокирование α-аминогрупп 25% 4-МеPip/DMF/ 0.1 M HOBt (5 + 10 мин), 3) конденсация 0.4 М раствора 4-кратного избытка ацилирующего агента (36.0 ммоль Fmoc-аминокислоты + 36.0 ммоль HOBt + 36.0 ммоль DIC) в DMF в течение 1.5 ч; 4) промывки DMF, 5) тест с нингидрином на остаточные аминогруппы. После заключительного отщепления Fmoc-группы и промывок пептидилполимер высушивали и деблокировали действием 350 мл смеси TFA/TIS/H2O (90 : 5 : 5 v/v) в течение 1.5 ч при комнатной температуре. Полимер отфильтровывали, промывали на фильтре деблокирующей смесью, фильтрат упаривали, продукт осаждали сухим диэтиловым эфиром. Осадок отфильтровывали, промывали эфиром, DCM, эфиром и высушивали. Пептид (I) очищали с помощью ВЭЖХ, как описано выше.
Получено 7.2 г инграмона-ацетата (выход 46.6% в расчете на стартовую аминокислоту), содержание уксусной кислоты, определенное ВЭЖХ, составило 4.0%, содержание воды, определенное методом К. Фишера, – 4.7%. Масс-спектр: найдено m/z 1411.8, вычислено для C59H98N18O22 М – 1411.52. Спектр 1Н-ЯМР см. табл. 1.
ТФС на 2-СТС-полимере
Автоматический синтез пептида (I) проводили на синтезаторе Tribute-UV с использованием полимерного носителя (Iris Biotech, ФРГ) с 2-хлортритилхлоридной (2-СТС) якорной группой, содержащего 1.2 экв. Cl/г 2-CTC-полимера. Для присоединения стартовой аминокислоты [16] к 1.5 г (1.8 ммоль) 2-CTC-полимера прибавляли раствор 1.43 г (3.6 ммоль) Fmoc-Thr(But)-OH в 15 мл DCM, в полученную суспензию приливали 1.53 мл (9.0 ммоль) DIEA и перемешивали 30 мин при 25°С, полимер отфильтровывали и промывали DCM 2 × 15 мл, DMF 2 × 15 мл, DCM 2 × 15 мл. Для отщепления остаточного хлора аминоацилполимер обрабатывали 15 мл смеси DCM–MeOH–DIEA (80 : 15 : 5 v/v) 2 × 10 мин, промывали DCM 4 × 15 мл, высушивали и сразу использовали. Содержание стартовой аминокислоты, определенное спектрофотометрически, составило 0.46 ммоль Fmoc-Thr(But)/г аминоацилполимера. ТФС пептида (I) проводили, исходя из 1.1 г (0.5 ммоль) аминоацилполимера. Цикл ТФС включал те же стадии, что и при получении пептидов (II) и (IV): 1) промывки Fmoc-аминоацил- или пептидилполимера DMF; 2) деблокирование α-аминогрупп 25% 4‑МеPip/ DMF/0.1 M HOBt (2 × 5 мин), 3) конденсация 0.4 М раствора 4-кратного избытка ацилирующего агента (2.0 ммоль Fmoc-аминокислоты + + 2.0 ммоль HOBt + 2.0 ммоль DIC) в DMF в течение 1 ч; 4) промывки DMF. Пептид отщепляли от полимерного носителя действием смеси TFA/TIS/H2O (90 : 5 : : 5 v/v) в течение 1.5 ч при комнатной температуре и очищали с помощью ВЭЖХ, как описано выше. Выход пептида (I) составил 54.2% в расчете на стартовую аминокислоту. Масс-спектр: найдено m/z 1411.9, вычислено для C59H98N18O22 М – 1411.52. Спектр 1Н-ЯМР см. табл. 1.
ЗАКЛЮЧЕНИЕ
На сегодняшний день доказана способность инграмона подавлять миграцию моноцитов и оказывать противовоспалительное действие как в экспериментальных, так и в клинических исследованиях (у здоровых добровольцев и больных с ишемической болезнью сердца). Возможность использования этого пептида для терапии сердечно-сосудистых заболеваний диктует необходимость разработки оптимизированных способов его синтеза. Оптимальная методика синтеза инграмона, позволяющая получать этот пептид с низким содержанием родственных примесей, предполагает использование для отщепления Fmoc-защит раствора 25% 4-МеPip/DMF с добавкой 0.1 M HOBt, карбодиимидного способа создания пептидных связей и полимерного носителя с 2-хлортритилхлоридной якорной группой. Разработанная методика воспроизводится при масштабировании синтеза инграмона. Эти результаты являются принципиально важными для организации в ФГБУ “НМИЦ кардиологии” Минздрава России экспериментального производства инграмона и его запланированных клинических испытаний.
Список литературы
Чазов Е.И., Сидорова М.В., Молокоедов А.С. Арефьева Т.И., Кухтина Н.Б., Красникова Т.Л., Беспалова Ж.Д., Бушуев В.Н. Пептид, обладающий способностью ингибировать миграцию моноцитарных клеток, стимулированную белком МСР-1. Патент РФ № 2260598. Заявл. 29.10.2003. Опубл. 20.09.2005. Бюл. № 26.
Сидорова М.В., Молокоедов А.С., Арефьева Т.И., Кухтина Н.Б., Красникова Т.Л., Беспалова Ж.Д., Бушуев В.Н. // Биоорганич. химия. 2004. Т. 30. С. 582–593. [Sidorova M.V., Molokoedov A.S., Aref’eva T.I., Kukhtina N.B., Krasnikova T.L., Bespalova Zh.D., Bushuev V.N. // Russ. J. Bioorg. Chem. 2004. V. 30. P. 523–533.]
Красникова Т.Л., Арефьева Т.И., Мелехов М. Г., Кухтина Н. Б., Сидорова М.В., Молокоедов А.С., Бушуев В.Н., Беспалова Ж.Д., Чазов Е.И. // ДАН. 2005. Т. 404. С. 551–554.
Чазов Е.И., Красникова Т.Л., Беспалова Ж.Д., Кухтина Н.Б., Мелехов М.Г., Арефьева Т.И., Сидорова М.В., Молокоедов А.С., Гвоздик Т.Е., Мартьянов Б.М., Поздеев В.В., Сергиенко В.Б., Бушуева Т.Л. // ДАН. 2006. Т. 411. С. 270–272.
Chazov E.I., Bespalova J.D., Arefieva T.I., Kukhtina N.B., Sidorova M.V., Provatorov S.I., Krasnikova T.L. // Can. J. Physiol. Pharmacol. 2007. V. 85. P. 332–340.
Кухтина Н.Б., Баштрыков П.П., Беспалова Ж.Д., Сидорова М.В., Арефьева Т.И., Соколов В.О., Красникова Т.Л. // Рос. физиолог. журн. им. И.М. Сеченова. 2008. Т.94. С. 27–37. [Kukhtina N.B., Bashtrykov P.P., Bespalova Zh. D., Sidorova M.V., Aref’eva T.I., Sokolov V.O., Krasnikova T.L. // Neurosci. and Behav. Physi. 2009. V. 39. P. 153–159.]
Красникова Т.Л., Никитин П.И., Ксеневич Т.И., Горшков Б.Г., Орлов А.В., Сидорова М.В., Азьмуко А.А., Арефьева Т.И., Мамочкина Е.Н., Ефремов Е.Е., Беспалова Ж.Д. // ДАН. 2010. Т. 433. С. 559–562.
Arefieva T.I., Krasnikova T.L., Potekhina A.V., Ruleva N.U., Nikitin P.I., Ksenevich T.I., Gorshkov B.G., Sidorova M.V., Bespalova Zh. D., Kukhtina N. B., Provatorov S.I., Noeva E.A., Chazov E.I. // Inflamm. Res. 2011. V. 60. P. 955–964.
Потехина А.В., Проваторов С.И., Арефьева Т.И., Кухтина Н.Б., Масенко В.П., Казначеева Е.И., Рулева Н.Ю., Ноева Е.А., Осяева М.К., Сидорова М.В., Беспалова Ж.Д., Красникова Т.Л. // Терапевтический архив. 2010. Т. 82. С. 58–63.
Lauer J.L. Fields C.G., Fields G.B. // Lett. Peptide Sci. 1994. V. 1. P. 197–205.
Stathopoulos P., Papas S., Kostidis S., Tsikaris V. // J. Peptide Sci. 2005. V. 11. P. 658–664.
Subirόs-Funosas R., El-Faham A., Albericio F. // Tetrahedron. 2011. V. 67. P. 8585–8593.
Tickler A.K., Barrow C.J., Wade J.D. // J. Peptide Science. 2001. V. 7. P. 488–494.
Michels T., Dölling R., Haberkorn U., Mier W. // Org. Lett. 2012. V. 14(20). P. 5218–5221.
Kaiser E., Colescott R.L., Bossinger C.D., Cook P.I. // Anal. Biochem. 1970. V. 34. P. 595–598.
Barlos K., Gatos D. // Convergent peptide synthesis // Fmoc Solid Phase Peptide Synthesis. A Practical Approach / Eds. Chan W.C., White P.D. Oxford Univ. Press, 2001. P. 215–228.
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия