

Биоорганическая химия, 2021, T. 47, № 6, стр. 775-784
Детекция белка PRAME на поверхности клеток меланомы с помощью флуоресцентно меченого моноклонального антитела
К. А. Сапожникова 1, А. В. Мисюрин 2, Н. Б. Пестов 3, 4, Е. Г. Мелешкина 1, 5, С. Д. Орешков 1, 6, Е. П. Ганжула 1, 7, А. С. Михайлова 1, 8, В. А. Коршун 1, 9, В. А. Мисюрин 10, *, В. А. Брылёв 1, **
1 ФГБУН “Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова” РАН
117997 Москва, ул. Миклухо-Маклая, 16/10, Россия
10 Национальный медицинский исследовательский центр онкологии имени Н.Н. Блохина
115478 Москва, Каширское шоссе, 23, Россия
2 ООО “Генотехнология”
117485 Москва, ул. Профсоюзная, 104, Россия
3 Московский физико-технический институт
141701 Долгопрудный, Институтский пер., 9, Россия
4 Федеральный научный центр исследования и разработки иммунобиологических препаратов им. М.П. Чумакова РАН
108819 Москва, поселение Московский, пос. Института полиомиелита, домовладение 8, корп. 17, Россия
5 Российский химико-технологический университет им. Д.И. Менделеева
125047 Москва, Миусская площадь, 9, Россия
6 ФГБУН “МГУ им. М.В. Ломоносова”, химический факультет
119991 Москва, Ленинские горы, 1/3, Россия
7 ФГБУН “Российский университет дружбы народов”
117198 Москва, ул. Миклухо-Маклая, 6, Россия
8 НИУ “Высшая школа экономики”
117312 Москва, ул. Вавилова, 7, Россия
9 Институт по изысканию новых антибиотиков им. Г.Ф. Гаузе
119021 Москва, ул. Б. Пироговская, 11, Россия
* E-mail: vsevolod.misyurin@gmail.com
** E-mail: v.brylev@yandex.ru
Поступила в редакцию 15.11.2020
После доработки 12.12.2020
Принята к публикации 13.12.2020
Аннотация
Определение уровня экспрессии белка-онкомаркера PRAME позволяет как прогнозировать течение заболевания, так и контролировать эффективность противоопухолевой терапии. Флуоресцентно меченое моноклональное антитело к белку PRAME получено периодатным окислением гликанов с последующей модификацией бифункциональным азидо-оксиаминовым реагентом и “клик”-реакцией с алкиновым производным сульфированного цианинового красителя Cy3. Предложен новый подход к синтезу бифункционального азидо-оксиаминового реагента с использованием этоксиэтилиденовой защитной группы для гидроксиламина. Полученное меченое антитело охарактеризовано спектрофотометрически, определена стехиометрия модификации. Продемонстрированы сохранение аффинности флуоресцентных антител и возможность их применения в качестве диагностического инструмента для определения остаточного маркера (белка PRAME) после проведения противоопухолевой терапии.
ВВЕДЕНИЕ
Раково-тестикулярный белок PRAME (preferentially expressed antigen in melanoma), сверхэкспрессирущийся при ряде онкологических заболеваний (меланоме, раке молочной железы, раке легких, раке почки, нейробластоме и др. [1–4]), нарушает передачу сигнала в ядерных рецепторах ретиноевой кислоты, выступающих лиганд-регулируемыми транскрипционными факторами, участвующими в процессах развития, гомеостаза и дифференцировки клеток [5]. Белок PRAME служит диагностическим и прогностическим маркером [6–11], в том числе при противоопухолевой терапии. Наличие белка PRAME наблюдалось на поверхности лейкозных клеток K562, что дает возможность использовать белок PRAME в качестве мишени для иммунотерапии [1, 9, 12]. Поскольку экспрессия этого белка не ограничивается лейкозами, установление его экспрессии в клетках солидных опухолей позволит разработать новые препараты также и для этих заболеваний.
Моноклональное антитело 6H8 к белку PRAME [13] может быть использовано как для детекции этого белка, так и для разработки терапевтических препаратов. В первую очередь представляла интерес прямая детекция связывания антитела с изучаемым белком, что можно осуществить с помощью проточной цитометрии, если антитела содержат флуоресцентную метку. Поэтому возникла необходимость разработки надежной процедуры флуоресцентного мечения моноклонального антитела 6H8, сохраняющей его аффинность к белку PRAME. В данной статье мы предлагаем использовать периодатное окисление углеводных фрагментов сайта гликозилирования моноклонального антитела 6H8 с последующей модификацией образующихся альдегидных групп бифункциональным азидо-оксиаминовым линкером. Такой метод позволяет вводить в антитела алифатические азидогруппы, по которым можно проводить дальнейшую модификацию антител алкиновыми производными флуоресцентных красителей в условиях Cu(I)-катализируемой “клик”-реакции.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Общий подход к мечению иммуноглобулина. Для флуоресцентного мечения был выбран подход, связанный с модификацией углеводных фрагментов антител. После окисления углеводной части иммуноглобулина 6H8 периодатом натрия [14] молекулу антитела последовательно модифицировали бифункциональным азидо-оксиаминовым реагентом (I) и алкиновым производным сульфированного цианинового красителя Cy3 (II) в условиях биоортогональной “клик”-реакции (схема 1 ). Этот подход основан на широко используемых методах – модификации карбонильных соединений O-алкилгидроксиламинами [15, 16] и Cu(I)-катализируемом циклоприсоединении терминальных алкинов к азидам [17]. Данный способ обладает несколькими преимуществами по сравнению с широко используемыми реакциями модификации амино- и тиольных групп антител. Кроме углеводного сайта другие фрагменты антител не окисляются и не меняют своих функций. Модификация гликана весьма удачна также потому, что сайт гликозилирования находится далеко от антиген-связывающего фрагмента. Таким образом, конъюгация антител с низкомолекулярными соединениями через углеводный участок не должна оказывать значительного влияния на аффинность антител к антигенам.

Схема 1 . Получение флуоресцентно меченого антитела. Схематически изображено окисление L-фукозы в альдегидокислоту [17].
Усовершенствованный метод синтеза бифункционального реагента (I). Синтез азидо-оксиаминового реагента (I) осуществляли из соответствующего азидоспирта (III) с использованием этоксиэтилиденовой защитной группы для гидроксиламина (схема 2 ). Ранее для превращения вещества (III) в реагент (I) использовали Boc- [18] и фталимидные [19, 20] защитные группы гидроксиламина. Мы обнаружили, что более удобной защитной группой служит этоксиэтилиденовая группа. Алкилирование этил-N-гидроксиацетимидата (IV) мезилатами спиртов с последующим легким деблокированием оказалось удобным методом получения соответствующих O-алкилгидроксиламинов [21]. Проведение реакции в смеси трет-бутанола и изопропанола (1 : 1) позволило получить защищенное производное (V) с общим выходом 80% после двух стадий. При проведении реакции алкилирования в метаноле наблюдалось образование значительных количеств простого метилового эфира азидоспирта (III). В результате обработки соединения (V) соляной кислотой в метаноле с последующим упариванием получили соединение (I) в виде гидрохлорида с выходом 92%.
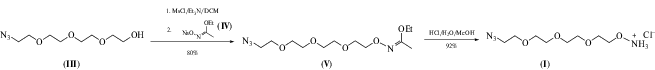
Схема 2 . Синтез реагента (I) с использованием этоксиэтилиденовой защитной группы.
Определение уровня экспрессии гена PRAME. В исследуемых клеточных линиях ген PRAME был стабильно активен, и уровень его экспрессии находился в диапазоне 6000–7000% в клетках линии K562 и 6000–7500% в клетках линии mel P. Таким образом, данные клеточные линии могут быть использованы для определения уровня экспрессии белка PRAME.
Конъюгация моноклонального антитела 6H8 с сульфированным красителем Cy3. Окисление углеводного фрагмента антитела 6H8 проходит довольно мягко в слабокислых условиях (pH 5.0) под действием периодата натрия. Количество образующихся карбонильных групп при окислении углеводных фрагментов антител может контролироваться тремя параметрами – временем, концентрацией периодата и pH реакционной среды. В нашем случае моноклональное антитело 6H8 окислялось 20 мМ раствором периодата натрия в течение 40 мин. Для остановки реакции использовали глицерин, быстро вступающий в реакцию с окислителем. Отделение низкомолекулярных продуктов реакции от окисленного продукта (антитела) удобно проводить с помощью гель-фильтрации на сефадексе G-50. Очищенное окисленное антитело затем может вступать в реакцию с оксиамином (I) также в слабокислых условиях (pH 5.0). Реакция соединения (I) с карбонильными группами антитела 6H8 протекает достаточно быстро (1 ч) и приводит к образованию модифицированного антитела, несущего в своем составе азидные группы, связанные ковалентно посредством тетраэтиленгликольного гидрофильного линкера с сайтом гликозилирования. Очищенное гель-фильтрацией азидированное антитело затем вводили в Cu(I)-катализируемую реакцию азид-алкинового циклоприсоединения [16] с алкиновым производным сульфированного красителя Cy3 (II). Для снижения неспецифического связывания использовали гидрофильный водорастворимый лиганд THPTA, образующий комплекс с Cu(I) и легко отделяющийся с помощью гель-фильтрации. Образующееся Cy3-меченое антитело удобно выделять на сефадексе G-50, поскольку оно окрашено, и протекание хроматографического разделения легко визуализируется.
Дальнейший анализ конъюгата производили спектрофотометрически. Для этого был получен спектр поглощения в УФ- и видимой областях. Поскольку на одну молекулу антитела после конъюгации приходилось несколько остатков красителя, то эти остатки красителя из-за близкого расположения начинали между собой взаимодействовать и образовывать H-агрегаты [22]. Такие взаимодействия отражались на форме спектра поглощения в области 550 нм. При этом агрегации антител не наблюдалось, и раствор конъюгата оставался длительное время стабильным без видимого выпадения в осадок меченого антитела. Для подавления агрегации остатков красителя и получения неискаженного спектра поглощения (рис. 1) был подобран буферный раствор, содержащий 10% додецилсульфата натрия. Далее по соотношению интенсивности полос поглощения при 280 и 550 нм была определена нагрузка N красителя (II) на одну молекулу антитела, которая составила величину 6, т.е. на одну молекулу иммуноглобулина приходилось шесть остатков молекул сульфированного красителя Cy3.
Рис. 1.
Спектр поглощения конъюгата антитела с сульфированным красителем Cy3 и определение стехиометрии (нагрузки, N) модификации антитела красителем.

Валидация антител, меченных сульфо-Cy3. Для определения способности антитела 6H8, меченного красителем сульфо-Cy3, распознавать белок PRAME использовали клетки линии K562. Антитела обнаруживались в цитоплазме более чем у 98% пермеабилизованных клеток (рис. 2). Эти же антитела использовали для окрашивания цитоплазмы клеток mel P, причем локализация меченого антитела 6H8 в цитоплазме также была зафиксирована более чем у 98% пермеабилизованных клеток (рис. 3). Таким образом, модифицированные антитела сохранили способность связываться с нативным белком PRAME, находящимся в цитоплазме клеток. Клетки меланомы мыши не окрашивались.
Рис. 2.
Пример окрашивания клеток K562 антителом 6H8, меченным сульфо-Cy3: (а) – неокрашенные клетки; (б) – окрашенные клетки.

Рис. 3.
Пример окрашивания клеток mel P антителом 6H8, меченным сульфо-Cy3: (а) – неокрашенные клетки; (б) – окрашенные клетки.

Окрашивание поверхности клеток линий K562 и mel P. Антитела 6H8, меченные красителем (II), связывались с поверхностью непермеабилизованных клеток линий K562 и mel P. Интенсивность связывания была очень низкой и практически не визуализировалась на скейтерограммах, однако связывание было доказано по изменению интенсивности флуоресценции клеток, выраженной в безразмерных единицах (рис. 4).
Рис. 4.
Интенсивность флуоресценции клеток K562 и mel P, окрашенных сульфо-Cy3-меченым антителом 6H8. Антитело добавляли в концентрациях 2000, 500 и 125 пг на пробу.

Интенсивность окрашивания снижалась при разведении антител. Таким образом, антитела связываются с поверхностью PRAME-экспрессирующих непермеабилизованных клеток линий K562 и mel P, что свидетельствует о наличии белка PRAME на мембране данных клеток.
Далее мы определили интенсивность флуоресценции CD20-экспрессирующих клеток хронического B-клеточного лимфоидного лейкоза, окрашенных модифицированными антителами, распознающими антиген CD20. Аномальность этих клеток была доказана при выявлении на их поверхности экспрессии антигенов CD22, CD19, CD5 и CD23, а также антителами к легкой цепи иммуноглобулина [23]. Интенсивность флуоресценции неокрашенных лейкозных B-клеток составила 183 ед. Окрашенные лимфоциты обладали интенсивностью флуоресценции на уровне 261 ед. Это позволяет сделать вывод, что известный антиген CD20, использующийся в качестве мишени, обладает сопоставимым с белком PRAME уровнем экспрессии на клеточной мембране.
Мы наблюдали еще одну особенность, которую нужно учитывать при определении поверхностной локализации PRAME – возможность принять цитоплазматический белок за мембранный. Это может произойти в случае повреждения мембраны погибших клеток во время центрифугирования при пробоподготовке. В этом случае цитоплазма, богатая белком PRAME, окрашивается, и такие клетки имеют интенсивный флуоресцентный сигнал (рис. 5).
Рис. 5.
Пример окрашивания клеток K562 антителом 6H8, меченным сульфо-Cy3. (а) – Окрашивание клеток 7aad; (б) – окрашивание 7aad-негативных клеток антителом 6H8; (в) – окрашивание 7aad-позитивных клеток антителом 6H8.

Избежать подобного можно при использовании ДНК-тропных интеркалирующих красителей. В нашем исследовании применялся краситель 7-аминоактиномицин D (7-aad), у которого пик эмиссии в спектре флуоресценции находится при 647 нм и отличается от пика эмиссии красителя сульфо-Cy3 (550 нм). Применение красителя 7-aad показало, что, действительно, все клетки, дающие интенсивный сигнал при окрашивании Cy3-меченым антителом 6H8, окрашивались 7-aad, что свидетельствует о проницаемости их мембран и опосредованного этим окрашивания цитоплазматического белка PRAME. Таким образом, при оценке активности PRAME на поверхности опухолевых клеток мы рекомендуем всегда использовать интеркалирующий краситель для исключения ложноположительного сигнала.
Проблема может быть также решена посредством создания антител, распознающих только мембранную форму белка PRAME, но не цитоплазматическую. Примером такого решения служит антиген CD3, мембранная экспрессия которого выявляется клоном антител HIT3a. При этом клон антител HIT3a не связывается с цитоплазматической детерминантой антигена CD3, которая может быть выявлена другим клоном – UCHT1. На настоящий момент, однако, неизвестно, каким образом лишенный трансмембранного домена белок PRAME удерживается на клеточной мембране, и невозможно определить аминокислотные последовательности белка, к которым можно разработать антитела для выявления мембранной локализации.
Помимо привлечения клеток-эффекторов лекарственные анти-PRAME-антитела могут быть использованы как средства доставки токсинов к опухоли. Данный способ терапии окажется особенно эффективным, если белок PRAME интернализуется после связывания с субстратом. К сожалению, в настоящее время потенциальная возможность использования мембранной формы белка PRAME в качестве мишени при терапии остается неисследованной.
Известно, что количество белков на поверхности клеток может быть различным. Чем больше это количество, тем более эффективным будет использование терапевтических антител. В работе Uckun et al. [24] установлено, что количество антигена CD19 на поверхности B-клеток составляет ~1.9 × 106 на клетку. Минимальное количество антигена, достаточное для развития иммунного ответа при помощи немодифицированных антител, составляет ~1 × 104 на клетку, а для коротких антител, связывающих T-лимфоциты с клетками-мишенями, составляет ~1 × 103 на клетку. Чтобы показать перспективность использования белка PRAME в качестве мишени, необходимо установить типичное количество данного белка, находящегося на мембране клеток. Согласно нашим наблюдениям, это количество небольшое, что может оказаться ограничением для разработки методов PRAME-направленной терапии. С другой стороны, значительное количество белка-мишени на поверхности клетки также может создавать трудности, поскольку каждая клетка будет захватывать очень много антител, и это потребует применения большой концентрации препарата [25].
Обнадеживающим результатом стало определение активности антигена CD20 на поверхности лейкозных клеток больного B-клеточным хроническим лимфоидным лейкозом. Оказалось, что активность белка PRAME и антигена CD20 на опухолевых клетках наблюдается на сопоставимом уровне. Поскольку CD20 – известная мишень для эффективного лекарственного препарата ритуксимаб, а также других, более продвинутых лекарственных препаратов на основе антител, мы предполагаем, что подобного эффекта можно добиться при использовании анти-PRAME-антител.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Общие методы. Для синтеза модифицирующего реагента (I) использованы реактивы: хлористый метилен и триэтиламин (Химмед, Россия), азид натрия и метансульфохлорид (Fluka, Швейцария), тетраэтиленгликоль (Aldrich, США). Метиленхлорид (DCM) перегоняли над CaH2. С остальными растворителями и реагентами работали без дополнительной очистки. Использовали алкиновое производное красителя Cy3 (II) (Lumiprobe, Россия). 11-Азидо-3,6,9-триоксаундеканол (III) [20] и натриевую соль этил-N-гидроксиацетимидата (IV) [21] получали, как описано в литературе. 1H- и 13C-ЯМР-спектры регистрировали на ЯМР-спектрометре Inova 300 МГц (Varian, Германия) с отнесением сигналов по пикам остаточных протонов в DMSO-d6 (2.50 м.д. для 1H и 39.52 м.д. для 13C). КССВ приведены в герцах (Гц) для соответствующих мультиплетов. Масс-спектры получены на спектрометре Q Exactive HF Hybrid Quadrupole-Orbitrap (Thermo Scientific, США). Тонкослойную хроматографию выполняли на алюминиевых пластинах TLC Silica gel 60 F254 (Merck, Германия).
Синтез бифункционального азидо-оксиаминового реагента (I). Этил N-(11-Азидо-3,6,9-триоксаундецилокси)-ацетимидат (V). К смеси исходного спирта (III) (200 мг, 0.43 ммоль) и триэтиламина (120 мкл, 0.86 ммоль) в сухом DCM (10 мл) добавляли метансульфонилхлорид (54 мг, 0.47 ммоль). Через 30 мин реакционную массу упаривали, перерастворяли в ТГФ (15 мл) и фильтровали через целлюлозный фильтр. После упаривания растворителя целевое вещество получали в виде желтоватого масла (521 мг, 0.43 ммоль). Его растворяли в смеси изопропилового и трет-бутилового спирта (1 : 1, v/v, 20 мл), затем к полученной смеси порциями прибавляли натриевую соль этил N‑гидроксиацетимидата (269 мг, 2.15 ммоль) при постоянном перемешивании. Реакционную массу кипятили 2 ч. Затем осадок мезилата натрия отфильтровывали, растворитель упаривали в вакууме. Целевое вещество выделяли с помощью колоночной хроматографии на силикагеле. Получали бесцветное масло; выход 105 мг (80%). Rf 0.4 (5% MeOH в DCM).
1H-ЯМР: 4.00–3.93 (м, 4H, N3CH2, N3CH2CH2), 3.77–3.57 (м, 12H, OCH2), 3.43–3.39 (м, 2H, СH2CH3), 1.89 (с, 3H, CH3C), 1.23 (т, J 7.5 Гц, 3H, СH2CH3).
13C-ЯМР: 162.17, 72.85, 70.36, 70.34, 70.31, 70.22, 69.73, 68.98, 62.27, 60.53, 14.67, 13.91.
Масс-спектр, m/z ([M + H]+): рассчитано для C12H25N4${\text{O}}_{5}^{ + }$ 305.1819; найдено 305.1816.
O-(11-Азидо-3,6,9-триоксаундецилокси)гидроксиламин гидрохлорид (I). Исходное соединение (V) (500 мг, 1.5 ммоль) растворяли в метаноле (20 мл). При комнатной температуре к раствору добавляли 37%-ный раствор соляной кислоты (350 мкл, 4.1 ммоль). Через 1 ч реакционную смесь упаривали. Получали бесцветное маслообразное вещество (355 мг, 92%). Rf 0.47 (20% MeOH в EtOAc).
1H-ЯМР: 11.04 (уш. с, 3H, ONH3), 4.16–4.13 (м, 4H, N3CH2, H3NOCH2), 3.65–3.58 (м, 8H, OCH2), 3.41–3.38 (м, 4H, OCH2).
13C-ЯМР: 73.99, 70.28, 70.23, 70.20, 70.17, 69.71, 68.19, 50.53.
Масс-спектр, m/z ([M + H]+): рассчитано для C8H19N4${\text{O}}_{4}^{ + }$ 235.1401; найдено 235.139.
Флуоресцентное мечение моноклонального антитела. Использовали ранее разработанные нами антитела 6H8 [26], распознающие PRAME. Эти антитела были мечены красителем сульфо-Cy3 (II). Концентрацию немеченого антитела определяли спектрофотометрически по поглощению на длине волны 280 нм. Раствор антитела 6H8 (0.50 мл, концентрация 1.7 мг/мл) переводили в ацетатный буфер (20 мМ AcONa, 150 мМ NaCl, pH 5) с помощью гель-фильтрации на колонке NAP-5 (Cytiva, США) по стандартному протоколу производителя. Получили раствор антитела 6H8 (700 мкл) с концентрацией 0.83 мг/мл. К этому раствору добавляли 100 мкл 160 мМ раствора NaIO4. Начальная концентрация периодата натрия в реакционной смеси составляла 20 мМ, начальная концентрация антитела – 0.73 мг/мл. Окисление углеводной части антитела проводили в темноте при комнатной температуре в течение 40 мин. Затем останавливали реакцию добавлением 100 мкл 20%-ного водного раствора глицерина. Полученную реакционную смесь (900 мкл) наносили на гель-фильтрационную колонку NAP-10 (Cytiva, США) и проводили обессоливание по стандартному протоколу в ацетатном буфере (20 мМ AcONa, 150 мМ NaCl, pH 5). Получили раствор окисленного моноклонального антитела (1.1 мл, концентрация 0.47 мг/мл). К аликвоте раствора окисленного антитела (370 мкл) добавляли 70 мМ водный раствор реагента (I) (0.7 мкл). В реакционной смеси начальная концентрация окисленного антитела составляла 3 мкМ, начальная концентрация оксиамина (I) – 150 мкМ. Реакционную смесь выдерживали 1 ч при комнатной температуре в темноте, наносили на гель-фильтрационную колонку с сефадексом G-50 (Pharmacia, Швеция) и элюировали полученный конъюгат натрий-фосфатным буфером. К полученному раствору азидированного антитела добавляли 50-кратный избыток (40 нмоль) алкинового производного сульфированного красителя Cy3 (II). Проводили медь-катализируемую реакцию азид-алкинового циклоприсоединения в присутствии 0.5 мМ эквимолярной каталитической смеси CuSO4-THPTA и 5 мМ аскорбиновой кислоты в течение 3 ч. Реакционную смесь очищали от избытка красителя (II) и низкомолекулярных соединений на сефадексе G-50 в натрий-фосфатном буфере, собирая окрашенную высокомолекулярную фракцию. Концентрацию полученного конъюгата и стехиометрию (нагрузку красителя на одну молекулу антитела) определяли спектрофотометрически по соотношению поглощения при 280 и 550 нм. Спектры поглощения регистрировали в натрий-фосфатном буферном растворе, содержащем 10% додецилсульфата натрия. Степень мечения антитела составляла величину 6.
Окрашивание клеток мечеными антителами. Для окрашивания цитоплазмы клеток K562 и mel P из коллекции клеточных культур НМИЦ онкологии им. Н.Н. Блохина Минздрава России [1, 5, 9] проводили их пермеабилизацию при помощи раствора для определения уровня экспрессии цитоплазматических белков методом проточной цитометрии (Sony Biotechnology, США) по инструкции, предоставленной разработчиком. В качестве отрицательного контроля использовали клетки линии меланомы мыши B16 из коллекции клеточных культур НМИЦ онкологии им. Н.Н. Блохина Минздрава России.
Связывание антител исследовали методом проточной цитометрии на приборе NovoCyte (ACEA Bioscience, США) с конфигурации оптической системы версии (лазеры синий 488 нм и красный 640 нм, детекторы 530/30 FITC, 572/28 PE, 675/30 PerCP/APC и 780/60 PE-Cy7/APC-Cy). Детекцию сигнала от сульфо-Cy3 проводили в канале PE. Окрашивание цитоплазмы клеток применялось для валидации работоспособности антител, меченных сульфо-Cy3. В качестве контроля использовали немеченые антитела 6H8 в сопоставимой концентрации, выявляемые вторичными антителами, распознающими иммуноглобулин мыши (SonyBiotechnology, США). Детекцию ДНК-связывающего красителя 7-aad [27] проводили в канале PE-Cy7.
Экспрессию мРНК гена PRAME проводили методом количественной ПЦР в реальном времени с использованием специфических праймеров и R6G-меченого зонда на приборе LightCycler (Roche, Швейцария), по методике, применявшейся нами ранее [13].
ЗАКЛЮЧЕНИЕ
Предложен новый подход к синтезу бифункционального азидо-оксиаминового реагента с использованием этоксиэтилиденовой защитной группы для гидроксиламина. Для получения флуоресцентно меченого моноклонального антитела 6H8 к белку PRAME проведено периодатное окисление углеводного фрагмента антитела с последующей модификацией бифункциональным азидо-оксиаминовым реагентом и “клик”-реакцией с алкиновым производным сульфированного цианинового красителя Cy3. Флуоресцентно меченое антитело охарактеризовано спектрофотометрически, определена стехиометрия модификации.
Впервые получены данные, подтверждающие наличие белка PRAME на поверхности клеток метастатической меланомы. Мембранная локализация характерна для всех клеток популяции, при этом количество белка PRAME на поверхности каждой из клеток сопоставимо с количеством белка CD20. Подобный уровень экспрессии характерен, например, для антигена CD23 на поверхности активированных B-лимфоцитов [23]. Столь низкий уровень экспрессии белка PRAME объясняет трудности его обнаружения в данном клеточном компартменте, в то время как данных о наличии этого белка в цитоплазме гораздо больше [1, 5, 9].
Мембранная локализация белка PRAME при меланоме также открывает возможность для разработки нового подхода иммунотерапии данного заболевания. Поскольку белок PRAME не представлен в нормальных соматических клетках, но присутствует на клетках меланомы, он может специфически распознаваться лекарственным антителом. Само антитело будет привлекать молекулы системы комплемента либо киллерные клетки, что вызовет лизис клеток меланомы. Для проверки этого предположения необходимо будет разработать гуманизированные антитела к антигену PRAME и провести доклинические исследования.
Список литературы
Wadelin F., Fulton J., McEwan P.A., Spriggs K.A., Emsley J., Heery D.M. // Mol. Cancer. 2010. V. 9. P. 226. https://doi.org/10.1186/1476-4598-9-226
Al-Khadairi G., DEcock J. // Cancers. 2019. V. 11. P. 984. https://doi.org/10.3390/cancers11070984
Wei R., Dean D.C., Thanindratarn P., Hornicek F.J., Guo W., Duan Z. // Cancer Lett. 2020. V. 479. P. 54–60. https://doi.org/10.1016/j.canlet.2019.10.024
Xu Y., Zou R., Wang J., Wang Z., Zhu X. // Cell Proliferat. 2020. V. 53. P. E12770. https://doi.org/10.1111/cpr.12770
Epping M.T., Wang L., Edel M.J., Carlée L., Hernandez M., Bernards R. // Cell. 2005. V. 122. P. 835–847. https://doi.org/10.1016/j.cell.2005.07.003
Field M.G., Decatur C.L., Kurtenbach S., Gezgin G., van der Velden P.A., Jager M.J., Kozak K.N., Harbour J.W. // Clin. Cancer Res. 2016. V. 22. P. 1234–1242. https://doi.org/10.1158/1078-0432.CCR-15-2071
Field M.G., Durante M.A., Decatur C.L., Tarlan B., Oelschlager K.M., Stone J.F., Kuznetsov J., Bowcock A.M., Kurtenbach S., Harbour J.W. // Oncotarget. 2016. V. 7. P. 59209–59219. https://doi.org/10.18632/oncotarget.10962
Gerami P., Yao Z., Polsky D., Jansen B., Busam K., Ho J., Martini M., Ferris L.K. // J. Am. Acad. Dermatol. 2017. V. 76. P. 114–120. https://doi.org/10.1016/j.jaad.2016.07.038
Pankov D., Sjöström L., Kalidindi T., Lee S.-G., Sjöström K., Gardner R., McDevitt M.R., O’Reilly R., Thorek D.L.J., Larson S.M., Veach D., Ulmert D. // Oncotarget. 2017. V. 8. P. 65917–65931. https://doi.org/10.18632/oncotarget.19579
Ferris L.K., Gerami P., Skelsey M.K., Peck G., Hren C., Gorman C., Frumento T., Siegel D.M. // Melanoma Res. 2018. V. 28. P. 478–482. https://doi.org/10.1097/CMR.0000000000000478
Wang W.-L., Gokgoz N., Samman B., Andrulis I.L., Wunder J.S., Demicco E.G. // Modern Pathol. 2020. https://doi.org/10.1038/s41379-020-00687-5
Gerber J.M., Qin L., Kowalski J., Smith B.D., Griffin C.A., Vala M.S., Collector M.I., Perkins B., Zahurak M., Matsui W., Gocke C.D., Sharkis S.J., Levitsky H.I., Jones R.J. // Am. J. Hematol. 2011. V. 86. P. 31–37. https://doi.org/10.1002/ajh.21915
Misyurin V.A., Finashutina Y.P., Turba A.A., Larina M.V., Solopova O.N., Lyzhko N.A., Kesaeva L.A., Kasatkina N.N., Aliev T.K., Misyurin A.V., Kirpichnikov M.P. // Dokl. Biochem. Biophys. 2020. V. 492. P. 135–138. https://doi.org/10.1134/S1607672920030072
Wolfe C.A.C., Hage D.S. // Anal. Biochem. 1995. V. 231. P. 123–130. https://doi.org/10.1006/abio.1995.1511
Kölmel D.K., Kool E.T. // Chem. Rev. 2017. V. 117. P. 10358–10376. https://doi.org/10.1021/acs.chemrev.7b00090
Zuberbühler K., Casi G., Bernardes G.J.L., Neri D. // Chem. Commun. 2012. V. 48. P. 7100–7102. https://doi.org/10.1039/C2CC32412A
Meldal M., Tornøe C.W. // Chem. Rev. 2008. V. 108. P. 2952–3015. https://doi.org/10.1021/cr0783479
Hudak J.E., Barfield R.M., de Hart G.W., Grob P., Nogales E., Bertozzi C.R., Rabuka D. // Angew. Chem. Int. Ed. 2012. V. 51. P. 4161–4165. https://doi.org/10.1002/anie.201108130
Kim C.H., Axup J.Y., Dubrovska A., Kazane S.A., Hutchins B.A., Wold E.D., Smider V.V., Schultz P.G. // J. Am. Chem. Soc. 2012. V. 134. P. 9918–9921. https://doi.org/10.1021/ja303904e
DeForest C.A., Tirrell D.A. // Nat. Mater. 2015. V. 14. P. 523–531. https://doi.org/10.1038/nmat4219
Khomutov M.A., Mandal S., Weisell J., Saxena N., Simonian A.R., Vepsalainen J., Madhubala R., Kochetkov S.N. // Amino Acids. 2010. V. 38. P. 509–517. https://doi.org/10.1007/s00726-009-0410-0
Hestand N.J., Spano F.C. // Chem. Rev. 2018. V. 118. P. 7069–7163. https://doi.org/10.1021/acs.chemrev.7b00581
Barna G., Reiniger L., Tátrai P., Kopper L., Matolcsy A. // Hematol. Oncol. 2008. V. 26. P. 167–170. https://doi.org/10.1002/hon.855
Uckun F.M., Jaszcz W., Ambrus J.L., Fauci A.S., Gajl-Peczalska K., Song C.W., Wick M.R., Myers D.E., Waddick K., Ledbetter J.A. // Blood. 1988. V. 71. P. 13–29. https://doi.org/10.1182/blood.V71.1.13.13
Powroźnik B., Kubowicz P., Pękala E. // Postepy Hig. Med. Dosw. 2012. V. 66. P. 663–673. https://doi.org/10.5604/17322693.1009980
Finashutina Yu.P., Misyurin A.V., Akhlynina T.V., Lyzhko N.A., Krutov A.A., Aksenova E.V., Misyurin V.A., Baryshnikov A.Yu. // Russ. J. Biother. 2015. V. 14. P. 29–36. https://doi.org/10.17650/1726-9784-2015-14-3-29-36
Chen Chiao Y.C., Gurudath Rao K., Hook J.W., Krugh T.R., Sengupta S.K. // Biopolymers. 1979. V. 18. P. 1749–1762. https://doi.org/10.1002/bip.1979.360180712
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия