

Биоорганическая химия, 2022, T. 48, № 2, стр. 129-161
Синтез олигосахаридов, структурно родственных фрагментам гиалуроновой кислоты
А. А. Гринькова 1, Н. Е. Устюжанина 1, *, Н. Э. Нифантьев 1
1 Институт органической химии им. Н.Д. Зелинского РАН
119991 Москва, Ленинский просп., 47, Россия
* E-mail: ustnad@gmail.com
Поступила в редакцию 13.11.2021
После доработки 20.11.2021
Принята к публикации 23.11.2021
- EDN: LAHYEV
- DOI: 10.31857/S0132342322020105
Аннотация
Гиалуроновая кислота (ГК) – природный полисахарид, построенный из чередующихся остатков β-D-глюкуроновой кислоты и N-ацетил-β-D-глюкозамина. ГК входит в состав гликопротеинов и протеогликанов, которые выполняют важные функции в живых организмах. Например, ГК играет ключевую роль в развитии таких процессов, как деление и миграция клеток, формирование сосудов, воспаление, онкогенез. В обзоре рассмотрены реализованные синтезы олигосахаридов, родственных цепям ГК, проанализированы стратегии сборки соответствующих углеводных цепей, а также оценены типы использованных гликозил-доноров и гликозил-акцепторов, рассмотрены стратегии выбора защитных групп в использованных реагентах. Рассмотрены также работы, проводившиеся с использованием ферментативного расщепления природных образцов ГК и хондроитинсульфатов с целью получения соответствующих низкомолекулярных олигосахаридов. Синтезированные олигосахариды и гликоконъюгаты на их основе – ценные модели для установления взаимосвязи структуры и биологических свойств ГК и для разработки подходов к созданию терапевтических препаратов на основе ГК (в том числе для заживления ран, лечения воспалительных процессов), а также ингибиторов некоторых этапов онкогенеза.
СОДЕРЖАНИЕ
ВВЕДЕНИЕ……………........................................129
СИНТЕЗ ОЛИГОСАХАРИДОВ, СТРУКТУРНО РОДСТВЕННЫХ ФРАГМЕНТАМ ГИАЛУРОНОВОЙ КИСЛОТЫ……………..........................130
ЗАКЛЮЧЕНИЕ……………..................................159
СПИСОК ЛИТЕРАТУРЫ………. .......................159
ВВЕДЕНИЕ
Синтетические олигосахариды строго определенного строения, структурно родственные фрагментам природных полисахаридов, выступают ценными моделями для выяснения взаимосвязи структуры и свойств этих биополимеров. В качестве яркого и успешного примера использования такого подхода можно считать серию работ французских, немецких, британских и американских ученых, посвященных направленному синтезу олигосахаридов, представляющих собой фрагменты природного гликозаминогликана гепарина, и изучению их биологических свойств [1–6]. В результате этих исследований были определены структуры минимальных активных участков цепи гепарина, ответственных за связывание с белками-мишенями (факторами коагуляции крови – тромбином и фактором Ха) и демонстрирующих значимую антикоагулянтную активность, но не обладающих побочными эффектами гепарина. Эти знания послужили основой для создания фармацевтического препарата Арикстра© (Санофи, Франция) – синтетического пентасахаридного гепариноида, который используется в настоящее время в медицинской практике как антикоагулянт. Примечательно, что Арикстра©, в отличие от природного гепарина, не вызывает серьезных побочных эффектов, таких как кровотечения и тромбоцитопения.
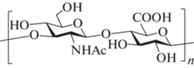
Гиалуроновая кислота (ГК), как и гепарин, – представитель полисахаридов, получивших название гликозаминогликаны. Этот тип полисахаридов широко распространен в различных организмах: от бактерий до млекопитающих [7–10]. Цепи ГК построены из дисахаридных блоков →3)-β-D-GlcNAc-(1→4)-β-D-GlcA-(1→ (рис. 1), эти цепи входят в состав различных гликопротеинов и протеогликанов. В организме высокомолекулярные цепи ГК, как правило, выполняют структурные функции, в то время как для низкомолекулярных фрагментов было показано регуляторное действие: они играют ключевую роль в процессах деления и миграции клеток, в формировании сосудов, воспалении, онкогенезе [11–17].
В обзоре рассмотрены реализованные синтезы олигосахаридов, родственных цепям ГК, проанализированы стратегии сборки соответствующих углеводных цепей, оценены типы использованных гликозил-доноров и гликозил-акцепторов, рассмотрены стратегии выбора защитных групп в использованных синтетических блоках, обозначены направления дальнейших биологических исследований полученных олигосахаридов и гликоконъюгатов на их основе.
СИНТЕЗ ОЛИГОСАХАРИДОВ, СТРУКТУРНО РОДСТВЕННЫХ ФРАГМЕНТАМ ГИАЛУРОНОВОЙ КИСЛОТЫ
Хотя строение ГК известно с середины XX века [18], детальное изучение взаимосвязи структуры и свойств этого биополимера с использованием синтетических олигосахаридов было начато лишь в конце XX века, что связано с разработкой именно к этому времени соответствующих методов химии углеводов. Для синтеза олигосахаридов, родственных ГК, необходимы методы, позволяющие выполнить следующие превращения: 1) проводить стереоизбирательное построение β-гликозидной связи (1,2-транс- в случае глюко-конфигурации); 2) обеспечить защиту атома азота в остатке D-глюкозамина оптимальной временной группой; 3) обеспечить защиту карбоксильной группы оптимальной временной группой; 4) эффективно проводить построения межзвеньевых гликозидных связей; 5) эффективно проводить блочную сборку углеводных цепей.
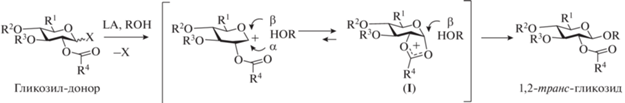
Высокая эффективность при построении 1,2‑транс-гликозидных связей достигается при использовании гликозил-доноров, содержащих соучаствующие O- и N-защитные группы при C-2 [19, 20]. Это связано с образованием в ходе реакции гликозилирования стабилизированного катиона (I), нуклеофильная атака аномерного центра в котором предпочтительна с противоположной стороны от заместителя при С-2 (рис. 2). Именно поэтому в большинстве рассматриваемых нами проведенных синтезов олигосахаридов использовались соучаствующие N-фталоильные или трихлорацетильные защитные группы при С-2 в производных глюкозамина, а в производных глюкозы или глюкуроновой кислоты – 2-О-ацильные заместители.
Рис. 2.
Пример стереоконтролирующего эффекта соучаствующего заместителя при О-2 в реакции гликозилирования. LA – кислота Льюиса.
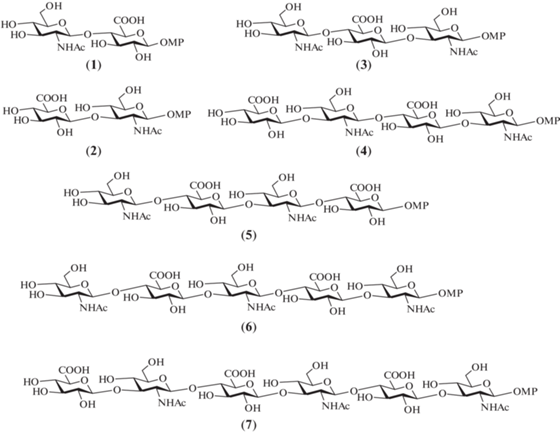
Одни из первых синтезов олигосахаридов, родственных ГК, были выполнены Vliegenthart et al. в 1990-х гг. [21–25]. Авторами были получены ди-, три-, тетра-, пента- и гексасахариды (1–7) в виде 4-метоксифенилгликозидов (рис. 3). В качестве защиты для атома азота в производных D‑глюкозамина была использована фталоильная группа. Для сборки углеводного скелета авторы выбрали производные D-глюкозы, содержащие ортогональную левулиновую защитную группу при О-6. Это позволило на финальных стадиях синтезов избирательно высвободить гидроксильную группу при С-6 и провести окисление гидроксиметильной группы до карбоксильной. В качестве гликозил-доноров были выбраны трихлорацетимидаты – высокореакционные производные D-глюкозы и D-глюкозамина, активируемые действием кислоты Льюиса [26].
Так, для получения целевого дисахарида (1) была проведена реакция гликозилирования моносахарида (8) трихлорацетимидатом (9) в присутствии эфирата трехфтористого бора (схема 1 ). В результате был получен требуемый β-связанный дисахарид (10) с выходом 81%. Далее в нем селективно высвободили гидроксильную группу при С-6 остатка глюкозы действием ацетата гидразина (86%), после чего окисляли гидроксиметильную группу до карбоксильной, что привело к получению соединения (11) с выходом 68%. Удаление ацильных групп при обработке метиламином в метаноле и последующее селективное N‑ацетилирование позволило получить целевой продукт (1) с выходом 93%.
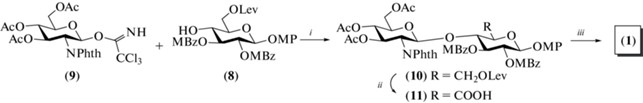
Схема 1 . Синтез целевого дисахарида (1). Реагенты и условия: i: BF3. Et2O, CH2Cl2, 81%; ii: a – N2H4 · AcOH, толуол/EtOH, 86%; b – (COCl)2, ДМСО; с – NaClO2, 68%; iii: a – MeNH2, EtOH; b – Ac2O, MeOH, 93%.
Синтез более крупных олигосахаридов был выполнен с использованием блочной сборки углеводного скелета. Сначала сочетанием моносахаридов (8) и (12) был синтезирован дисахарид (13), в котором далее была селективно удалена аллилоксикарбонильная защитная группа с образованием гликозил-акцепторного блока (14) (схема 2 ). Кроме того, из дисахарида (10) был получен гликозил-донор (15). Для этого удаляли метоксифенильную группу с последующей обработкой трихлорацетонитрилом в присутствии основания ДБУ. Сочетание дисахаридов (14) и (15) привело к образованию тетрасахарида, однако очистка продукта была выполнена лишь на следующей стадии после удаления изопропилиденовой защиты и ацетилирования (→16). Выход тетрасахарида (16) составил 77%. Стереоизбирательное построение β-гликозидной связи было обеспечено наличием при О-2 в гликозил-доноре (15) соучаствующей метилбензоильной группы. Далее проводили избирательное удаление левулиновой защитной группы при О-6 во всех остатках глюкозы (→17), а затем окисляли гидроксиметиленовые группы до карбоксильных действием оксалилхлорида и хлората натрия. На завершающем этапе проводили удаление всех защитных групп с образованием целевого тетрасахарида (5).
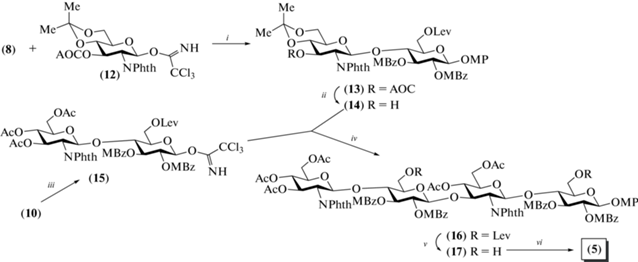
Схема 2 . Синтез целевого тетрасахарида (5). Реагенты и условия: i: BF3. Et2O, CH2Cl2, 90%; ii: Pd(PPh3)4, ТГФ, морфолин, 94%; iii: а – (NH4)2Ce(NO3)6, 96%; b – CCl3CN, ДБУ, 93%; iv: TMSOTf; v: CF3COOH, CH2Cl2; Ac2O, DMAP в Py, 77% на две стадии; vi: a – N2H4 · AcOH, толуол/EtOH, 87%; b – (COCl)2, ДМСО, i-Pr2NEt; c – NaClO2, NaH2PO4, 76% на две стадии; d – MeNH2, EtOH; e –Ac2O, MeOH; f – MeONa, MeOH, 61%.
Для получения олигосахаридов (2–4) и (7), содержащих остаток N-ацетил-D-глюкозамина на восстанавливающем конце, был использован моносахаридный гликозил-акцептор (18) (схема 3 ). Сочетанием моносахаридов (18) и (19) в присутствии TMSOTf был синтезирован дисахарид (20) с выходом 81%, после деблокирования которого получали целевой дисахарид (2). При взаимодействии моносахаридов (18) и (22) получали дисахарид (23) (87%), в котором селективно удаляли изопропилиденовую защиту, а затем ацетилировали свободные гидроксильные группы, после чего удаляли аллилоксикарбонильную защиту с образованием гликозил-акцептора (24). Сочетанием дисахарида (24) и моносахарида (9) получали β-связанный трисахарид (25) с выходом 81%. Селективное высвобождение первичной гидроксильной группы в остатке D-глюкозы в соединении (25), последующее окисление продукта, метилирование и деблокирование приводили к получению целевоого трисахарида (3).
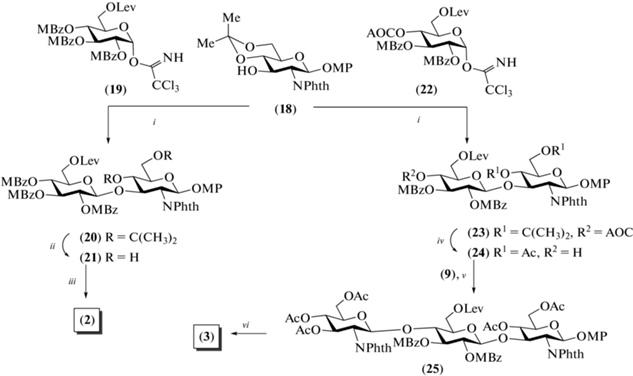
Схема 3 . Синтез дисахарида (2) и трисахарида (3). Реагенты и условия: i: TMSOTf, CH2Cl2, 81% для соединения (20) и 87% для соединения (23); ii: CF3COOH, CH2Cl2, 84%; iii: a – Ac2O, Py, 97%; b – N2H4 · AcOH, толуол/EtOH, 98%; c – (COCl)2, ДМСО; d – NaClO2, 70% на две стадии; e – CH3NH2, EtOH; f – Ac2O, MeOH; g – MeONa, MeOH, 65%; iv: a – CF3COOH, CH2Cl2, 88%; b – Ac2O, Py, 98%; c – Pd(PPh3)4, ТГФ, морфолин, 95%; v: BF3. Et2O, CH2Cl2, 81%; vi: a – N2H4 · AcOH, толуол/EtOH, 88%; b – (COCl)2, ДМСО; c – NaClO2, 95% на две стадии; d – CH3NH2, EtOH; e – Ac2O, MeOH, 79% на две стадии.
Для сборки углеводных скелетов тетра-, пента- и гексасахаридов (4), (6) и (7) были разработаны схемы блочного синтеза [1+3], [2+3] и [1+5] соответственно (схема 4 ). Трисахаридный гликозил-акцептор (27) был получен сочетанием соединений (12) и (24) (69%) и последующим удалением аллилоксикарбонильной защитной группы (89%). Гликозилирование по схеме [1+3] приводило к получению тетрасахарида (28) (87%), который был далее трансформирован в целевой тетрасахарид (4) так, как описано для превращения (17)→(5).
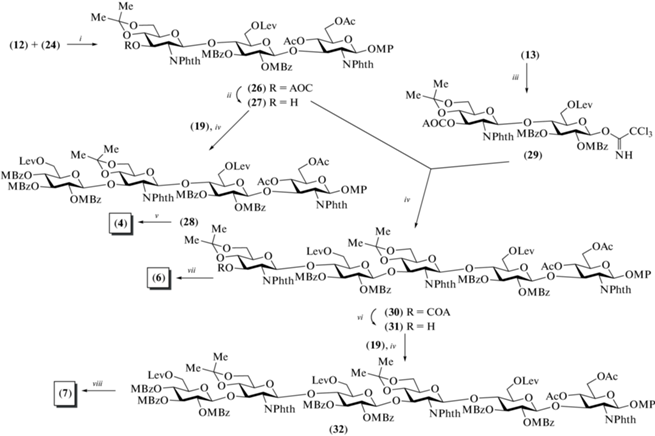
Cхема 4. Синтез целевых тетра-, пента- и гексасахаридов (4), (6) и (7). Реагенты и условия: i: BF3. Et2O, CH2Cl2, 69%; ii: Pd(PPh3)4, ТГФ, морфолин, 89%; iii: a – (NH4)2Ce(NO3)6; b – СCl3CN, ДБУ, 67%; iv: TMSOTf, 87% для соединения (28), 81% для соединения (30), 62% для соединения (32); v: a – CF3COOH, CH2Cl2, 85%; b – Ac2O, Py, 96%; c – N2H4 · AcOH, толуол/EtOH, 76%; d – (COCl)2, ДМСО; e – NaClO2, 86% на две стадии; f – CH3NH2, EtOH; g – Ac2O, MeOH, 82% на две стадии; vi: Pd(PPh3)4, ТГФ, морфолин, 89%; vii: a – CF3COOH, CH2Cl2; b – Ac2O, Py, 90% на две стадии; c – N2H4 · AcOH, толуол/EtOH, 74%; d – PDC, Ac2O, 70%; е – CH3NH2, EtOH; f – Ac2O, MeOH, 64% на две стадии; viii: a – CF3COOH, CH2Cl2; b – Ac2O, Py, 84% на две стадии; c – N2H4 · AcOH, толуол/EtOH, 75%; d – PDC, Ac2O, 58%; e – NH2CH2CH2NH2, 1-BuOH; f – Ac2O, Py, DMAP; g – 2 M NaOH (водн.) в ТГФ, 73%.
В свою очередь, сочетание дисахарида (29) и трисахарида (27) приводило к образованию пентасахарида (30) с выходом 81%. Селективное удаление аллилоксикарбонильной группы в соединении (30) позволило получить пентасахаридный гликозил-акцептор (31) (89%). Реакция по схеме [1+5] прошла с выходом 62%, что вполне приемлемо для таких крупных молекул. Далее следовали стадии удаления изопропилиденовых защит в соединениях (30) и (32), ацетилирования и селективного высвобождения первичных гидроксильных групп в остатках D-глюкозы. Окисление двух и трех гидроксильных групп в пента- и гексасахаридах было выполнено действием PDC с выходами 70 и 58% соответственно. Последующая обработка метиламином продуктов окисления и селективное N-ацетилирование приводили к получению целевых соединений (6) и (7).
В 1994–1996 гг. Jacquinet et al. осуществили синтез метил-гликозидов ди-, тетра-, гекса- и октасахаридов (33–36), родственных ГК [27–29] (рис. 4). В отличие от работ Vliegenthart et al., авторы использовали трихлорацетильную защитную группу для атома азота в остатках глюкозамина и производные D-глюкуроновой кислоты вместо производных D-глюкозы, что позволило избежать реакций окисления на финальных стадиях синтезов. Так же, как и в работах Vliegenthart et al., в качестве гликозил-доноров были использованы высокореакционные трихлорацетимидатные гликозил-доноры.
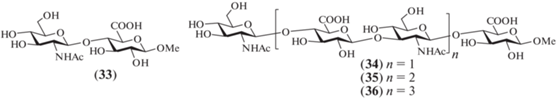
Для синтеза дисахарида (33) сначала провели реакцию гликозилирования соединения (38) моносахаридом (37) в присутствии TMSOTf, в результате чего был получен дисахарид (39) с выходом 91% (схема 5 ). Стереоизбирательное построение β-гликозильной связи было обеспечено наличием соучаствующей N-трихлорацетильной группы при C-2 в гликозил-доноре (37). Примечательно, что наличие электроноакцепторной карбометоксильной группы в соединении (38) не снижало эффективность реакции гликозилирования. После удаления защитных групп в соединении (39) получали целевой дисахарид (33). Кроме того, из соединения (39) селективным удалением хлорацетильной группы действием тиомочевины получали дисахарид (40) (96%), содержащий свободную гидроксильную группу при С-3 остатка D-глюкозамина.
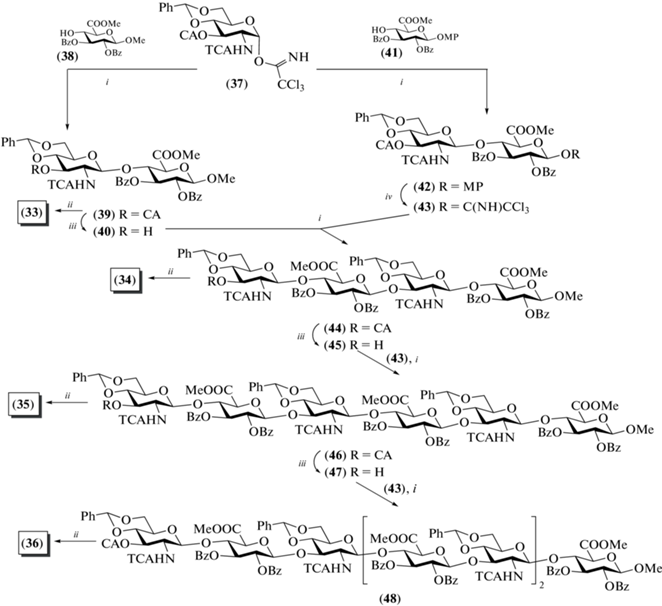
Схема 5 . Синтез олигосахаридов (33–36). Реагенты и условия: i: TMSOTf, CH2Cl2, 91% для соединения (39), 89% для соединения (42), 87% для соединения (44), 93% для соединений (46) и (48); ii: а – Bu3SnH, AIBN, 81% для соединения (33), 88% для соединения (34), 91% для соединения (35) и 92% для (36); b – AcOH; c – 3 M NaOH в MeOH, 80–83%; iii: (NH2)2CS, 96% для соединения (40), 95% для соединения (45), 83% для соединения (47); iv: a – (NH4)2Ce(NO3)6, b – СCl3CN, ДБУ, 78%.
Сборка углеводных скелетов тетра-, гекса- и октасахаридов (34–36) была выполнена с использованием блочных схем [2+2], [2+4] и [2+6] соответственно. Ключевой дисахаридный гликозил-донор (43) получен сочетанием моносахаридов (37) и (41) (89%) с последующим удалением метоксифенильного агликона и переводом образующегося в результате этого превращения полуацеталя в трихлорацетимидат. Гликозилирование по схеме [2+2] проходило с высоким выходом (87%) и протекало стереоизбирательно с образованием β-гликозидной связи в тетрасахариде (44) благодаря соучаствующему эффекту 2-О-бензоильной группы в гликозил-доноре (43). В результате селективного удаления хлорацетильной группы в соединении (44) получили тетрасахаридный гликозил-акцептор (45), гликозилирование которого имидатом (43) приводило к образованию гексасахарида (46) по схеме [2+4] также с высоким выходом (93%). Удаление хлорацетильной группы в гексасахариде (46) и последующее удлинение цепи на два остатка при гликозилировании донором (43) приводили к получению октасахарида (48) (93%). После удаления защитных групп в продуктах (44), (46) и (48) были получены целевые олигосахариды (34–36) соответственно. При этом трансформацию N-трихлорацетильных групп в N-ацетильные выполняли действием три-бутилолова гидрида и азоизобутиронитрила, удаляли бензилиденовые защиты в условиях гидролиза действием уксусной кислоты, а омыление сложноэфирных групп проводили действием 3 М NaOH, что позволило получить целевые соединения с общими выходами 80–83%. Подводя итог синтезов соединений (34–36), можно отметить высокую эффективность предложенного дисахаридного донора (43), как и достаточно удачную комбинацию использованных О- и N-защитных групп.
В 2007 г. L. Huang и X. Huang описали синтез полностью незащищенных метил-гликозидов ди-, тетра-, пента- и гексасахаридов (49–53), родственных ГК [30] (рис. 5). В ходе синтеза авторы использовали N-фталоильную защиту в остатках D-глюкозамина. Для построения углеводного скелета были выбраны производные D-глюкозы, содержащие пара-метоксибензильную защитную группу при О-6, которые на финальных стадиях синтеза были переведены в остатки D-глюкуроновой кислоты.
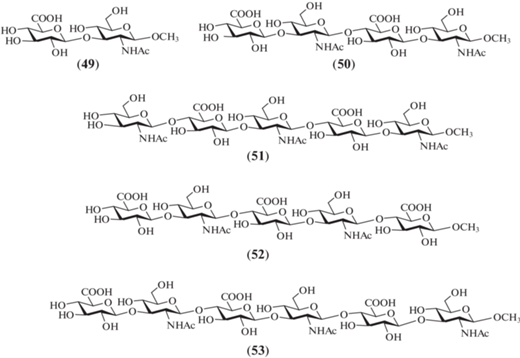
В отличие от предыдущих синтезов, в которых в качестве гликозил-доноров использовали трихлорацетимидаты, в данной работе с этой целью применяли толилтиогликозиды. В частности, был использован моносахаридный донор (54), содержащий комбинацию избирательно удаляемых защитных групп во всех положениях: соучаствующую бензоильную группу при О-2, бензильную при О-3, трет-бутил-ди-метилсилильную при О-4 и пара-метоксибензильную при О-6 (схема 6 ). Сочетанием соединения (54) и метил-гликозида (55) в присутствии AgOTf, р-TolSCl, TTBP и молекулярных сит AW300 получали β-связанный дисахарид (56) с высоким выходом 88%. Селективное удаление силильной защиты в соединении (56) действием HF.Py приводило к получению дисахарида (57), который был использован далее в синтезе более крупных олигосахаридов. В свою очередь, селективное удаление пара-метоксибензильной защиты в соединении (56) и последующие окисление карбометоксильной группы в карбоксильную и удаление защит по стандартным методикам приводили к получению целевого дисахарида (49).
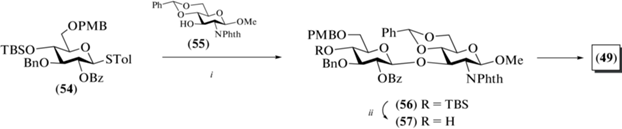
Схема 6 . Синтез целевого дисахарида (49). Реагенты и условия: i: AgOTf, p-TolSCl, –65°C, AW300, 88%; ii: HF·Py, 87%.
Для синтеза крупных тетра-, пента- и гексасахаридов были разработаны однореакторные (“one-pot”) протоколы, позволяющие проводить сборку от тетра- до гексасахаридов (50–53) из моно- и дисахаридных предшественников. Так, сначала выполняли конденсацию соединений (54) и (58), при которой активация тиогликозида (54) проводилась действием р-TolSCl в присутствии AgOTf, а взаимодействие с гликозил-акцептором (58) осуществлялось в присутствии основания TTBP (схема 7 ). Затем полученный продукт (59), содержащий тиотолильный агликон, подвергали тем же реакциям в аналогичной последовательности: активировали действием р-TolSCl и AgOTf, а затем добавляли дисахаридный блок (57) в присутствии ТТВР. Таким образом был синтезирован тетрасахарид (60) (64%), из которого получали незащищенный целевой тетрасахарид (50). Проведение схожей последовательности превращений с использованием блоков (61), (62) и (57) приводило к образованию пентасахарида (64) (65%), из которого затем получали незащищенный пентасахарид (51).
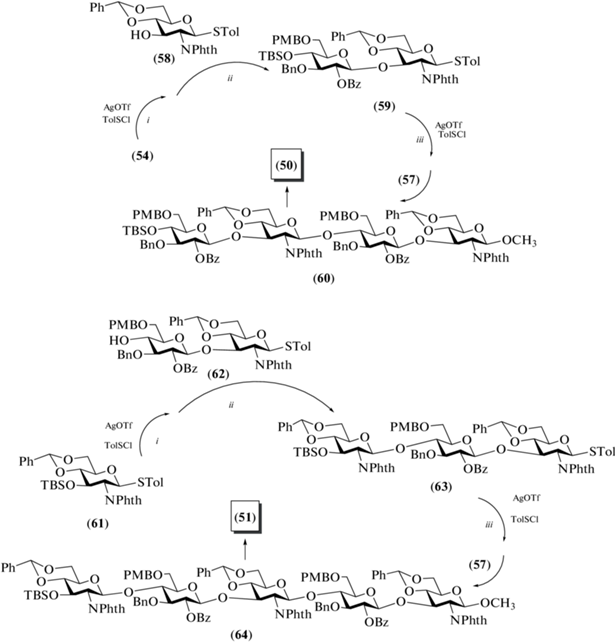
Схема 7 . Синтез тетрасахарида (50) и пентасахарида (51). Реагенты и условия: i: AgOTf, p-TolSCl, –65°C, 10 мин; ii: акцептор, TTBP, 0°C, 90 мин; iii) акцептор, ТТВР, AgOTf, p-TolSCl, –65°C → 0°C в течение 90 мин, 64% для соединения (60) и 65% для соединения (64).
Для сборки пента- и гексасахаридов (67) и (68), предшественников целевых соединений (52) и (53), был разработан иной “one-pot” протокол, предполагающий последовательную активацию трех тиотолилгликозидов (схема 8 ). Синтез соединения (67) начинался с активации моносахарида (54), затем к нему добавляли моносахарид (58), полученный тиогликозид (59) снова активировали и добавляли к нему дисахарид (62). Из полученного тиотолильного тетрасахарида (65) были синтезированы пентасахарид (67) добавлением моносахарида (66), а гексасахарид (68) – добавлением дисахарида (57). Общие выходы проведенных последовательностей реакций для соединений (67) и (68) составили 55 и 54% соответственно. Реализованная схема сборки олигосахаридов выглядит привлекательно для получения крупных молекул. Однако, несмотря на кажущуюся простоту исполнения “one-pot” превращений, представленный синтез не выглядит оптимальным из-за сложностей выделения продуктов гликозилирования, которые обладают схожей хроматографической подвижностью и незначительно различаются по размерам. Это затрудняет проведение эффективной очистки продуктов методами адсорбционной и гель-проникающей хроматографии.
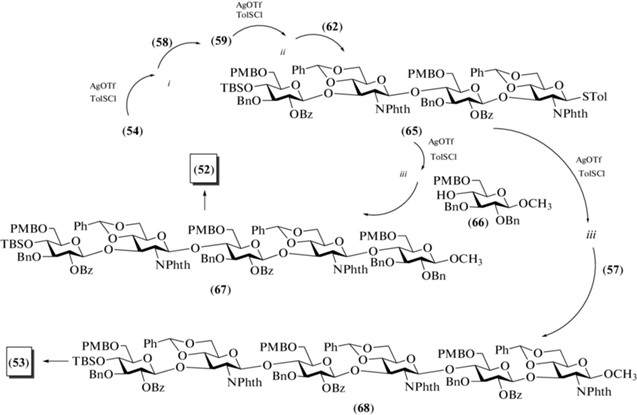
Схема 8 . Синтез пентасахарида (52) и гексасахарида (53). Реагенты и условия: i: AgOTf, p-TolSCl, –65°C, 10 мин; ii: акцептор, TTBP, 0°C, 90 мин; iii: акцептор, ТТВР, AgOTf, p-TolSCl, –65°C →0°C в течение 90 мин, 55% для соединения (67) и 54% для соединения (68).
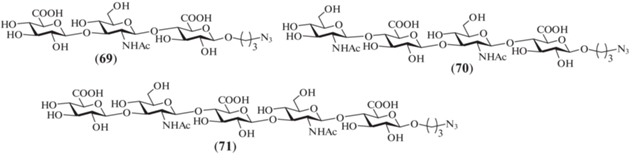
B 2007 г. под руководством van der Marel был выполнен синтез три-, тетра- и пентасахаридов (69–71), родственных ГК, несущих в качестве агликона азидопропильный спейсер (рис. 6) [31]. В данной работе в качестве моносахаридных предшественников были использованы производные D-глюкуроновой кислоты и N-трихлорацетил-D-глюкозамина.
В рамках выбранной стратегии полуацеталь (72) активировали системой Ph2SO/Tf2O/TTBP, а затем добавляли гликозил-акцептор (73), в результате образовывался дисахарид (74) с выходом 60% (схема 9 ). Примечательно, что выбранные условия генерации гликозил-донора оказались применимы для успешного введения в реакцию даже малоактивного производного глюкуроновой кислоты (72). Далее было проведено гликозилирование дисахаридом (74) спейсерированного моносахарида (75) в присутствии ангидрида трифторметансульфоновой кислоты и TTBP, в результате чего был получен полностью защищенный трисахарид (76) с выходом 50%.
Действием гидразина в смеси Рy/AcOH проводили селективное удаление левулиновой защитной группы в соединении (76) с образованием продукта (77), содержавшего свободную гидроксильную группу при С-4. Трисахарид (77) был использован в синтезе тетра- и пентасахаридов (79) и (80). Так, по схеме [1+3] был получен тетрасахарид (79) с выходом 60%, а по схеме [2+3] – пентасахарид (80) с выходом 40%. Деблокирование соединений (76), (79) и (80) приводило к получению целевых три-, тетра- и пентасахаридов (69–71) соответственно.
Характерная особенность реализованной синтетической схемы – активирование полуацеталя D-глюкуроновой кислоты в дегидратирующих условиях с последующим добавлением гликозил-акцептора, содержащего фенилтио-агликон. Средние выходы в проведенных реакциях гликозилирования сильно сказываются на общем выходе целевых олигосахаридов. Необходимо рассчитывать точное количество используемого основания для нейтрализации образующейся трифторметансульфоновой кислотой in situ. Введение избытка основания приводит к нежелательному образованию ортоэфира, а при недостатке основания – наблюдается частичное удаление бензилиденовой защитной группы.
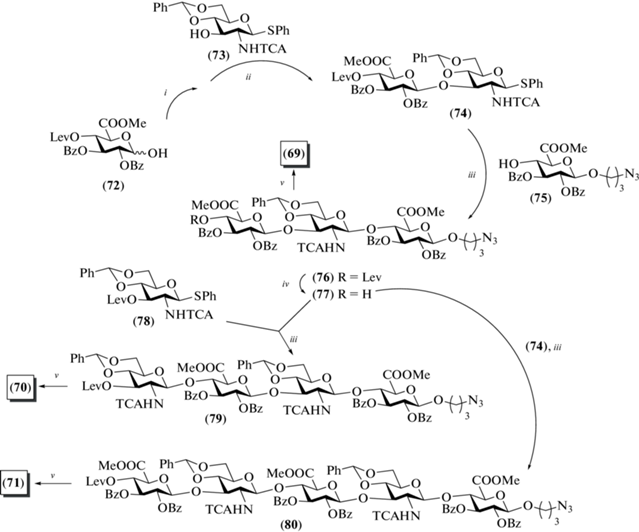
Схема 9 . Синтез целевых олигосахаридов (69–71). Реагенты и условия: i: Ph2SO, TTBP, CH2Cl2, –60°C; ii: Tf2O, –40°C, 60 мин, акцептор, 0°C, 56%; iii: a – Ph2SO, TTBP, CH2Cl2, –60°C; b – Tf2O, –40°C, 60 мин, акцептор, 0°C, 47% для соединения (76), 62% для соединения (79) и 48% для соединения (80); iv: N2H4, AcOH, Py, 96%; v: a – pTsOH, MeOH; b – H2O, THF, KOH; c – Ac2O, MeOH, 58% для соединения (69), 54% для соединения (70) и 48% для соединения (71).
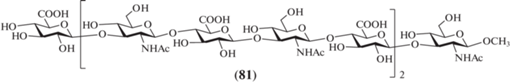
В 2009 г. X. Huang et al. описали синтез декасахарида (81), родственного ГК, в виде метилгликозида [32] (рис. 7). Данная работа – продолжение рассмотренных выше синтезов, выполненных в 2007 г. [30]. Несмотря на успешный синтез гексасахарида (53) (см. схему 8 ), получить по такой же схеме декасахарид (81) не удалось. По использованному в случае продукта (53) “one-pot” протоколу был синтезирован и полностью защищенный декасахарид, однако авторы не смогли удалить пять фталоильных защитных групп в присутствии пяти О-бензильных защит в молекуле из-за образования малореакционных липосомоподобных полупродуктов. Поэтому была разработана новая схема синтеза, которая предусматривала использование N-трихлорацетильной защиты в остатке D-глюкозамина вместо фталоильной.
Так, гликозилирование моносахаридных производных (82) и (83) тиогликозидом (54) в присутствии AgOTf и p-TolSCl приводило к образованию требуемых дисахаридов (84) и (85) с выходами 80 и 70% соответственно (схема 10 ). Избирательное удаление пара-метоксибензильной группы при О-6 в остатках D-глюкозы в соединениях (84) и (85), окисление С-6 действием PDC и последующее бензилирование карбоксильной группы приводили к образованию дисахаридов (86) и (87) соответственно. Удаление TBS-защиты в соединениях (86) и (87) действием HF·Py позволило получить дисахаридные блоки (88) и (89) соответственно, которые были необходимы для сборки целевого декасахарида.
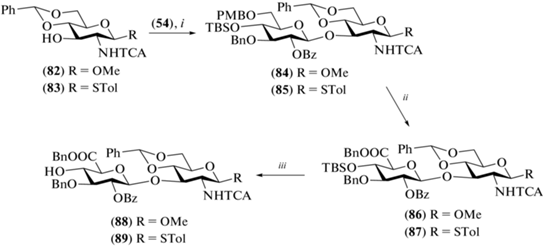
Схема 10 . Синтез дисахаридных блоков (86–89). Реагенты и условия: i: p-TolSCl, AgOTf, –78°C, ТТВP, 80% для соединения (85); ii: a – DDQ, CH2Cl2/Na2CO3 (водн.); b – PDC, DMF; c – PhCHN2, CH2Cl2, 65% на три стадии для соединения (87); iii: HF/Py, 90% для соединения (89) и 48% на пять стадий для соединения (88).
Так же, как и в синтезе 2007 г., авторы использовали схему с предварительной активацией тиогликозидного гликозил-донора действием p-TolSCl и AgOTf. Гликозилирование акцептора (89) донором (87) в этих условиях приводило к образованию тетрасахаридa (90) с выходом 82% (схема 11 ). Активация тиогликозида (90) и добавление к нему дисахаридного блока (88) позволили получить гексасахарид (91) с выходом 71%. Далее в этом соединении селективно удаляли силильную защитную группу, высвобождая гидроксильную группу при С-4 концевого остатка D-глюкозы. Последующая сборка углеводного скелета по схеме [4+6] приводила к получению требуемого декасахарида (93) с высоким выходом 77%. Действием HF·Py при комнатной температуре была удалена TBS-защита в соединении (93) (79%). Далее удаляли пять N-трихлорацетильных групп и омыляли пять бензиловых эфиров при щелочной обработке (20 экв. КОН) в течение 5 недель, после чего следовали стадии N-ацетилирования и удаления бензилиденовых защит с образованием целевого декасахарида (81) с общим выходом 35%.
В 2009 г. под руководством van der Marel был выполнен еще один синтез олигосахаридов, родственных ГК. В отличие от рассмотренной выше серии соединений (69–71), полученных в этой лаборатории, новые продукты – три- (94), пента- (95) и гептасахарид (96) – содержали остаток N‑ацетил-D-глюкозамина на восстанавливающем конце (рис. 8) [33]. Как и в предыдущей серии синтезов, авторы использовали синтетические блоки на основе производных N-трихлорацетил-D-глюкозамина и D-глюкуроновой кислоты.
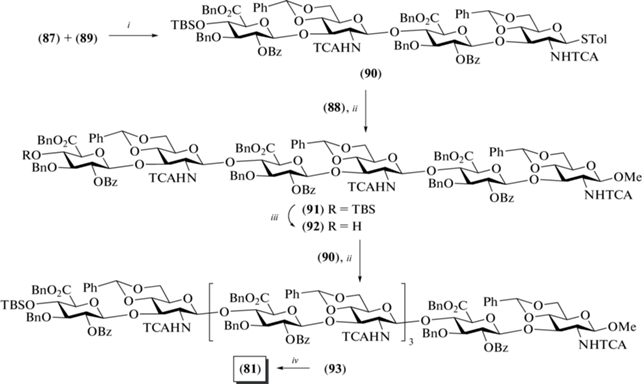
Схема 11 . Синтез декасахарида (81). Реагенты и условия: i: AgOTf, Et2O, –78°C, p-TolSCl, TMSOTf, 82%; ii: AgOTf, Et2O, –78°C, p-TolSCl, (88), TMSOTf, 71% для соединения (91), 77% для соединения (93); iii: HF⋅Py, 90%; iv: a – HF⋅Py, 79%; b – KOH, H2O/ТГФ; c – Ac2O, MeOH, TEA; d – Pd(OH)2, H2, AcOH/MeOH/ТГФ, 35% на 3 стадии.
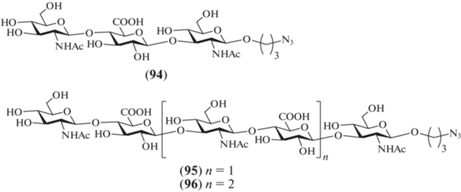
Для защиты О-4 и О-6 в остатках N-трихлорацетил-D-глюкозамина была выбрана ди-трет-бутилсилилиденовая группа. Взаимодействием N-фенил-трифторацетимидата (97) и гликозил-акцептора (98) в присутствии TfOH был получен дисахаридный тиогликозид (99) с выходом 90% (схема 12 ). Его сочетание с моносахаридом (100), содержащим азидопропильный спейсер, приводило к образованию трисахарида (101) с выходом 75%. Деблокированием соединения (101) по отработанным методикам получали целевой трисахарид (94).
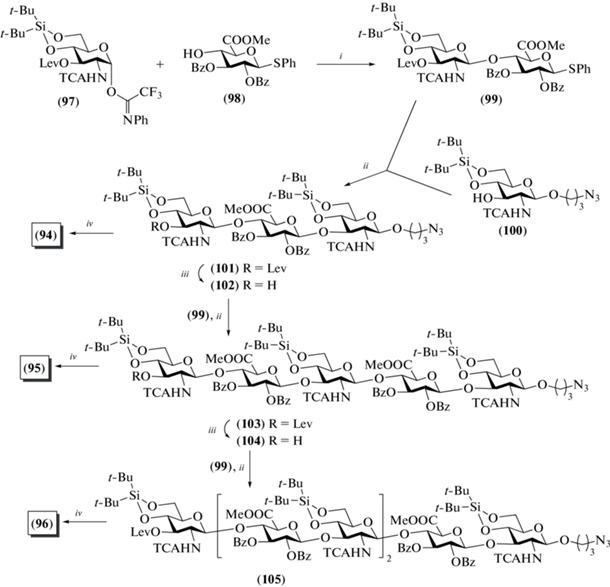
Схема 12 . Синтез целевых олигосахаридов (94–96). Реагенты и условия: i: TfOH, 0°C, 90%; ii: NIS, TfOH, CH2Cl2, 75% для соединения (101), 98% для соединения (103), 61% для соединения (105); iii: N2H4, H2O, Py, AcOH, 94% для соединения (102), 91% для соединения (104); iv: a – Et3N/3HF, ТГФ; b – KOH, ТГФ, H2O; c – Ac2O, MeOH; d – H2O, LiOH, 47% для соединения (94), 31% для соединения (95), 46% для соединения (96).
Селективное удаление левулиновой защиты при О-3 концевого остатка в соединении (101) приводило к получению трисахаридного гликозил-акцептора (102) (94%). Далее по схеме [2+3] с использованием блоков (99) и (102) был получен пентасахарид (103) с выходом 98%. Удаление левулиновой защиты в соединении (103) приводило к образованию пентасахаридного гликозил-акцептора (104) (91%). По схеме [2+5] с использованием блоков (99) и (104) был получен уже гептасахарид (105) с выходом 61%. Удаление защитных групп в соединениях (103) и (105) приводило к получению целевых пента- (95) и гептасахаридов (96) соответственно.
В 2010 г. Brase et al. опубликовали синтез ди-, тетра-, гекса- и октасахаридов (106–109) – защищенных олигосахаридов, родственных ГК (рис. 9) [34]. Частичное удаление защитных групп было выполнено только для тетрасахарида (107). Однако данная работа представляет интерес как еще один пример сборки углеводного скелета олигосахаридов, родственных ГК. В качестве гликозил-донора использовали этилтио-глюкозид (110), описанный ранее [34], содержавший соучаствующую 2-О-бензоильную защитную группу и ортогональную кислотолабильную пара-метоксибензильную группу при О-6 (схема 13 ). В качестве акцептора использовали производное аллил-N-трихлорацетил-D-глюкозамина (111). Взаимодействием моносахаридов (110) и (111) в присутствии NIS и TMSOTf получали дисахарид (112) с выходом 72%, в котором далее удаляли PMB-группу при О-6 действием (NH4)2Ce(NO3)6. Окисление С-6 в полученном промежуточном моногидроксильном продукте было проведено перйодной кислотой и CrO3 с последующим переводом образующейся карбоксильной группы в соответствующий метиловый эфир. Общий выход дисахарида (106) на четыре стадии составил 45%.
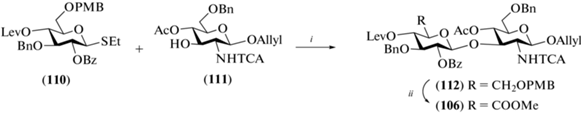
Схема 13 . Синтез целевого дисахарида (106). Реагенты и условия: i: NIS, TMSOTf, CH2Cl2, –30°C, 72%; ii: a – (NH4)2Ce(NO3)6, CH2Cl2; b – H5IO6, CrO3, CH3CN/H2O, 0°C; c – СH2N2, CH2Cl2, 45% общий выход.
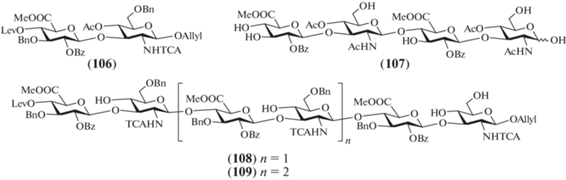
Из соединения (106) были получены ключевые дисахаридные блоки, необходимые для сборки крупных олигосахаридов (схема 14 ). Так, действием хлорида палладия в уксусной кислоте удалили аллильную защитную группу в соединении (106) с последующим переводом полученного полуацеталя в трихлорацетимидат (113) (45%). Селективное удаление левулиновой защитной группы в соединении (106) действием ацетата гидразина приводило к получению дисахаридного акцептора (114) с выходом 90%.
Взаимодействием дисахаридных блоков (113) и (114) был получен тетрасахарид (115) с выходом 51%. Далее в этом соединении действием гидразина проводили высвобождение гидроксильной группы при С-4 терминального остатка D-глюкозы с образованием тетрасахаридного акцептора (116). Его последующее гликозилирование трихлорацетимидатом (113) приводило к получению гексасахарида (108) с выходом только 45%. Удалением левулиновой защитной группы в соединении (108) получали гексасахаридный гликозил-акцептор (117). Сборка октасахарида (109) по схеме [2+6] с использованием соединений (113) и (117) прошла с умеренным выходом (32%). Постепенное уменьшение выхода с ростом цепи на стадиях гликозилирования связано, возможно, с более сложной пространственной организацией гликозил-акцептора, в результате чего подход гликозил-донора к свободной гидроксильной группе концевого остатка становится пространственно затруднительным.
Из всех полученных защищенных олигосахаридов авторы выполнили частичное удаление защитных групп лишь в тетрасахариде (115). Действием Zn в смеси AcOH и диоксана в соединении (115) провели восстановительное дегалогенирование N‑трихлорацетильной группы в остатках D-глюкозамина, которое протекало с низким выходом 20%. Удаление аллильной защиты было осуществлено при действии PdCl2 в смеси водной уксусной кислоты, а снятие бензильных групп выполняли гидрогенолизом над Pd(OH)2/C.
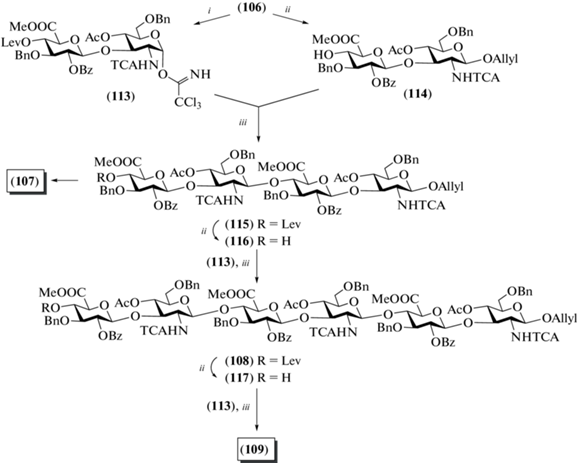
Схема 14 . Синтез целевых олигосахаридов (107–109). Реагенты и условия: i: a – PdCl2, NaOAc, AcOH/H2O, 79%; b – CCl3CN, ДБУ, CH2Cl2, 0°C, 52%; ii: N2H4, AcOH, CH2Cl2, MeOH, 90% для соединений (114), (116) и (117); iii: TMSOTf, CH2Cl2, 0°C, соединение (113), 51% для соединения (115), 45% для соединения (108), 32% для соединения (109).
В 2011 г. van der Marel et al. осуществили синтез ди- (118) и тетрасахаридного 4-метилумбеллиферилгликозида (119) (рис. 10) [35]. Эти соединения содержали метильную группу при О-4 остатка D‑глюкуроновой кислоты. Такие производные были необходимы для изучения субстратной специфичности фермента гиалуронидазы [36], которое планировали выполнить в будущем.
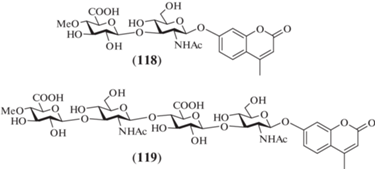
В качестве гликозил-донора в работе van der Marel et al. было использовано производное D-глюкуроновой кислоты (120), содержавшее метильную защитную группу при О-4 (схема 15 ), а в качестве гликозил-акцептора – N-ацетил-β-D-глюкозаминид (121) с 4-метилумбеллиферильным агликоном. Взаимодействием соединений (120) и (121) в присутствии Ph2SO и Tf2O получали дисахарид (122) с выходом 53%. Удаление в нем защитных групп приводило к получению целевого продукта (118).
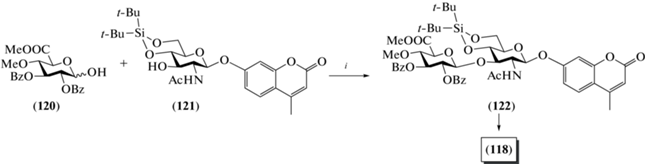
Схема 15 . Синтез целевого дисахарида (118). Реагенты и условия: i: Ph2SO, Tf2O, CH2Cl2, –20°C, 53%; ii: a – NaOMe, MeOH, CH2Cl2; b – HF·Py; c – Na2CO3 (водн.), 63% на 3 стадии.
Для синтеза тетрасахарида (119) в качестве гликозил-донора использовали дисахаридный N‑фенил-трифторацетимидат (123) (схема 16 ). Его сочетание с моносахаридом (121) приводило к образованию трисахарида (124) с выходом 60%. Селективное удаление левулиновой защитной группы в соединении (124) позволило получить трисахаридный гликозил-акцептор (125) с практически количественным выходом. Взаимодействие соединений (125) и (126) приводило к тетрасахариду (127) с выходом 85%. Последний далее переводили в целевой тетрасахарид (118).
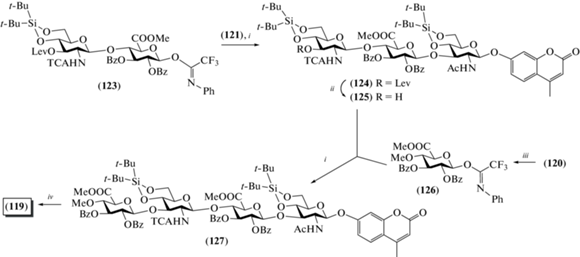
Схема 16 . Синтез целевого тетрасахарида (119). Реагенты и условия: i: TfOH, CH2Cl2, 0°C, 60% для соединения (124), 85% для соединения (127); ii: N2H4 ⋅ AcOH, Py, AcOH, 99%; iii: Cs2CO3, ClC(NPh)CF3, (CH3)2CO, 65%; iv: a – Zn порошок, AcOH; b – KI, (CH3)2CO; c – Zn порошок, AcOH; d – NaOMe, MeOH, CH2Cl2; e – (HF)3·Et3N, Py; f – Na2CO3 (водн.), 43% на 6 стадий.
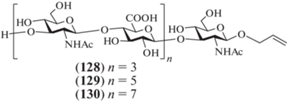
В 2012 г. van der Marel, Codée et al. описали синтез гепта- (128), ундека- (129) и пентадекасахарида (130), родственных ГК и содержащих аллильный агликон (рис. 11) [37]. Особенность данной работы – использование метода автоматического синтеза на твердой подложке для сборки углеводного скелета.
Для синтеза соединений (128–130) использовали полученные ранее моно- и дисахаридные блоки (97) (схема 12 ) и (123) (схема 16 ). На первой стадии моносахарид (97) иммобилизировали на смоле Меррифилда, затем селективно удаляли левулиновую защиту и проводили реакцию с дисахаридным гликозил-донором (123) (схема 17 ). Проведение циклов удаления левулиновой защитной группы и гликозилирования дисахаридом (123) 3, 5, 7 раз и последующее извлечение продуктов с твердофазного носителя позволило получить гепта- (131), ундека- (132) и пентадекасахарид (133) исключительно в автоматическом режиме. После удаления защитных групп целевые олигосахариды (128–130) были выделены с суммарными выходами 26, 32 и 18% соответственно, что представляется весьма хорошим результатом для таких крупных соединений. Стоит отметить, что описанный синтез – первый пример автоматизированного получения олигосахаридов, родственных фрагментам ГК и в целом гликозаминогликанов с использованием углеводного синтезатора.
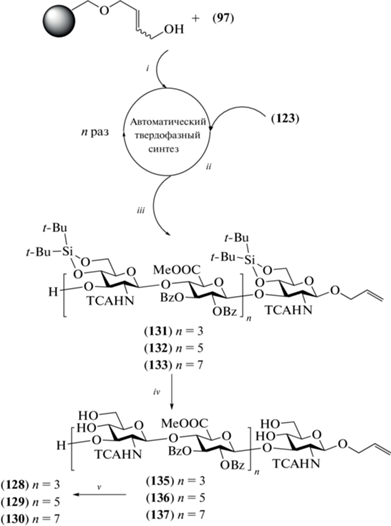
Схема 17 . Синтез целевых олигосахаридов (128–130). Реагенты и условия: i: N2H4, Py, AcOH, 2 × 10 мин; ii: a – TfOH, CH2Cl2, 0°C, 3 × 30 мин; b – N2H4, Py, AcOH, 2 × 10 мин; iii: катализатор Граббса, Cl3CC(=O)NH2, H2C=CH2, CH2Cl2; iv: 3HF·Et3N, ТГФ; v: a – KOH, H2O, ТГФ, 3–4 дня; b – Ac2O, NaHCO3, H2O, 26% для соединения (128) на 10 стадий, 32% для соединения (129) на 14 стадий, 18% для соединения (130) на 18 стадий.
В 2013 и 2015 гг. Giannis et al. описали синтез аллилгликозидных тетрасахаридов (137) и (138), родственных ГК [38], и гексасахарида (139) в виде 3-азидо-2-гидроксипропильного производного [39] (рис. 12). В тетрасахариде (138) остаток D‑глюкуроновой кислоты включал 13С-метки для последующего использования этого соединения при изучении связывания ГК с белками-мишенями, включая IL-8 и факторы роста, методами ЯМР-спектроскопии.
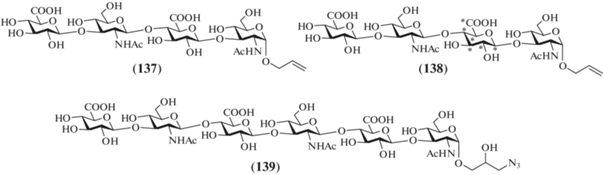
Для проведенных синтезов в качестве гликозил-донора был выбран трихлорацетимидат (140) [40], использованный ранее при получении олигосахаридов, родственных фрагментам хондроитинсульфата. Взаимодействие тиогликозида (141) с моносахаридом (140) в присутствии TMSOTf приводило к образованию дисахаридного гликозил-донора (142) с выходом 90% (схема 18 ). Для получения дисахаридного гликозил-акцептора (145) сначала проводили сочетание моносахаридов (140) и (143) (62%), а затем в образующемся продукте (144) удаляли силильную защитную группу, что протекало с выходом лишь 35%. Сборка блоков (142) и (145) по схеме [2+2] приводила к тетрасахариду (146) (59%), полное удаление защитных групп в котором приводило к получению целевого продукта (137). Избирательное же удаление силильной защитной группы в тетрасахариде (146) и последующее гликозилирование продукта (147) дисахаридом (142) приводили к гексасахариду (148). Модификация аллильного агликона и удаление защитных групп в (148) позволили получить целевой продукт (139).
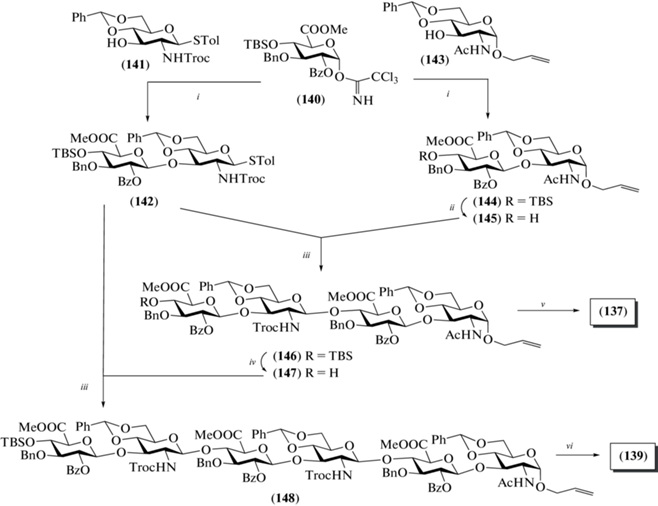
Схема 18 . Синтез целевых олигосахаридов (137) и (139). Реагенты и условия: i: TMSOTf, –20°C, 90% для соединения (142), 62% для соединения (144); ii: TBAF, ТГФ, 35%; iii: NIS, TfOH, 59% для соединения (146), 49% для соединения (148); iv: HF·Py, 78%; v: a – AcOH, Zn; b – Ac2O, Py, 43% на две стадии; с – HF·Py, 46%; d – MeONa, MeOH, Amberlite 120; e – NaOH, H2O, Dowex 50, 63% на 3 стадии; vi: a – AcOH, Zn, 50%; b – HF·Py; c – Ac2O, Py, 70%; d – DMSO, (CH3)2CO, –78°C; e – NaN3, DMF, 70% на две стадии.
Для получения тетрасахарида (138), содержащего 13С-метки в остатке D-глюкозы, был использован 13С-меченый моносахаридный предшественник (149) (схема 19 ). Его взаимодействие с моносахаридом (143) приводило к образованию дисахарида (150) (80%), в котором селективно удаляли пара-метоксибензильную группу, окисляли С-6, метилировали образовавшуюся карбоксильную группу, после чего действием TBAF удаляли силильную защиту (35%). Полученный дисахаридный блок (153) был использован в синтезе целевого тетрасахарида (138) так же, как и соединение (145) в синтезе тетрасахарида (137).
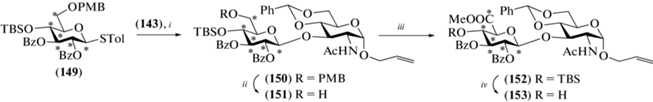
Схема 19 . Получение дисахаридного блока (153). Реагенты и условия: i: NIS, AgOTf, 80%; ii: DDQ, CH2Cl2, насыщ. водн. NaHCO3, 67%; iii: a – BAIB, TEMPO, CH2Cl2, H2O; b – MeI, K2CO3, 90% на две стадии; iv: TBAF, ТГФ, 35%.
В 2013 г. Jacquinet и Lopin-Bon et al. выполнили синтез 2-нафтилметилгликозидных ди- (154), тетра- (155) и пентасахарида (156), родственных ГК (рис. 13) [41]. Необычность данного синтеза заключается в использовании в качестве ключевого предшественника дисахарида (157), который был получен из коммерчески доступного природного хондроитинсульфата (схема 20 ) [42].
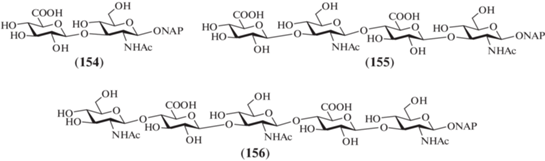
Природный хондроитинсульфат построен из дисахаридных повторяющихся звеньев →3)-β-D-GalNAc-(1→4)-β-D-GlcA-(1→, в которых сульфатные группы располагаются, как правило, при О-4 или О-6 в остатке N-ацетил-D-галактозамина [43]. Авторами ранее был разработан метод получения избирательно защищенного дисахарида (157) из хондроитинсульфата [42], который включал десульфатирование, направленное расщепление (1→4)-β-гликозидных связей, метилирование карбоксильных групп и расстановку защитных групп.
Дальнейшая трансформация дисахарида (157) заключалась в удалении бензилиденовой защитной группы, селективном 6-О-бензоилировании в остатке D-галактозамина с образованием продукта (158) (схема 20 ). Обращение конфигурации при С-4 в остатке D-галактозамина было выполнено генерацией 4-трифлата действием Tf2O c последующим нуклеофильным замещением TfO-группы на гидроксильную и ацетилированием. Таким образом, превращение остатка GalN в GlcN прошло с суммарным выходом 63%, что представляется хорошим результатом в данном случае.
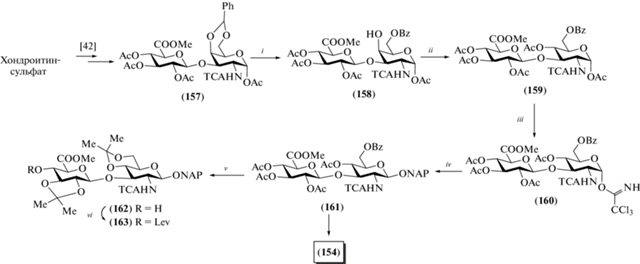
Схема 20 . Синтез целевого дисахарида (154). Реагенты и условия: i: a – TFA : H2O (3 : 1), CH2Cl2, 0°C, 3 ч, 82%; b – BzCN, Py, 3 ч, 88%; ii: a – (CF3SO2)2O, CH2Cl2, Py, 0°C, 2 ч, 95%; b – NBu4NO2, толуол, 4 ч, 82%; с – Ac2O, Py, 16 ч, 81%; iii: a – N2H4·AcOH, DMF, 40 мин; b – CCl3CN, ДБУ, CH2Cl2, 30 мин, 61% на две стадии; iv: NAPOH, TMSOTf, CH2Cl2, 30 мин, 94%; v: a – MeONa, MeOH, 5 ч, 67%; b – (CH3)2C(OCH3)2, CSA, DMF, 2 ч, 68%; vi: LevOH, DCC, DMAP, CH2Cl2, 3 ч, 94%.
Из полностью защищенного дисахарида (159) получали гликозил-донор (160), который вводили в реакцию с 2-нафтилметанолом и получали гликозид (161) с выходом 94%. Удаление защит в этом соединении приводило к образованию целевого дисахарида (154). Дезацетилирование дисахарида (161) действием метилата натрия в метаноле и последующее введение изопропилиденовых защит позволили получить дисахарид (162), в который далее вводили левулиновую группу при О-4 остатка D-глюкозы (→163) действием левулиновой кислоты в присутствии DCC.
Полученный дисахарид (163) использовали для синтеза блоков, необходимых для сборки крупных олигосахаридов (схема 21 ). Удалением ацетильных защитных групп и введением на их место бензоильных заместителей получали соединение (164) с выходом 72%. Селективное удаление 2-нафтилметильной группы действием DDQ и последующий перевод полученного полуацеталя в трихлорацетимидат приводили к получению гликозил-донора (165) (45% на две стадии). Селективное удаление левулиновой защиты в соединении (164) действием гидразина давало гликозил-акцептор (166) (84%). Сочетание дисахаридов (165) и (166) в присутствии TMSOTf приводило к тетрасахариду (167) с выходом 43%. После удаления всех защитных групп в последнем получали целевой тетрасахарид (155), а при селективном снятии только левулиновой группы – гликозил-акцептор (168). В результате сборки цепи по схеме [1+4] с использованием блоков (169) и (168) получали пентасахарид (170) (50%), удаление защитных групп в котором приводило к образованию целевого пентасахарида (156).
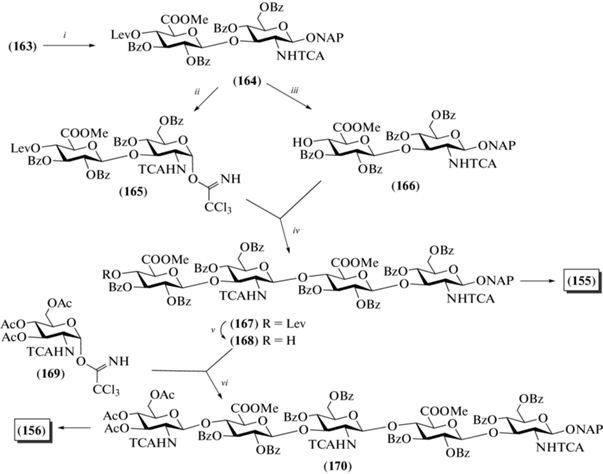
Схема 21 . Синтез целевых олигосахаридов (155) и (156). Реагенты и условия: i: а – 60% AcOH, 100°C, 1 ч; b – BzCl, Py, 0°C, 2 ч, 72% на две стадии; ii: a – DDQ, CH2Cl2/MeOH, 24 ч; b – СCl3CN, ДБУ, CH2Cl2, 30 мин, 45% на две стадии; iii: N2H4, Py, 8 мин, 84%; iv: TMSOTf, CH2Cl2, 30 мин, 43%; v: N2H4, Py, 8 мин, 72%; vi: TMSOTf, CH2Cl2, 30 мин, 50%.
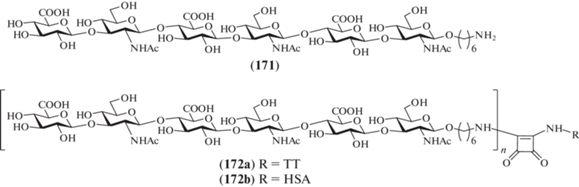
В 2013 г. Pinto et al. синтезировали спейсерированный гексасахарид (171), родственный ГК (рис. 14) [44]. На основе этого соединения были получены гликоконъюгаты (172a) и (172b) с белками-носителями: столбнячным анатоксином (tetanus toxoid, TT) и сывороточным альбумином человека (human serum albumin, HSA). Гликоконъюгаты были использованы в качестве иммуногенов для наработки антител к ГК [45]. Для защиты атома азота в остатках D-глюкозамина авторы использовали фталоильную группу. Для построения углеводного скелета были выбраны производные D-глюкозы, содержащие временную левулиновую защиту при О-6. В качестве гликозил-доноров использовали реакционноспособные трихлорацетимидаты.
Рис. 14.
Гексасахарид, родственный ГК, и гликоконъюгаты на его основе, синтезированные Pinto et al. [44].
Взаимодействием моносахаридов (173) и (174) в присутствии TMSOTf был получен дисахарид (175) с выходом 81% (схема 22 ), в котором удаляли монохлорацетильную группу при О-3 остатка D-глюкозамина при обработке тиомочевиной с образованием моногидроксильного продукта (176) (73%). Гликозилирование дисахарида (176) трихлорацетимидатом (177) приводило к получению трисахарида (178) (82%), который затем переводили в гликозил-донорный блок (179) (67%). Удалением метоксифенильной группы в соединении (175) и последующим превращением образующегося полуацеталя в трихлорацетимидат получали гликозил-донор (180) с выходом 70%. Его взаимодействие со спейсерированным моносахаридом (181) и последующее селективное удаление хлорацетильной защитной группы в образующемся продукте (182) приводило к получению гликозил-акцепторного блока (183). Сборка цепи по схеме [3+3] с использованием блоков (179) и (183) позволила получить гексасахарид (184) с выходом 70%.
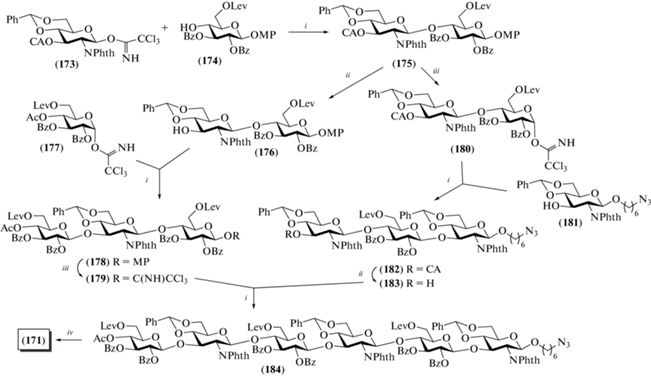
Схема 22 . Синтез целевого гексасахарида (171). Реагенты и условия: i: TMSOTf, CH2Cl2, –40°C → 0°C, 81% для соединения (175), 82% для соединения (178), 83% для соединения (182), 70% для соединения (184); ii: CS(NH2)2, 2,6-лутидин, CH2Cl2/MeOH, 73% для соединения (176), 78% для соединения (183); iii: a – (NH2)2Ce(NO3)6, CH3CN/H2O, 0°C; b – СCl3CN, ДБУ, CH2Cl2, 67% на две стадии для соединения (179), 70% для соединения (180); iv: a – 80% AcOH, 70°C; b – Ac2O, Py, 78%; c – N2H4, AcOH, 2 : 1 EtOH−C6H5CH3, 89%; d – PDC/Ac2O, CH2Cl2, 67%; e – H2NCH2CH2NH2, 1-бутанол; f – Ac2O, Py; g – 1 н. LiOH, ТГФ, 0°C, 72%; h – 10% Pd/C, NaBH4 в 0.05 М NaOH, 90%.
Дальнейшее превращение соединения (184) проводили с использованием стандартных методов. Сначала действием 80%-ной уксусной кислоты были удалены три бензилиденовые защиты, а затем ацетилировали высвободившиеся гидроксильные группы при С-4 и С-6 остатков глюкозамина. Далее следовало селективное удаление левулиновых защит, после чего свободные гидроксильные группы при С-6 остатков глюкозы были окислены дихроматом пиридиния. Действием этилендиамина проводили удаление N-фталоильных защитных групп, после чего выполняли N-ацетилирование действием уксусного ангидрида. После этого удаляли бензоильные защиты, восстанавливали азидную группу в спейсере и получали спейсерированный гексасахарид (171).
Получение конъюгатов лиганда (171) с белками-носителями (конъюгаты (172a, b)) проводили в два этапа: реакцией лиганда (171) с диэтилскваратом в буферном растворе получали производное моноэтилскварата, которое после гель-хроматографии далее конъюгировали с белком-носителем в 0.1 М карбонатном буфере при pH 10.
Эксперименты на мышах показали высокую иммуногенность конъюгата (172a) с белком TT. Это свидетельствует о том, что гексасахаридный лиганд достаточен, чтобы вызвать значимую выработку антител к ГК. Полученные антитела использовали для мониторинга в плазме и моче уровня низкомолекулярных цепей ГК – маркеров таких заболеваний, как цирроз печени, ревматоидный артрит, а также состояния сепсиса [45].
В 2014 г. Nieto et al. описали синтез метоксифенилгликозидных три- (185) и тетрасахарида (186), родственных ГК (рис. 15) [46]. Для защиты атома азота использовали трихлорацетильную группу, для защиты карбоксильной группы в моносахаридных предшественниках – бензильную группу, а в качестве гликозилирующих агентов были выбраны трихлорацетимидаты.
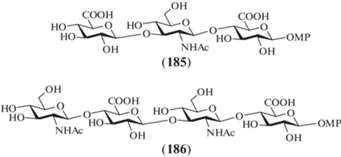
При получении соединений (185) и (186) сравнивали два подхода, в первом из которых использовали обычные наборы защитных групп и методов выделения продуктов реакций, а во втором в структуру гликозил-акцептора включали ацильный полифторированный остаток, что позволяло проводить выделение продуктов на специальном хроматографическом носителе. Так, из коммерчески доступной диацетон-D-глюкозы был получен моносахаридный блок (187), который затем переводили как в бензиловый эфир глюкуроновой кислоты (188), так и в производное (189), содержащее фторированную ацильную группу при О-6 (схема 23 ). Последовательность удлинения цепи включала сочетание моносахаридного акцептора с трихлорацетимидатом (190), после чего в полученном дисахариде удаляли левулиновую защиту и снова проводили гликозилирование трихлорацетимидатом (195). Удаление защитных групп в полученных таким образом трисахаридах (196) и (198) приводило к образованию целевого продукта (185). Сравнение выходов превращений, проведенных двумя способами, показало, что наличие фторированного фрагмента не улучшает выход продукта, а наоборот, снижает его. Авторы объясняли это необратимой сорбцией части соединения с фторированным ацилом на носителе, именно поэтому синтез целевого тетрасахарида (186) был выполнен без использования фторированного ацила. Для этого в трисахариде (196) удаляли левулиновую защиту, а полученный продукт (197) гликозилировали имидатом (190). В образующемся тетрасахариде (199) далее удаляли защитные группы с помощью обычных методов и получали тетрасахарид (186).
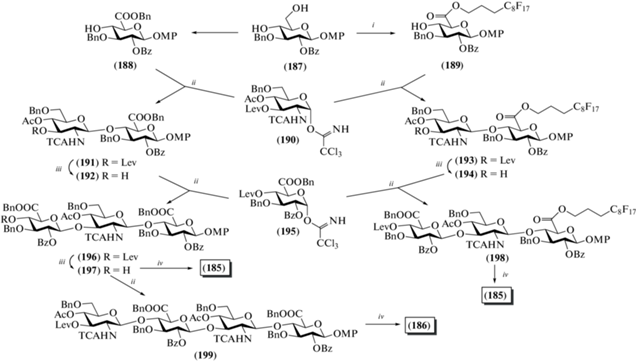
Схема 23 . Синтез целевых олигосахаридов (185) и (186). Реагенты и условия: i: a – TEMPO, Ca(ClO)2, Bu4NBr, KBr, NaHCO3, CH2Cl2/H2O, 0°C; b – C8F17-(CH2)3-I, DMF, 60°C, 55%; ii: TMSOTf, CH2Cl2, 0°C, 72% для соединения (191), 41% для соединения (196), 12% на три стадии для соединения (198), 64% для соединения (199); iii: N2H4, Py/AcOH, CH2Cl2, 93% для соединения (192), 71% для соединения (197); iv: а – LiOH, H2O2, TГФ; b – NaOH, MeOH; Ac2O, MeOH, Et3N; c – H2, Pd(OH)2, H2O/MeOH, 95–97%.
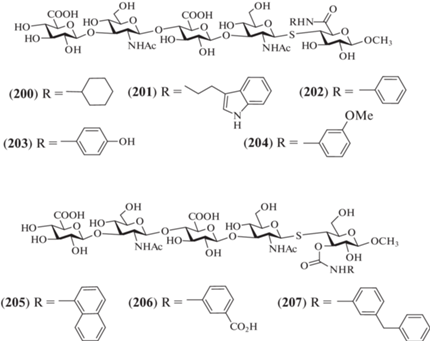
В 2015 г. X. Lu и X. Huang провели синтез пентасахаридов (200–207) – производных и аналогов пентасахарида, родственного фрагменту ГК (рис. 16) [47]. Соединения (200–204) содержали гидрофобные заместители при карбоксильной группе концевого остатка D-глюкуроновой кислоты, а соединения (205–207) – при О-3 этого же остатка. Кроме этого, в соединениях (200–207) один из кислородных межзвеньевых мостиков заменен на серный, который более устойчив к ферментативному расщеплению. Такие производные были необходимы для изучения эффективности связывания ГК с CD44-рецептором на поверхности большинства типов клеток млекопитающих, который отвечает за процессы клеточной адгезии, миграции и деления [48]. Например, предполагалось, что наличие гидрофобных заместителей в соединениях (200–207) будет способствовать лучшему связыванию олигосахаридов с белком CD44.
Проведенный синтез был основан на использовании в качестве одного из исходных соединений тетрасахарида (208), образующегося при ферментативном гидролизе ГК (схема 24 ) [49, 50]. Авторам удалось из 80 г ГК получить 50 г тетрасахарида (208). Далее действием смолы Dowex (TBA+) соединение (208) переводили в аммонийную соль, после чего метилировали карбоксильные группы и ацетилировали гидроксильные группы. Полностью защищенный тетрасахарид (209) в виде α- и β-изомеров был получен с выходом 35%. Далее из него получали α-бромид (210), который затем переводили в тиоацеталь (211).
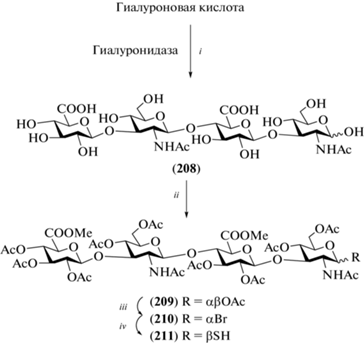
Схема 24 . Синтез тетрасахаридного блока (211). Реагенты и условия: i: гиалуронидаза, 37°С, рН 5.2, NaOAc; ii: a – Dowex (TBA+); b – MeI, DMF; c – Ac2O, Py/DMF, 35% на три стадии; iii: HBr/AcOH, Ac2O, CH2Cl2; iv: а – TBASAc, HSAc, CH3CN, 30%; b – NaOMe, –40°C, 100%.
Кроме соединения (211) в качестве исходных для получения пентасахаридов (200–207) использовали гликозил-акцепторные блоки (213) и (214), содержащие трифторметансульфонатную группу при С-4 (схема 25 ). Они были получены из метил-галактозида (212) в 6 стадий, как показано на схеме 25 . Взаимодействие моносахарида (213) с тетрасахаридом (211) в присутствии основания, удаление силильной защиты, ацетилирование и последующее удаление левулиновой защиты приводили к получению пентасахарида (215) с выходом 19%. Взаимодействие последнего с серией изоцианатов (R-NCO) в присутствии основания и последующее удаление защитных групп приводили к получению целевых олигосахаридов (205–207), содержащих гидрофобный заместитель при О-3 глюкозного остатка.
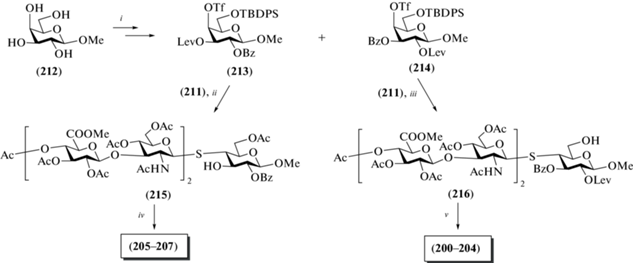
Схема 25 . Синтез целевых олигосахаридов (200–207). Реагенты и условия: i: a – (OMe)C6H4C(OMe)2, CSA, DMF; b – LevOH, CH2Cl2, DMAP, DCC; c – BzCl, DMAP; d – TsOH, MeOH; e – TBDPSCl, имидазол; f – Tf2O, Py; ii: a – TEA, DMF; b – HF·Py; c – Ac2O, Py; d – H2NNH2, Py/AcOH, 19%; iii: a – TEA, DMF; b – HF·Py; c – Ac2O, Py, 29% на две стадии; iv: a – R-NCO, DMAP; b – NaOMe, H2O/MeOH, pH 9.5; v: a – BAIB, TEMPO; b – RNH2, EDC, DMAP; c – NaOMe, H2O/MeOH, pH 9.5.
Взаимодействие моносахарида (214) с тетрасахаридом (211) в присутствии основания и последующее удаление силильной защиты приводили к образованию пентасахарида (216) с выходом 29%. Окисление гидроксиметильной группы до карбоксильной и последующая конденсация с серией аминов в присутствии EDC, а затем удаление защитных групп приводили к целевым олигосахаридам (200–204), содержащим гидрофобный заместитель при карбоксильной группе остатка глюкуроновой кислоты на “восстанавливающем” конце.
Полученные продукты (200–207) были изучены в качестве потенциальных ингибиторов взаимодействия ГК и белка CD44 методом конкурентного иммуноферментного анализа (ИФА). Наибольшую активность продемонстрировало соединение (207), содержащее мета-бензил-фенилкарбаматный фрагмент при О-3 глюкозного звена. Полученные результаты могут быть использованы в дальнейшем при разработке фармацевтических препаратов, влияющих на процессы клеточной адгезии и миграции клеток.
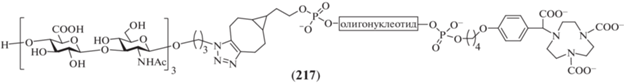
Как уже отмечалось выше, синтезы олигосахаридов, родственных ГК, часто проводятся для получения моделей, необходимых в исследовании клеточных процессов с участием этого полисахарида. Так, в 2016 г. Virta et al. осуществили синтез сложного гликоконъюгата (217), включающего в качестве углеводного лиганда тример повторяющегося звена ГК (рис. 17) [51]. Это соединение было получено в качестве основы для последующего создания контрастера для позитронно-эмиссионной томографии (ПЭТ КТ). Помимо гексасахаридной части, конъюгат (217) включает олигонуклеотидный фрагмент anti-miR-15b (22 основания), а также хелатирующий фрагмент NOTA для комплексования иона 68Ga.
Рис. 17.
Гликоконъюгат, содержащий тример повторяющегося звена ГК, синтезированный Virta et al. [51].
Сборка углеводного скелета гексасахарида, требуемого для получения гликоконъюгата (217), была выполнена с использованием моносахаридных блоков (218) и (219) (схема 26 ). Их взаимодействием в присутствии TMSOTf получали дисахарид (220) с выходом 81%. Удалением силильного агликона и последующим переводом образующегося полуацеталя в трихлорацетимидат получали дисахаридный гликозил-донор (221) (83%). Этим соединением гликозилировали азидопропанол с последующим удалением в образующемся продукте левулиновой защиты и получали дисахаридный гликозил-акцептор (224).
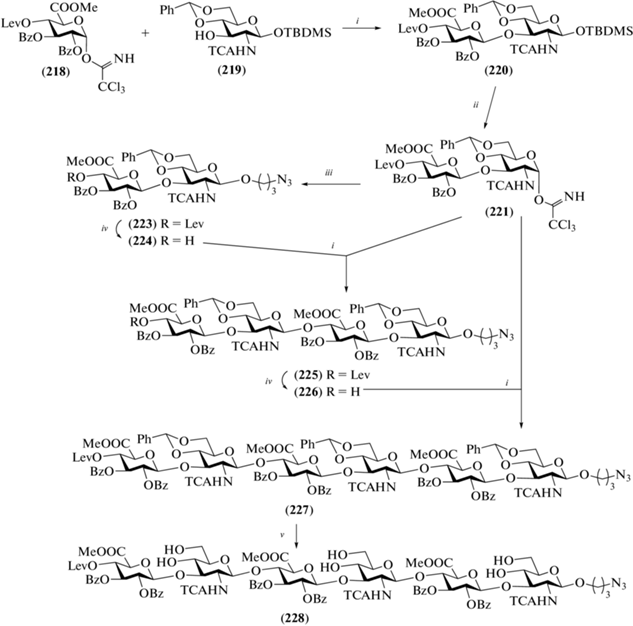
Схема 26 . Синтез предшественника (228). Реагенты и условия: i: TMSOTf, CH2Cl2, 0°C, 81% для соединения (220), 33% для соединения (225); ii: a – Et3N·3HF, ТГФ; b – CCl3CN, ДБУ, 0°C, 83%; iii: TMSOTf, 3-азидопропанол, CH2Cl2, 0°C, 40%; iv: N2H4, Py, AcOH, 75% для соединения (224), 78% для соединения (226); v: p-TSA, MeOH, 16%, начиная с соединения (226).
Далее по схеме [2+2] проводили сборку блоков (221) и (224) с образованием тетрасахарида (225), которая протекала с умеренным выходом 33%. Удаление левулиновой защиты в соединении (225) и удлинение цепи на два остатка снова с использованием донора (221) приводили к получению гексасахарида (227), в котором затем были удалены бензилиденовые защиты с образованием продукта (228). Именно это соединение было использовано для конъюгации, а удаление защитных групп в гексасахариде было выполнено на финальных стадиях получения гликоконъюгата (217), которые в данном обзоре не рассматриваются. Однако отметим, что применение 68Ga-контрастера на основе конъюгата (217) на модели инфаркта миокарда у крыс позволило более эффективно визуализировать поврежденную область миокарда, чем при использовании контрастера, не содержащего олигосахаридный фрагмент.
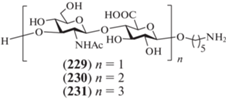
В 2020 г. Hung et al. синтезировали ди- (229), тетра- (230) и гексасахарид (231) с аминопентильным спейсерным агликоном (рис. 18) [52]. В отличие от всех описанных выше работ этого типа, авторы использовали азидную группу при С-2 в остатках D-глюкозамина, которая, строго говоря, выступает не защищенной аминогруппой, а ее донором. Для построения углеводного скелета авторы выбрали производные D-глюкозы с временной ацетильной группой при О-6, которые на финальных стадиях синтеза были переведены в остатки D-глюкуроновой кислоты.
В качестве гликозил-доноров авторы использовали трихлорацетимидаты и тиогликозиды. Так, гликозилированием моносахарида (233) α-трихлорацетимидатом (232) в присутствии BF3.OEt2 получали β-связанный дисахарид (234) (81%) (схема 27 ), хотя 2-азидогруппа не является соучаствующей для построения 1,2-транс-связи. Авторы предполагают, что реакция в этом случае проходила по механизму SN2 с обращением конфигурации при аномерном центре. Далее действием TMSSTol и ZnI2 соединение (234) было переведено в тиогликозид (235), гликозилирование которым производного аминопентанола (236) приводило к получению дисахарида (237) (88%).
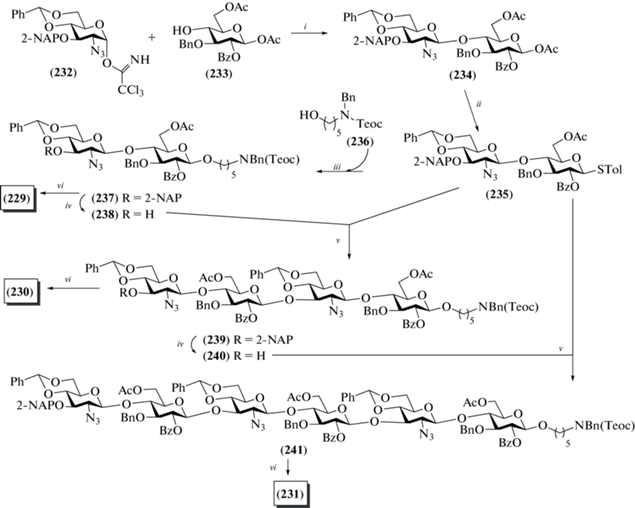
Схема 27 . Синтез целевых олигосахаридов (229–231). Реагенты и условия: i: BF3·OEt2, CH2Cl2, –78°С, 81%; ii: ZnI2, TMSSTol, CH2Cl2, 0°С, 72%; iii: NIS, AgOTf, CH2Cl2, –60°С → –20°С, 88%; iv: DDQ, H2O/CH2Cl2, 78% для соединения (238), 69% для соединения (240); v: NIS, TfOH, CH2Cl2, –78°С → –40°С, 79% для соединения (237), 77% для соединения (241); vi: a – Mg(OMe)2, CH2Cl2, 0°С; b – TEMPO/BAIB, CH3CN/H2O; c – CH2N2, CH2Cl2; d – NaOH, MeOH, CHCl3; e – PMe3⋅ТГФ; f – Ac2O, Et3N, MeOH; g – H2, Pd(OH)2/C; h – 1 M TBAF в ТГФ, CH3CN.
Для получения тетра- и гексасахаридов авторы использовали блочную сборку углеводного скелета. Сначала в дисахариде (237) действием DDQ была селективно удалена 2-нафтилметильная защитная группа с образованием гликозил-акцепторного блока (238). Сборка цепи по схеме [2+2] с использованием блоков (235) и (238) приводила к тетрасахариду (239) с выходом 79%. Удаление 2‑NAP-группы привело к образованию тетрасахаридного гликозил-акцептора (240), взаимодействие которого с гликозил-донором (235) приводило к получению гексасахарида (241) с выходом 77%.
Селективное высвобождение гидроксильных групп при С-6 остатков глюкозы проводили при обработке Mg(OMe)2. Далее действием TEMPO/BAIB в ацетонитриле гидроксиметиленовые группы окисляли до карбоксильных с последующим их переводом в соответствующие метиловые эфиры. Восстановление азидогруппы проводили действием триметилфосфина, а удаление бензильных и бензилиденовых защит было выполнено в условиях гидрогенолиза.
Было изучено взаимодействие полученных соединений (229–231) с белком CD44 методом калориметрического титрования. Дисахарид (229) не был активен, тогда как более крупные соединения, тетра- (230) и гексасахарид (231), показали значимое связывание с белком CD44, что позволило сделать вывод о размере соответствующего эпитопа.
ЗАКЛЮЧЕНИЕ
Природный полисахарид гиалуроновая кислота, построенная из дисахаридных повторяющихся звеньев →3)-β-D-GlcNAc-(1→4)-β-D-GlcA-(1→, играющая важную роль в протекании различных процессов клеточного узнавания, в том числе при развитии заболеваний, продолжает привлекать большой интерес исследователей. Это относится в том числе и к углеводным химикам, которые проводят синтезы олигосахаридов и гликоконъюгатов, структурно родственных фрагментам ГК. В данном обзоре рассмотрены различные стратегии сборки олигосахаридов, родственных ГК, включающие вариации защитных групп в остатках D-глюкуроновой кислоты и D-глюкозамина, различные типы блочной сборки углеводного скелета, использование автоматизированного твердофазного синтеза, а также применение в качестве исходных олигосахаридных блоков продуктов ферментативного расщепления природных полимеров гиалуроновой кислоты и хондроитинсульфата. Синтезированные олигосахариды и гликоконъюгаты на их основе – модельные соединения, а подчас и незаменимые инструменты для установления взаимосвязи структуры и биологических свойств ГК. Описанные в обзоре гликоконъюгаты на основе синтетических олигосахаридов с разной длиной цепи уже были использованы в качестве субстратов при изучении специфичности фермента гиалуронидазы, а также модельных гаптенов для оценки эффективности связывания ГК с различными белками-мишенями (например, с IL-8, факторами роста, CD44). Синтетические олигосахариды, родственные ГК, использованы при создании иммуногенов для получения антител, распознающих ГК, а также для дизайна векторированных контрастеров для компьютерной томографии. Кроме того, в последнее время активно развивается направление, связанное с доставкой лекарственных препаратов с использованием векторов на основе ГК [53–55]. В связи с этим представляется актуальной дальнейшая разработка эффективных методов синтеза олигосахаридов, родственных фрагментам ГК, необходимого строения. Весьма перспективно в этой связи использование гликозил-доноров на основе 2-азидо-2-дезокси-1-селено-D-глюкозы. Предложенные в последнее время препаративные методы получения такого рода соединений путем азидофенилселенилирования гликалей [56–58], в том числе при использовании проточной технологии (flow-technology) [59], сделали доступными такого рода гликозил-доноры, которые уже активно используются в реакциях гликозилирования [60–66], в том числе с построением 1,2-транс-гликозидных связей [60]. Таким образом, можно предположить, что подходы к получению олигосахаридов, родственных ГК, будут продолжать развиваться, как и гликобиологические и гликотехнологические исследования с использованием таких соединений.
Список литературы
Mende M., Bednarek C., Wawryszyn M., Sauter P., Biskup M.B., Schepers U., Bräse S. // Chem. Rev. 2016. V. 116. P. 8193–8255. https://doi.org/10.1021/acs.chemrev.6b00010
Baytas S.N., Linhardt R.J. // Drug Discov. Today. 2020. V. 25. P. 2095–2109. https://doi.org/10.1016/j.drudis.2020.09.011
Orgueira H.A., Bartolozzi A., Schell P., Litjens R., Palmacci E.R., Seeberger P.H. // Chem. Eur. J. 2003. V. 9. P. 140–169. https://doi.org/10.1002/chem.200390009
Lindahl U. // Haemostasis. 1990. V. 20. P. 146–153. https://doi.org/10.1159/000216173
Chang C.H., Lico L.S., Huang T.Y., Lin S.Y., Chang C.L., Arco S.D., Hung S.C. // Ang. Chem. Int. Ed. 2014. V. 53. P. 9876–9879. https://doi.org/10.1002/anie.201404154
Petitou M., Imberty A., Duchaussoy P., Driguez P.A., Ceccato M.L., Gourvenec F., Sizun P., Herault J.P., Perez S., Herbert J.M. // Chem. Eur. J. 2001. V. 7. P. 858–873. https://doi.org/10.1002/1521-3765(20010216)7:4<858: :aid-chem858>3.0.co;2-n
Fallacara A., Baldini E., Manfredini S., Vertuani S. // Polymers. 2018. V. 10. P. 701–737. https://doi.org/10.3390/polym10070701
Abbruzzese F., Basoli F., Costantini M., Giannitelli S.M., Gori M., Mozetic P., Rainer A., Trombetta M. // J. Biol. Regul. Homeost. Agents. 2017. V. 31. P. 9–22.
Abdallah M.M., Fernández N., Matias A.A., Bronze M.R. // Carbohydr. Polym. 2020. V. 243. P. 116441. https://doi.org/10.1016/j.carbpol.2020.116441
Jiang D.H., Liang J.R., Noble P.W. // Physiol. Rev. 2009. V. 91. P. 221–264. https://doi.org/10.1152/physrev.00052.2009
Aya K.L., Stern R. // Wound Rep. Reg. 2014. V. 22. P. 579–593. https://doi.org/10.1111/wrr.12214
Jiang D., Liang J., Noble P.W. // Annu. Rev. Cell Dev. Biol. 2007. V. 23. P. 435–461. https://doi.org/10.1146/annurev.cellbio.23.090506.123337
Ponta H., Sherman L., Herrlich P.A. // Nat. Rev. Mol. Cell Biol. 2003. V. 4. P. 33–45. https://doi.org/10.1038/nrm1004
Haylock D.N., Nilsson S.K. // Regen. Med. 2006. V. 1. P. 437–445. https://doi.org/10.2217/17460751.1.4.437
Belting M. // Thrombosis Res. 2014. V. 133. P. S95–S101. https://doi.org/10.1016/S0049-3848(14)50016-3
Tan J.-X., Wang X.-Y., Su X.-L., Li H.-Y., Shi Y., Wang L., Ren G.-S. // PLoS One. 2011. V. 6. P. e22836. https://doi.org/10.1371/journal.pone.0022836
Takabe P., Bart G., Ropponen A., Rilla K., Tammi M., Tammi R., Pasonen-Seppänen S. // Exp. Cell Res. 2015. V. 337. P. 1–15. https://doi.org/10.1016/j.yexcr.2015.07.026
Weissmann B., Meyer K. // J. Am. Chem. Soc. 1954. V. 76. P. 1753–1757.
Nukada T., Bereces A., Zgierski M.Z., Whitfield D. // J. Am. Chem. Soc. 1998. V. 120. P. 13291–13295.
Токатлы А.И., Винницкий Д.З., Устюжанина Н.Е., Нифантьев Н.Э. // Биоорг. химия. 2021. Т. 47. С. 57–75. [Tokatly A.I., Vinnitskiy D.Z., Ustuzhanina N.E., Nifantiev N.E. // Russ. J. Bioorg. Chem. 2021. V. 47. P. 53–70.] https://doi.org/10.31857/S0132342321010255
Slaghek T.M., Nakahara Y., Ogawa T. // Tetrahedron Lett. 1992. V. 33. P. 4971–4974. https://doi.org/10.3762/bjoc.11.67
Slaghek T.M., Nakahara Y., Ogawa T., Kamerling J.P., Vliegenthart J.F.G. // Carbohydr. Res. 1994. V. 255. P. 61–85. https://doi.org/10.1016/s0008-6215(00)90971-6
Slaghek T.M., Hypponen T.K., Ogawa T., Kamerling J.P., Vliegenthart J.F.G. // Tetrahedron Lett. 1993. V. 34. P. 7939–7942.
Slaghek T.M., Hypponen T.K., Ogawa T., Kamerling J.P., Vliegenthart J.F.G. // Tetrahedron: Asymmetry. 1994. V. 5. P. 2291–2301.
Halkes K.M., Slaghek T.M., Hyppönen T.K., Kruiskamp P.H., Ogawa T., Kamerling J.P., Vliegenthart J.F. // Carbohydrate Res. 1998. V. 309. P. 161–174. https://doi.org/10.1016/s0008-6215(98)00116-5
Dullenkopf W., Castro-Palomino J.C., Manzoni L., Schmidt R.R. // Carbohydr. Res. 1996. V. 296. P. 135–147. https://doi.org/10.1016/s0008-6215(96)00237-6
Blatter G., Beau J.-M., Jacquinet J.-C. // Carbohydr. Res. 1994. V. 260. P. 189–202. https://doi.org/10.1016/0008-6215(94)84038-5
Coutant C., Jacquinet J.-C. // J. Chem. Soc. Perkin Trans. 1995. V. 1. P. 1573–1581.
Blatter G., Jacquinet J.-C. // Carbohydr. Res. 1996. V. 288. P. 109–125. https://doi.org/10.1016/s0008-6215(96)90785-5
Huang L., Huang X. // Chem. Eur. J. 2007. V. 13. P. 529–540. https://doi.org/10.1002/chem.200601090
Dinkelaar J., Code J.D., van den Bos L.J., Overkleeft H.S., van der Marel G.A. // J. Org. Chem. 2007. V. 72. P. 5737–5742. https://doi.org/10.1021/jo070704s
Lu X., Kamat M.N., Huang L., Huang X. // J. Org. Chem. 2009. V. 74. P. 7608–7617. https://doi.org/10.1021/jo9016925
Dinkelaar J., Gold H., Overkleeft H.S., Code J.D., van der Marel G.A. // J. Org. Chem. 2009. V. 74. P. 4208–4216. https://doi.org/10.1021/jo9003713
Virlouvet M., Gartner M., Koroniak K., Sleeman J.P., Brase S. // Adv. Synth. Catal. 2010. V. 352. P. 2657–2662.
Gold H., Munneke S., Dinkelaar J., Overkleeft H.S., Aerts J.M., Code J.D., van der Marel G.A. // Carbohydr. Res. 2011. V. 346. P. 1467–1478. https://doi.org/10.1016/j.carres.2011.03.042
Jin P., Kang Z., Zhang N., Du G., Chen J. // Sci. Rep. 2014. V. 4. P. 4471. https://doi.org/10.1038/srep04471
Walvoort M.T.C., Volbeda A.G., Reintjens N.R., van den Elst H., Plante O.J., Overkleeft H.S., van der Marel G.A., Codée J.D. // C. Org. Lett. 2012. V. 14. P. 3776–3779. https://doi.org/10.1021/ol301666n
Rigol S., Xia L., Giannis A. // Bioorg. Med. Chem. 2013. V. 21. P. 733–741. https://doi.org/10.1016/j.bmc.2012.11.025
Bantzi M., Rigol S., Giannis A. // Beilstein J. Org. Chem. 2015. V. 11. P. 604–607. https://doi.org/10.3762/bjoc.11.67
Tully S.E., Mabon R., Gama C.I., Tsai S.M., Liu X., Hsieh-Wilson L.C. // J. Am. Chem. Soc. 2004. V. 126. P. 7736–7737. https://doi.org/10.1021/ja0484045
Lopez A.F., Jacquinet J.-C., Lopin-Bon C. // Eur. J. Org. Chem. 2013. V. 2013. P. 6934–6947.
Lopin C., Jacquinet J.-C. // Angew. Chem. Int. Ed. 2006. V. 45. P. 2574–2578. https://doi.org/10.1002/anie.200503551
Ji Y., Zhang S., Qiao M., Jiao R., Li J., Song P., Zhang X., Huang H. // Carbohydr. Polym. 2020. V. 248. P. 116796. https://doi.org/10.1016/j.carbpol.2020.116796
Gu G., Adabala P.J.P., Szczepina M.G., Borrelli S., Pinto B.M. // J. Org. Chem. 2013. V. 78. P. 8004–8019. https://doi.org/10.1021/jo4012442
Laurent T.C., Laurent U.B.G., Fraser J.R.E. // Ann. Med. 1996. V. 28. P. 241–253. https://doi.org/10.3109/07853899609033126
Macchione G., de Paz J.L., Nieto P.M. // Carbohydr. Res. 2014. V. 394. P. 17–25. https://doi.org/10.1016/j.carres.2014.05.007
Lu X., Huang X. // Glycoconj. J. 2015. V. 32. P. 549–556. https://doi.org/10.1007/s10719-015-9597-3
Lesley J. // J. Biol. Chem. 2000. V. 275. P. 26967–26975. https://doi.org/10.1074/jbc.M002527200
Tawada A., Masa T., Oonuki Y., Watanabe A., Matsuzaki Y., Asari A. // Glycobiology. 2002. V. 12. P. 421–426. https://doi.org/10.1093/glycob/cwf048
Mahoney D.J., Aplin R.T., Calabro A., Hascall V.C., Day A.J. // Glycobiology. 2001. V. 11. P. 1025–1033. https://doi.org/10.1093/glycob/11.12.1025
Jadhav S., Käkelä M., Mäkilä J., Kiugel M., Liljenbäck H., Virta J., Poijärvi-Virta P., Laitala-Leinonen T., Kytö V., Jalkanen S., Saraste A., Roivainen A., Lönnberg H., Virta P. // Bioconj. Chem. 2016. V. 27. P. 391–403. https://doi.org/10.1021/acs.bioconjchem.5b00477
Yeh C.-J., Zulueta M.M.L., Li Y.-K., Hung S.-C. // Org. Biomol. Chem. 2020. V. 18. P. 5370–5391. https://doi.org/10.1039/d0ob01048k
Luo Y., Ziebell M.R., Prestwich G.D. // Biomacromolecules. 2000. V. 1. P. 208–218. https://doi.org/10.1021/bm000283n
Luo, Y., Bernshaw N.J., Lu Z.R., Kopecek J., Prestwich G.D. // Pharm. Res. 2002. V. 19. P. 396–402. https://doi.org/10.1023/a:1015170907274
Peer D., Margalit R. // Neoplasia. 2004. V. 6. P. 343–353. https://doi.org/10.1593/neo.03460
Mironov Y., Sherman A., Nifantiev N. // Tetrahedron Lett. 2004. V. 45. P. 9107–9110. https://doi.org/10.1016/j.tetlet.2004.10.022
Fomitskaya P.A., Argunov D.A., Tsvetkov Y.E., Lalov A.V., Ustyuzhanina N.E., Nifantiev N.E. // Eur. J. Org. Chem. 2021. V. 2021. P. 5897–5904. https://doi.org/10.1002/ejoc.202101167
Qin C., Liu Z., Ding M., Cai J., Fu J., Hu J., Seeberger P.H., Yin J. // J. Carbohydr. Chem. 2020. V. 39. P. 374–397. https://doi.org/10.1080/07328303.2020.1839479
Guberman M., Pieber B., Seeberger P.H. // Org. Process Res. Dev. 2019. V. 23. P. 2764–2770. https://doi.org/10.1021/acs.oprd.9b00456
Khatuntseva E.A., Sherman A.A., Tsvetkov Y.E., Nifantiev N.E. // Tetrahedron Lett. 2015. V. 57. P. 708–711. https://doi.org/10.1016/j.tetlet.2016.01.013
Hagen B., Ali S., Overkleeft H.S., van der Marel G.A., Codée J.D.C. // J. Org. Chem. 2017. V. 82. P. 848–868. https://doi.org/10.1021/acs.joc.6b02593
Hagen B., van Dijk J.H.M., Zhang Q., Overkleeft H.S., van der Marel G.A., Codée J.D.C. // Org. Lett. 2017. V. 19. P. 2514–2517. https://doi.org/10.1021/acs.orglett.7b00747
Kazakova E.D., Yashunsky D.V., Krylov V.B., Bouchara J.P., Cornet M., Valsecchi I., Fontaine T., Latgé J.-P., Nifantiev N.E. // J. Am. Chem. Soc. 2020. V. 142. P. 1175–1179. https://doi.org/10.1021/jacs.9b11703
Kazakova E.D., Yashunsky D.V., Khatuntseva E.A., Nifantiev N.E. // Pure Appl. Chem. 2020. V. 92. P. 1047–1056. https://doi.org/10.1515/pac-2020-0105
Zhang Y., Gómez-Redondo M., Jiménez-Osés G., Arda A., Overkleeft H.S., van der Marel G.A., Jiménez-Barbero J., Codée J.D.C. // Angew. Chem. Int. Ed. 2020. V. 59. P. 12746–12750. https://doi.org/10.1002/anie.202003951
Kazakova E.D., Yashunsky D.V., Nifantiev N.E. // Molecules. 2021. V. 26. P. 5887. https://doi.org/10.3390/molecules26195887
Дополнительные материалы отсутствуют.
Инструменты
Биоорганическая химия