

Электрохимия, 2019, T. 55, № 11, стр. 1371-1380
Парадокс вариации объемного сопротивления мембран калиевых ионоселективных электродов в области нернстовского потенциометрического отклика
Е. О. Кондратьева a, Е. В. Соловьева a, Г. А. Хрипун a, К. Н. Михельсон a, *
a Санкт-Петербургский государственный университет
Санкт-Петербург, Россия
* E-mail: konst@km3241.spb.edu
Поступила в редакцию 28.01.2019
После доработки 23.04.2019
Принята к публикации 20.05.2019
Аннотация
Объемное сопротивление и другие электрохимические свойства мембран K+-селективных электродов (ИСЭ), содержащих валиномицин, измерены методами хронопотенциометрии и электрохимического импеданса. Показано, что объемное сопротивление мембран в области выполнения нернстовского потенциометрического отклика возрастает по мере снижения концентрации KCl в растворе. Аналогичные результаты сообщались ранее для Ca2+ и ${\text{NO}}_{{\text{3}}}^{ - }$ ИСЭ. Это непостоянство объемного сопротивления не согласуется с принятыми взглядами на механизм отклика ИСЭ. С осторожностью, данный парадокс приписан гетерогенности мембран вследствие поглощения ими воды из растворов.
ВВЕДЕНИЕ
Семейство химических сенсоров, основанных на применении ионофоров: липофильных молекул или ионов, способных к селективному связыванию различных аналитов, постоянно расширяется. Ионоселективные электроды (ИСЭ) на основе ионофоров – потенциометрические сенсоры, известны уже в течение десятилетий [1], и были первыми, где использовались достоинства этих агентов. ИСЭ активно используются для широкого набора практических применений, прежде всего в клинической химии, мониторинге окружающей среды и в промышленности [2–5]. Начиная с 1990-х годов, ионофоры используются также в оптических сенсорах (оптодах) [2] и кондуктометрических сенсорах [6, 7]. Недавно были предложены электрохимические сенсоры на базе ионофоров, использующие новые принципы регистрации сигнала. Это селективные вольтамперометрические [8–12] и кулонометрические сенсоры [13–16]. Для этих новых сенсоров на основе ионофоров существенно применение твердоконтактной конструкции с электрополимером, способным к окислению и восстановлению. Наиболее важные свойства вольтамперометрических и кулонометрических сенсоров на основе ионофоров получили теоретическое описание [10, 11, 15]. Теория ИСЭ разработана более детально. Помимо нернстовской части, объяснение получили нелинейные отклики, а также нижний и верхний пределы электродной функции. Простейшими являются равновесные модели [1–4, 17]. Более продвинутые модели учитывают эффекты диффузионных процессов вблизи границы раздела мембрана/раствор [3, 18, 19], а также трансмембранные потоки необменно сорбированного электролита [20, 21]. Дальнейшее обобщение позволило моделировать отклик ИСЭ в реальном времени и пространстве путем решения системы уравнений Нернста–Планка и Пуассона [22–25].
Важно, что вне пределов нернстовского отклика, когда потенциалы ИСЭ отклоняются от линейности вследствие необменной сорбции водного электролита при контакте электродов с концентрированными растворами, объемное сопротивление мембран ИСЭ снижается по мере увеличения концентрации раствора [21]. Однако в пределах линейного нернстовского отклика ИСЭ, в рамках названных теорий (будь то упрощенных или обобщенных), объемное сопротивление мембран должно быть постоянным. Дело в том, что линейный нернстовский отклик ИСЭ предполагает, что необменная сорбция пренебрежимо мала, и процесс ионного обмена на границе мембрана/раствор доминирует в формировании межфазного потенциала. Кроме того, в нернстовской области изменяется только межфазный потенциал на границе раздела мембрана/раствор, тогда как другие вклады в полный мембранный потенциал и в э. д. с. гальванической ячейки постоянны. В свою очередь, межфазный потенциал подчиняется уравнению:
(1)
${{\varphi }_{{\text{b}}}} = \frac{{\mu _{{\text{I}}}^{{0,{\text{aq}}}} - \mu _{{\text{I}}}^{{0,{\text{mem}}}}}}{F} + \frac{{RT}}{{{{z}_{{\text{I}}}}F}}\ln \frac{{a_{{\text{I}}}^{{{\text{aq}}}}}}{{a_{{\text{I}}}^{{{\text{mem}}}}}}.$(2)
${{\varphi }_{{\text{b}}}} = \varphi _{{\text{b}}}^{0} + \frac{{RT}}{{{{z}_{{\text{I}}}}F}}\ln a_{{\text{I}}}^{{{\text{aq}}}}$Калиевый ИСЭ с валиномицином в качестве нейтрального ионофора известен в течение десятилетий и тщательно исследован. Поэтому данный ИСЭ представляет собой подходящую модельную систему для исследования. Более того, наше раннее исследование выявило зависимость сопротивления электрода от концентрации KCl в растворе [26]. Поэтому, в данной работе мы исследуем электрохимические свойства K+-ИСЭ подробно, следуя тем же экспериментальным протоколам, что в наших недавних работах по Ca2+ и ${\text{NO}}_{{\text{3}}}^{ - }$ ИСЭ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Поливинилхлорид (ПВХ) С-66 (высокомолекулярный) был производства Охтален (Россия), нейцтральный ионофор валиномицин, катионообменник тетракис-(п-Cl-фенилборат) калия (KClТФБ), липофильный электролит тетракис-(п-Cl-фенилборат) тетрадодециламмония (ETH 500) и пластификатор бис(2-бутилпентил)адипинат (ББПА) были от Fluka (Швейцария). Тетрагидрофуран (ТГФ) и циклогексанон (ЦГ) были от Вектон (Россия) и перегонялись перед использованием. Неорганические соли (х. ч.) были от Реактив (Россия). Все водные растворы готовились на деионизованной воде с удельным сопротивлением 18.2 МОм см (Milli-Q Reference, Millipore, Франция). Мембранные коктейли готовили растворением ПВХ (460 мг), ББПА (920 мг), валиномицина (20.4 мг), KClТФБ (4.6 мг) и (если требовалось, см. табл. 1) также ETH 500 (10.5 мг) в 6.3 мл ТГФ. Для получения мембран коктейли перемешивали в течение 30 мин на роллер-миксере Movil Rod (Selecta, Испания) и затем выливали в стеклянные чашки Петри диаметром 50 мм. Чашки закрывали фильтровальной бумагой для замедления испарения ТГФ, полное испарение ТГФ занимало 1 сут. Так получали мастер-мембраны толщиной около 0.7 мм.
Таблица 1.
Составы мембран (массовые проценты и моляльности компонентов)
| Компонент | Мембрана 1 | Мембрана 2 |
|---|---|---|
| Валиномицин | 1.46%, 0.02 m | 1.44%, 0.02 m |
| KClТФБ | 0.33%, 0.01 m | 0.32%, 0.01 m |
| ETH 500 | 0 | 0.75%, 0.01 m |
| ББПА | 65.47% | 64.99% |
| ПВХ | 32.74% | 32.50% |
Составы мембран приведены в табл. 1. Эти составы типичны для K+-ИСЭ на основе валиномицина [3, 4, 29].
Электроды готовили, вырезая из мастер-мембран диски диаметром 12 мм, и приклеивая их к корпусам из трубок ПВХ с внешним диаметром 12 мм и внутренним 8 мм. Клеем служил раствор ПВХ в ЦГ. Внутренним электродом сравнения служила хлорированная серебряная проволока в полипропиленовом корпусе. Предварительно ИСЭ заполнили 0.01 M и кондиционировали в таком же растворе в течение недели.
Бестоковые потенциометрические измерения проводили с помощью 8-канальной потенциометрической станции Экотест-120 (Эконикс, Россия). Электродом сравнения был однокамерный хлоридсеребряный электрод ЭВЛ-1-М3 (Измеритель, Беларусь), заполненный 3.5 M KCl, с солевым мостиком, заполненным таким же раствором. Истечение KCl из мостика было очень ограничено, поэтому загрязнение растворов ионами K+ и Cl– из мостика было незначительным даже при низких концентрациях. Калибровки в растворах KCl проводили от 0.1 M вниз до 10–8 M KCl, используя разбавитель 700 Dosino под управлением контроллера 711 Liquino (Metrohm, Швейцария).
Хронопотенциометрические кривые и спектры электрохимического импеданса снимали с помощью потенциостата-гальваностата Autolab 302N с модулем частотного анализатора FRA 2 (Metrohm, Швейцария). Хронопотенциометрические и импедансные измерения проводили в симметричных ячейках с растворами одинаковых составов снаружи и внутри ИСЭ, используя Ag/AgCl-электроды (хлорированные серебряные проволочки), погруженные в эти растворы. Вспомогательным электродом был стержень из стеклоуглерода.
В хронопотенциометрических измерениях потенциал электрода сначала регистрировали в течение 10 с без тока. Затем подавали импульс тока магнитудой 10–8 A и длительностью 60 c и регистрировали потенциал. Среднеквадратичное значение площади поверхности ИСЭ было 0.75 см2, поэтому плотность тока была 1.33 × 10–8 A/см2. После этого ток выключали и записывали потенциал еще 60 с. Разрешение по времени в хронопотенциометрических измерениях составляло 0.2 с. Импедансные измерения проводили в потенциостатических условиях с магнитудой возбуждающего сигнала ±5 мВ относительно потенциала открытой цепи, в диапазоне частот от 100 кГц до 0.01 Гц.
Все измерения проводили в полипропиленовом сосуде объемом 50 мл при комнатной температуре (22 ± 1°C). В работе использовали по 3 электрода-репликанта, соответсвенно, ИСЭ 1, 2 и 3 с мембраной 1 и ИСЭ 4, 5 и 6 с мембраной 2.
Активности катионов K+ и Na+ в растворах вычисляли по уравнению 2-го приближения теории Дебая–Хюккеля:
(3)
$\lg {{\gamma }_{{\text{I}}}} = - \frac{{0.512\sqrt J }}{{1 + a_{{\text{I}}}^{{{\text{Kiel}}}}0.328\sqrt J }}.$РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
K+-ИСЭ с мембранами на основе валиномицина детально охарактеризованы в плане их потенциометрического отклика и селективности [1–5]. Поэтому наши потенциометрические исследования были ограничены проверкой линейной области отклика ИСЭ в растворах KCl и их K+/Na+ селективностью. Кривые отклика, полученные при последовательном 10-кратном разбавлении 50 мл 0.1 M раствора KCl деионизованной водой до 10–8 M, приведены на рис. 1. Когда концентрация KCl достигла наименьшего значения: 10–8 M, из сосуда отобрали 5 мл раствора и заменили на 5 мл 1 M NaCl, создав концентрацию ионов Na+ 0.1 M. Так мы получили данные о селективности ИСЭ.
Рис. 1.
Кривые отклика ИСЭ при последовательном разбавлении 0.1 M KCl деионизованной водой: (а) ИСЭ 1–3, (б) ИСЭ 4–6. Вертикальные линии относятся к моментам разрыва контакта при разбавлениях.
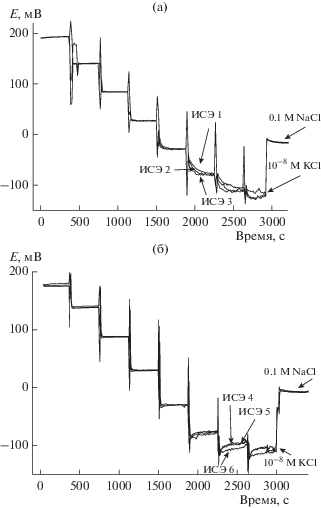
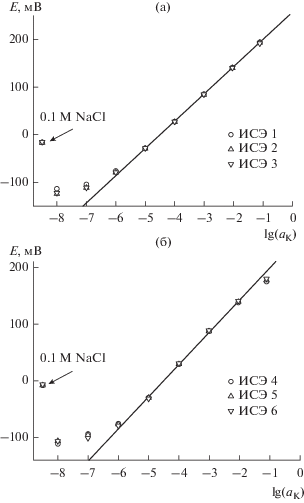
В каждом растворе э. д. с. записывали в течение 300 с, среднее значение за последние 100 с использовали для построения калибровочных кривых, приведенных на рис. 2. ИСЭ проявили линейный отклик на K+ от 0.1 до 10–6 M KCl, нижний предел обнаружения был lg(aK) = –6.6. Дальнейшее снижение концентрации KCl приводит к потере отклика. Угловые коэффициенты в линейной области для ИСЭ 1–3 и ИСЭ 4–6 составили, соответственно, 57.3 ± 0.4 мВ/lg(aK) и 56.8 ± 0.4 мВ/lg(aK), т.е. близко к теоретическому нернстовскому значению для температуры 22 ± ± 1°C. Коэффициенты K+/Na+ селективности (метод раздельных растворов) для этих электродов составили lgKK/Na ≈ –4.77 и lgKK/Na ≈ –4.64. Полученные значения угловых коэффициентов, пределов обнаружения и коэффициентов селективности типичны для K+-ИСЭ на основе валиномицина [4, 29, 30], таким образом, электроды были пригодны к дальнейшим исследованиям.
Рис. 2.
Калибровочные кривые, полученные в растворах KCl, и величины э. д. с., измеренные в 0.1 M NaCl: (а) ИСЭ 1–3, (б) ИСЭ 4–6.
Сопротивление и другие электрохимические параметры ИСЭ исследовали путем хронопотенциометрических и импедансных измерений. Мембраны контактируют с двумя водными растворами: внутренним и внешним, и обе соответствующие границы раздела дают вклады в измеряемые сигналы. Поэтому, корректная интерпретация хронопотенциометрической кривой или спектра импеданса может быть проведена только для симметричной ячейки: внутренний и внешний растворы должны быть одинаковы, как и соответствующие электроды сравнения (хлорированные серебряные проволочки) [21, 27, 28, 31]. Концентрацию KCl варьировали от 10–5 до 10–1 M, т.е. в той области, где ИСЭ проявляют линейный нернстовский отклик.
ИСЭ выдерживали в соответствующих растворах в течение 1 недели до измерений, чтобы быть уверенными, что мембраны пришли в равновесие с водной фазой. Поляризация Ag/AgCl-электродов в контакте с 10–3 M и более концентрированными растворами KCl была пренебрежимо малой по сравнению с поляризацией мембран. В случае растворов 10–5 и 10–4 M KCl, хронопотенциометрические кривые, полученные с ИСЭ, корректировали на поляризацию Ag/AgCl-электродов и сопротивление растворов. Для этого проводили измерения с ПВХ мембранами и без них. Пример измеренных и скорректированных хронопотенциометрических кривых приведен на рис. 3. Видно, что без коррекции результаты были бы смещены вследствие поляризации хлоридсеребряных электродов и за счет сопротивления раствора. Вклад этих эффектов возрастает по мере разбавления водной фазы (вставка в рис. 3).
Рис. 3.
Хронопотенциометрические кривые, полученные в 10–5 M KCl. Данные для ИСЭ 1 (без ETH 500) и ИСЭ 4 (с ETH 500), измеренные, а также скорректированные с учетом кривых, полученных для Ag/AgCl-электродов без мембраны. Вставка показывает хронопотенциометрические кривые Ag/AgCl-электродов в 10–5–10–1 M KCl.

В качестве примера, хронопотенциометрические кривые, полученные с ИСЭ 1 (скорректированные на сопротивление раствора и поляризацию хлоридсеребряных электродов), приведены на рис. 4а. До включения тока потенциал был близок к нулю вследствие симметрии ячейки. По включении тока наблюдался омический скачок потенциала в положительном направлении и последующая поляризационная кривая. При выключении тока наблюдали омический скачок потенциала в отрицательном направлении и последующую кривую релаксации. Мы полагаем, что омические скачки потенциала в хрнопотенциометрических измерениях обусловлены объемными сопротивлениями ИСЭ. Импедансные измерения (см. ниже) подтверждают эту интерпретацию. Величины положительного и отрицательного омических скачков (и вычисленные из них значения объемного сопротивления мембран) совпадали с точностью ±1%. Объемные сопротивления ИСЭ 1–3 (без ETH 500), приведенные в равновесие с 10–1 M KCl, составили 2.72 ± 0.09 МОм, а таковые для ИСЭ 4–6 (с ETH 500) были 1.03 ± ± 0.09 МОм. В равновесии с 10–2 M KCl сопротивления были несколько выше, соответственно 2.88 ± 0.07 и 1.13 ± 0.10 МОм. По мере снижения концентрации KCl эти величины возрастали, и наибольший рост объемного сопротивления был получен при замене 10–4 M раствора KCl на раствор 10–5 M KCl. В контакте с этим раствором сопротивления ИСЭ достигли, соответственно, 4.81 ± 0.12 и 1.77 ± 0.07 МОм. Данные приведены в табл. 2.
Рис. 4.
(а) Хронопотенциометрические кривые, полученные с ИСЭ 1 в равновесии с растворами KCl 10–5 – 10–1 M. Врезки показывают поляризационную и релаксационную части кривой, записанной в 10–2 M KCl. (б) Кинетика изменения объемного сопротивления ИСЭ 1 при замене 10–2 M KCl на 10–4 M KCl, и обратно.

Таблица 2.
Объемное сопротивление ИСЭ, измеренное методами хронопотенциометрии и импеданса. Средние значения и среднеквадратичные отклонения для трех электродов-репликантов
| Электролит | ИСЭ 1–3, Rbulk, MОм | ИСЭ 4–6, Rbulk,MОм | ||
|---|---|---|---|---|
| хронопотенциометрия | импеданс | хронопотенциометрия | импеданс | |
| KCl 10−1 M | 2.72 ± 0.09 | 2.80 ± 0.06 | 1.03 ± 0.09 | 1.01 ± 0.07 |
| KCl 10−2 M | 2.88 ± 0.07 | 2.81 ± 0.10 | 1.13 ± 0.08 | 1.12 ± 0.08 |
| KCl 10−3 M | 3.17 ± 0.07 | 3.20 ± 0.07 | 1.11 ± 0.09 | 1.20 ± 0.09 |
| KCl 10−4 M | 3.33 ± 0.06 | 3.29 ± 0.05 | 1.37 ± 0.08 | 1.36 ± 0.10 |
| KCl 10−5 M | 4.81 ± 012 | 4.80 ± 0.11 | 1.77 ± 0.07 | 1.83 ± 0.07 |
| KSal 10−1 M | 2.12 ± 0.10 | 2.33 ± 0.08 | ||
| KSal 10−2 M | 2.78 ± 0.08 | 2.72 ± 0.09 | ||
| KPi 10−2 M | 2.12 ± 0.08 | 1.99 ± 0.09 | ||
| KPi 10−3 M | 3.03 ± 0.09 | 2.98 ± 0.08 | ||
Кинетику изменения объемного сопротивления ИСЭ при изменении концентрации раствора исследовали так же, как ранее в случае Ca2+ электродов [27]. Сначала выдерживали ИСЭ в контакте с 10–4 M KCl (заполненные тем же раствором) в течение 24 ч, после чего снимали хронопотенциометрические кривые. После этого растворы: внешний и внутренний, заменяли на 10–2 M KCl. Манипуляции по смене растворов занимали примерно 1 мин. Хронопотенциометрические кривые в 10–2 M KCl снимали через 3 мин после смены растворов, и далее несколько раз в течение 10 ч. Как ранее в случае Ca2+-ИСЭ [27], основное изменение сопротивления происходило в течение 3 мин после смены растворов, а после этого происходили только незначительные изменения. Такое же поведение было зарегистрировано при обратной смене растворов, когда 10–2 M KCl заменили на 10–4 M KCl. Результаты приведены на рис. 4б.
Как ранее в случаях Ca2+-ИСЭ [27] и ${\text{NO}}_{{\text{3}}}^{ - }$-ИСЭ [28], так же и в случае K+-ИСЭ поляризационная и релаксационная части хронопотенциометрической кривой для всех электродов во всех растворах успешно описываются следующим выражением:
(4)
$\eta = i{{R}_{{{\text{expon}}}}}\left( {1 - \exp \left( { - {t \mathord{\left/ {\vphantom {t \tau }} \right. \kern-0em} \tau }} \right)} \right) + iN\sqrt t .$
Экспоненциальные части поляризационной и релаксационной кривых для данного ИСЭ в данном растворе оказались очень близки. Соответственно, значения ${{R}_{{{\text{expon}}}}},$ полученные из поляризационной или релаксационной кривой, совпадали в пределах 10% относительной погрешности. Эти величины хаотично разбросаны в пределах от 1 до 5 кОм для ИСЭ с или без ETH 500, без какой-либо регулярной зависимости от концентрации KCl в растворе. Значения характеристического времени составляли $\tau = 4 \pm 2$ с. Величины соответствующих емкостей, вычисленные как ${{C}_{{{\text{expon}}}}} = {\tau \mathord{\left/ {\vphantom {\tau {{{R}_{{{\text{expon}}}}}}}} \right. \kern-0em} {{{R}_{{{\text{expon}}}}}}},$ были очень велики: от 20 до 50 мкФ. Отсутствие регулярной зависимости ${{R}_{{{\text{expon}}}}}$ от концентрации KCl наряду с очень большой ${{C}_{{{\text{expon}}}}}$ означает, что эти величины не могут быть приписаны сопротивлению переноса заряда и емкости двойного слоя на границе мембрана/раствор. Осторожная интерпретация их природы предлагается ниже.
Второе слагаемое в уравнении (4) линейно зависит от квадратного корня из времени. Это предполагает, что данное слагаемое обусловлено транспортными ограничениями. Согласно соотношению Стокса–Эйнштейна, коэффициенты диффузии частиц обратно пропорциональны кубическому корню из их масс: ${{{{D}_{A}}} \mathord{\left/ {\vphantom {{{{D}_{A}}} {{{D}_{B}}}}} \right. \kern-0em} {{{D}_{B}}}}$ ≈ $\sqrt[3]{{{{{{M}_{B}}} \mathord{\left/ {\vphantom {{{{M}_{B}}} {{{M}_{A}}}}} \right. \kern-0em} {{{M}_{A}}}}}}.$ Таким образом, можно ожидать, что комплексы ионов K+ с валиномицином должны быть приблизительно на 36% менее подвижными, чем анионы ClTPB–. Поскольку речь в любом случае идет об очень грубой оценке, мы не пытались учитывать эту “ожидаемую” разницу в 36% и принимали подвижности частиц обоих видов равными. Удельное сопротивление мембраны ρ, а также диффузионная поляризация зависят от cion – концентрации отдиссоциированных ионов: K[Val]+ и ClTPB–, и от Dion – их коэффициентов диффузии как $\rho \sim {{\left( {{{c}_{{{\text{ion}}}}}{{D}_{{{\text{ion}}}}}} \right)}^{{ - 1}}}$ и ${{d\eta } \mathord{\left/ {\vphantom {{d\eta } {d{{t}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}} \right. \kern-0em} {d{{t}^{{{1 \mathord{\left/ {\vphantom {1 2}} \right. \kern-0em} 2}}}}}}$ ∼ $c_{{{\text{ion}}}}^{{ - 1}}D_{{{\text{ion}}}}^{{{{ - 1} \mathord{\left/ {\vphantom {{ - 1} 2}} \right. \kern-0em} 2}}}.$ Это позволяет оценить cion и Dion [32, 33]:
(5)
${{c}_{{{\text{ion}}}}} = \frac{{32RT}}{{\pi {{F}^{2}}}}\rho {{i}^{2}}{{\left( {{{d\eta } \mathord{\left/ {\vphantom {{d\eta } {dt}}} \right. \kern-0em} {dt}}} \right)}^{{ - 2}}},$(6)
${{D}_{{{\text{ion}}}}} = \frac{\pi }{{64}}{{\rho }^{{ - 2}}}{{i}^{{ - 2}}}{{\left( {{{d\eta } \mathord{\left/ {\vphantom {{d\eta } {dt}}} \right. \kern-0em} {dt}}} \right)}^{2}}.$Оценка коэффициентов диффузии составила ${{D}_{{{\text{ion}}}}} = \left( {1.0 \pm 0.5} \right) \times {{10}^{{ - 8}}}$ см2/с, что согласуется с величинами, полученными для мембран из пластифицированного ПВХ методом радиоактивных меток [34]. Наша оценка ниже полученных другими авторами (также электрохимически): около 2.5 × 10–8 см2/с [35] или около 3.0 × 10–8 см2/с [36]. Причина этого различия может быть в следующем. Авторы работ [35, 36] основывались на значениях коэффициентов диффузии анионов, оцененных отдельно, тогда как наша оценка основана на допущении равенства коэффициентов диффузии катионных комплексов и анионов (см. выше).
Оценки концентрации ионов составили ${{c}_{{{\text{ion}}}}}$ = (2 ± 1) × 10–3 M для мембран без ETH 500, и ${{c}_{{{\text{ion}}}}}$ = (3 ± 1) × 10–3 для мембран с ETH 500. Эти величины указывают, что степень диссоциации электролитов в мембранах составляет 15–20%.
Спектры импеданса измеряли после приведения ИСЭ в равновесие с растворами KCl концентрациями от 10–5 до 0.1 M. Спектры ИСЭ 4–6 содержали полуокружность в диапазоне частот от 100 кГц до 1 Гц и прямую линию на меньших частотах. В качестве примера, спектры импеданса ИСЭ 6 приведены на рис. 6а. Спектры импеданса ИСЭ 1–3 (без ETH 500) содержали только полуокружность, см. рис. 6б.
Рис. 6.
Спектры импеданса, полученные в растворах KCl: (а) ИСЭ 1 (без ETH 500), (б): ИСЭ 6 (с ETH 500).

Форма спектров, записанных для растворов KCl 10–4 и 10–5 M, предполагает отрицательные значения Zreal на наиболее высоких частотах. По-видимому, это артефакт, вызванный поляризацией Ag/AgCl-электродов при низкой концентрации Cl−-анионов. Спектры фитинговали к контурам, содержащим резистор R параллельно постоянно-фазовому элементу Q, и последовательно им импедансу Варбурга. Значения Q составили $\left( {6 \pm 2} \right) \times {{10}^{{ - 11}}}F{{s}^{{n - 1}}}$ с фактором n от 0.88 дo 0.94 (т.е., близко к 1), и не показали регулярной зависимости от концентрации KCl. Значения соответствующих постоянных времени были около 0.1–0.3 мс. Эти величины указывают, что R и Q отвечают объемному сопротивлению и геометрической емкости мембран. Тангенс наклона прямой на частотах ниже 1 Гц составлял около 0.6–0.7, существенно ниже, чем для “чистого” импеданса Варбурга. Фитинг этой части спектра к контуру, содержащему резистор, емкость и дифузионное сопротивление (по аналогии с уравнением (4) для хронопотенциометрии), не имел смысла из-за малости низкочастотной части в спектрах.
Значения объемного сопротивления, полученные из измерений импеданса, хорошо согласуются с таковыми, измеренными хронопотенциометрически, см. табл. 2 и рис. 7. Таким образом, результаты измерений импеданса подтверждают закономерности объемного сопротивления мембран, полученные из хронопотенциометрических измерений.
Рис. 7.
Объемное сопротивление K+-ИСЭ без ETH 500 (незаполненные символы) и с ETH 500 (заполненные символы) в растворах KCl.

Объемное сопротивление ИСЭ возрастает приблизительно на 8–16%, когда концентрация KCl снижается с 10–1 до 10–3 M. Дальнейшее разбавление KCl до 10−5 M вызывает рост объемного сопротивления мембран на 52–59%. В принципе, зависимость объемного сопротивления мембран от концентрации раствора может быть вызвана необменной сорбцией водного электролита [21]. Однако, необменная сорбция должна вызывать противоположную тенденцию. В случае необменной сорбции объемное сопротивление должно быть постоянным в разбавленных растворах и снижаться при контакте ИСЭ с более концентрированным KCl. Для K+-ИСЭ без фонового электролита ETH 500 известно, что необменная сорбция салицилата калия (KSal) и пикрата калия (KPi) вызывает анионный отклик вместо катионного в 10–1 M KSal или 10–2 M KPi [21]. Измерения объемного сопротивления ИСЭ 1–3 в контакте с этими растворами показали существенно меньшие значения, чем в контакте с KCl. Однако, в контакте с 10–2 M KSal или с 10–3 KPi сопротивление ИСЭ практически такое же, как в случае KCl (табл. 2). Эти данные подтверждают наш вывод, полученный для Ca2+- и ${\text{NO}}_{{\text{3}}}^{ - }$-ИСЭ [27, 28], о том, что необменная сорбция электролита водного раствора не может быть причиной непостоянства объемного сопротивления мембран в области выполнения нернстовского потенциометрического отклика ИСЭ.
ЗАКЛЮЧЕНИЕ
Полученные результаты показывают, что объемное сопротивление K+-ИСЭ зависит от концентрации раствора в области нернстовского потенциометрического отклика. Это согласуется с нашими результатами, полученными с Ca2+- и ${\text{NO}}_{{\text{3}}}^{ - }$-ИСЭ [27, 28], и не согласуется со сложившимися представлениями о механизме отклика ИСЭ и, в частности, о роли ионного обмена на границе мембрана/раствор. Происхождение зависимости объемного сопротивления мембран от концентрации раствора требует дальнейшего изучения. С осторожностью, мы относим эти результаты к гетерогенности реальных мембран, вызванной поглощением воды из раствора и образованию в мембранах капелек воды размером порядка 15–20 нм [37–40]. Кажется возможным, что межфазный потенциал устанавливается между водной фазой и собственно органической фазой мембраны, тогда как сопротивление относится к мембране как материалу. Капельки воды могут затруднять перенос заряда сквозь мембраны, например как ловушки для ионов. Более того, сообщалось, что содержание воды в мембранах возрастает по мере снижения концентрации раствора [40]. В этом случае объемное сопротивление мембраны ИСЭ может изменяться в пределах нернстовского отклика ИСЭ. Можно ожидать увеличения полярности мембран по мере роста поглощения воды, и потому некоторого закономерного изменения геометрической емкости мембран по мере разбавления водной фазы. Однако вследствие сильного разброса оценок этого параметра, полученных фитингом спектров импеданса, мы не можем сделать каких-либо ясных выводов о поведении геометрической емкости.
Двойные электрические слои вокруг водных капель, в совокупности, могут образовать распределенный конденсатор, что может быть причиной неожиданно высоких значений емкости, полученных при фитинге поляризационных и релаксационных кривых в хронопотенциометрических измерениях.
Список литературы
Morf, W.E., The Principles of Ion-Selective Electrodes of Membrane Transport, Budapest, Akademiai.
Bakker, E., Bühlmann, P., and Pretsch, E., Chem. Rev., 1997, vol. 97, p. 3083.
Bobacka, J., Ivaska, A., and Lewenstam, A., Chem. Rev., 2008, vol. 108, p. 329.
Mikhelson, K.N., Ion-selective Electrodes (Lecture Notes in Chemistry, vol. 81), Heidelberg–New York–Dordrecht–London, Springer, 2013.
Mikhelson, K.N. and Peshkova, M.A., Russ. Chem. Rev, 2015, vol. 84, p. 555.
Shul'ga, A.A., Ahlers, B., and Cammann, K., J. Electroanalyt. Chem., 1995, vol. 395, p. 305.
Cammann, K., Ahlers, B., Henn, D., Dumschat, C., and Shul’ga, A.A., Sens. Actuat. B., 1996, vol. 35, p. 26.
Kim, Y. and Amemiya, S., Anal. Chem., 2008, vol. 80, p. 6056.
Greenawalt, P.J. and Amemiya, S., Anal. Chem., 2016, vol. 88, p. 5827.
Yuan, D., Cuartero, M., Crespo, G.A., and Bakker, E., Anal. Chem., 2017, vol. 89, p. 586.
Yuan, D., Cuartero, M., Crespo, G.A., and Bakker, E., Anal. Chem., 2017, vol. 89, p. 595.
Jarolimova, Z., Bosson, J., Labrador, G.M., Lacour, J., and Bakker, E., Electroanalysis., 2018, vol. 30, p. 650.
Vanamo, U., Hupa, E., Yrjänä, V., and Bobacka, J., Anal. Chem., 2016, vol. 88, p. 4369.
Han, T., Vanamo, U., and Bobacka, J., ChemElectroChem., 2016, vol. 3, p. 2071.
Jarolímová, Z., Han, T., Mattinen, U., Bobacka, J., and Bakker, E., Anal. Chem., 2018, vol. 90, p. 8700.
Jaworska, E, Pawłowski, P., Michalska, A., and Maksymiuk, K., Electroanalysis, 2018, vol. 30, p. 1. (статья пока доступна только online, )https://doi.org/10.1002/elan.201800649
Mikhelson, K.N., Electroanalysis, 2003, vol. 15, p. 1236.
Егоров, В.В., Новаковский, А.Д., Здрачек, Е.А. Электрохимия. 2018. Т. 54. С. 437. [Egorov, V.V., Novakovskii, A.D., and Zdrachek, E.A., Russ. J. Electrochem., 2018, vol. 54, p. 381.]
Egorov, V.V., Novakovskii, A.D., and Zdrachek, E.A., Anal. Chem., 2018, vol. 90, p. 1309.
Morf, W.E., Badertscher, M., Zwickl, T., de Rooij, N.F., and Pretsch, E., J. Electroanal. Chem., 2002, vol. 526, p.19.
Ivanova, A.D., Koltashova, E.S., Solovyeva, E.V., Peshkova, M.A., and Mikhelson, K.N., Electrochim. Acta, 2016, vol. 213, p. 439.
Sokalski, T. and Lewenstam, A., Electrochem. Commun., 2001, vol. 3, p. 107.
Kucza, W., Danielewski, M., and Lewenstam, A., Electrochem. Commun., 2006, vol. 8, p. 416.
Jasielec, J.J., Sokalski, T., Filipek, R., and Lewenstam, A., Anal. Chem., 2015, vol. 87, p. 8665.
Szyszkiewicz, K., Jasielec, J.J., Danielewski, M., and Lewenstam, A., J. Electrochem. Soc., 2017, vol. 164, p. E3559.
Shvarev, A.E., Rantsan, D.A., and Mikhelson, K.N., Sens. Actuators B, 2001, vol. 76, p. 500.
Kondratyeva, Y.O., Solovyeva, E.V., Khripoun, G.A., and Mikhelson, K.N., Electrochim. Acta, 2018, vol. 259, p. 458.
Ivanova, A. and Mikhelson, K., Sensors., 2018, vol. 18, p. 2062.
Mikhelson, K.N., Bobacka, J., Lewenstam, A., and Ivaska, A., Electroanalysis, 2001, vol. 13, p. 876.
Bühlmann, P., Pretsch, E, and Bakker, E., Chem. Rev., 1998, vol. 98, p. 1593.
Mikhelson, K.N., Bobacka, J., Ivaska, A., Lewenstam, A., and Bochenska, M., Anal. Chem., 2002, vol. 74, p. 518.
Михельсон, К.Н., Лутов, В.М., Сулко, К., Стефанова, О.К. Электрохимия. 1988. Т. 24. С. 1487. [Mikhelson, K.N., Lutov, V.M., Sulko, K., and Stefanova, O.K., Sov. Electrochem., 1988, vol. 24, p. 1369.]
Peshkova, M.A., Korobeynikov, A.I., and Mikhelson, K.N., Electrochim. Acta, 2008, vol. 53, p. 5819.
Oesch, U. and Simon W., Helv. Chim. Acta, 1979, vol. 62, S. 754.
Bodor, S., Zook, J.M., Lindner, E., Tóth, K., and Gyurcsányi, R.E., J. Solid State Electrochem., 2009, vol. 13, p. 171.
Zook, J., Bodor, S., Lindner, E., Tóth, K., and Gyurcsányi, R.E., Electroanalysis, 2009, vol. 21, p. 1923.
Chan, A.D.C., Li, X., and Harrison, J.D., Anal. Chem., 1992, vol. 64, p. 2512.
Li, Z, Li, X., Petrovic, S., and Harrison, D.J., Anal. Chem., 1996, vol. 68, p. 1717.
Lindfors, T., Sundfors, F, Höfler, L., and Gyurcsányi, G.E., Electroanalysis, 2009, vol. 21, p. 1914.
He, N. and Lindfors, T., Anal. Chem., 2013, vol. 85, p. 1006.
Дополнительные материалы отсутствуют.