

Электрохимия, 2019, T. 55, № 11, стр. 1363-1370
Электрохимическая полимеризация пиррола в присутствии сульфокислотных полиэлектролитов
О. Л. Грибкова a, *, В. А. Кабанова a, А. А. Некрасов a
a Институт физической химии и электрохимии им. А.Н. Фрумкина РАН
119071 Москва, Ленинский просп., 31, корп. 4, Россия
* E-mail: oxgribkova@gmail.com
Поступила в редакцию 31.01.2019
После доработки 25.04.2019
Принята к публикации 20.05.2019
Аннотация
Проведено сравнительное исследование электрохимической полимеризации пиррола в присутствии солевых и кислотных форм гибкоцепных сульфокислотных полиэлектролитов. Электрохимическими и спектральными методами контроля хода синтеза показано, что электросинтез поли-пиррола в присутствии полиэлектролитов протекает с большими скоростями и при меньших концентрациях мономера, чем в водных растворах с неорганическими анионами. Обнаружено, что при использовании Н+-формы полиэлектролитов скорость полимеризации пиррола выше чем в соответствующих Na+-солях. Так же были изучены электрохимические, спектроэлектрохимические свойства в видимой и ближней ИК-областях и морфология полученных гибридных пленок полипиррола.
ВВЕДЕНИЕ
Полипиррол (ПП) является одним из наиболее широко изучаемых проводящих полимеров благодаря высокой проводимости и стабильности в проводящей форме. Он обладает уникальным сочетанием физико-химических, электрохимических и оптических характеристик, которые делают его перспективным для использования в химических и биологических сенсорах, в качестве электродных материалов в аккумуляторах и конденсаторах, электрохромных устройствах, материалов для антикоррозионной защиты, “искусственных мускулов” и т.д. [1].
Основными методами синтеза ПП являются химическая и электрохимическая полимеризация пиррола. Метод электрохимического синтеза пленок проводящих полимеров удобен тем, что на металлических и оптически прозрачных подложках с проводящим слоем происходит формирование равномерных слоев без примесей окислителя. Их толщину и морфологию можно регулировать изменением количества электричества, затраченного на электрохимический синтез. Таким образом, методика электрохимической полимеризации одновременно решает задачу синтеза полимера и нанесения его в виде тонкой пленки на проводящие подложки.
На электросинтез ПП влияют режимы его проведения, а так же состав электрополимеризационной среды (растворитель, фоновый электролит, концентрация мономера, pH) [2]. ПП получают электрохимически как в органических так и в водных растворах. В литературе отмечается, что электросинтез ПП в водных растворах приводит к получению более пористых пленок [3], при этом электрополимеризация пиррола в режиме циклирования потенциала начинается при менее положительных потенциалах, чем в ацетонитриле [3], и протекает с большей скоростью [4]. При использовании объемных полимерных анионов наблюдается увеличение скорости синтеза ПП [5]. Предполагается, что они легче адсорбируются на поверхности электрода, что ускоряет первую стадию электрополимеризации. Кроме этого объемные органические анионы повышают стабильность пленок ПП и их термическую устойчивость [5]. Полимерный анион включается в состав полимерной пленки и не может быть полностью выведен из нее при дедопировании [6, 7].
При электрополимеризации пиррола в основном использовали солевые формы таких полиэлектролитов, как поливинилсульфонат [7], полистиролсульфонат [5–9], и кислотную форму поли-2-акриламидо-2-метил-1-пропансульфоната [10]. При этом о различиях в характере синтеза ПП в присутствии разных (кислотной или солевой) форм полиэлектролита упоминалось только в [6]. Следует отметить, что большинство исследований было проведено при достаточно высоких концентрациях мономера (0.1–0.05 М). Только в работах [11, 12] для электросинтеза ПП в водных растворах с неорганическими анионами было изучено влияние концентрации мономера на полимеризацию пиррола.
Таким образом, проведенные до настоящего времени исследования в основном ограничиваются констатацией факта возможности проведения электросинтеза ПП в присутствии полиэлектролитов и не рассматривают влияние их химического строения и природы противоиона на характер синтеза и свойства пленок ПП.
В настоящей работе было проведено сравнительное исследование электрохимической полимеризации пиррола из водных растворов гибкоцепных полимерных сульфокислотных электролитов различной структуры, а также изучено влияние природы компенсирующего противоиона полиэлектролита на электрохимический синтез и свойства образующегося ПП. Процесс электросинтеза ПП был изучен методом in situ спектроскопии в УФ-видимой и ближней ИК-областях спектра с одновременным контролем электрохимических параметров синтеза. Впервые были исследованы спектроэлектрохимические свойства пленок ПП, полученных в присутствии полиэлектролитов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
ПП получали электрохимической полимеризацией пиррола в водных растворах сульфокислотных полиэлектролитов: поли-(2-акриламидо-2-метил-1-пропансульфоновой) кислоты (ПАМПСК), полистиролсульфокислоты (ПССК) и их солей. ПАМПСК использовали фирмы “Aldrich”, 15% водный раствор, Mw ≈ 2 × 106. Натриевую соль ПАМПСК (ПАМПСNa) получали нейтрализацией поликислоты эквимолярным водным раствором NaOH. ПССNa – “Aldrich”, 25%-ный водный раствор, Mw ≈ 1 × 106. Из Nа+- в Н+-форму ПССК переводили на ионообменной колонке, заполненной катионитом КУ-2-8чС. Все используемые полиэлектролиты очищали диализом относительно воды (целлюлозная мембрана ZelluTrans, MWCO 8000–10 000).
Пиррол перегоняли в атмосфере аргона, использовали свежеперегнанный продукт. Растворы полиэлектролитов требуемой концентрации готовили за день до синтеза. Растворение пиррола проводили при интенсивном перемешивании в течение 2 ч.
pH реакционных растворов (табл. 1) измеряли с помощью рН-метра OP-208/1 (Radelkis). Полимеризацию пиррола проводили в трехэлектродной ячейке, в качестве противоэлектрода использовали платиновую фольгу, электрода сравнения – насыщенный хлоридсеребряный электрод (н. х. с. э.). Все потенциалы в настоящей работе представлены относительно данного электрода.
Таблица 1.
pH растворов синтеза и электрохимические параметры, наблюдаемые при различных режимах электрополимеризации пиррола. EПД – потенциал начала окисления мономера в первом цикле ПД-режима, TПС – продолжительность индукционного периода при ПС-синтезе, ЕГС – потенциал синтеза в ГС-режиме
| pH | EПД, В | ЕГС, В | TПС, с | |
|---|---|---|---|---|
| ПССК | 2.3 | 0.65 | 0.64 | 43 |
| ПССNa | 4.1 | 0.69 | 0.65 | 67 |
| ПАМПСК | 2.4 | 0.66 | 0.65 | 48 |
| ПАМПСNa | 4.2 | 0.71 | 0.65 | 112 |
| ПАМПСNa | 5.5 | 0.76 | 0.77 | 160 |
| H2SO4 | 1.9 | 0.7 | 0.71 | 300 |
| Na2SO4 | 5.5 | 0.8 | Рост | 820 |
| Пиррол/Na2SO4 = 0.05 М/0.1 M | 6.0 | 0.76 | 0.6 | 121 |
Электроосаждение пленок ПП проводили в потенциостатическом (0.65 В), гальваностатическом (100 мкА/см2) и потенциодинамическом (в диапазоне –0.8–0.8 В при скорости развертки 50 мВ/с) режимах на электроде SnO2–стекло, площадью 2 см2. Молярное отношение пиррол/ сульфокислотные группы всегда поддерживали равным 1 молекуле пиррола на 2 сульфокислотные группы полиэлектролита. Концентрацию пиррола варьировали от 0.0025 до 0.015 М. Для сравнения также была проведена электрохимическая полимеризация пиррола в водных растворах, содержащих 0.01 М пиррола и 0.02 М серной кислоты или сульфата натрия, а также 0.05 М пиррола и 0.1 М сульфата натрия. Заряд, затраченный на синтез пленок ПП, составлял 50 мКл/см2. Пленки после синтеза промывали дистиллированной водой и сушили на воздухе. Толщина пленок ПП, полученного в присутствии полиэлектролитов, составляла 140 ± 10 и 80 ± 10 нм для ПП, полученного в неорганических электролитах. Для измерения толщины пленок использовали микроинтерферометр МИИ-4 (ЛОМО).
В процессе полимеризации пиррола проводили регистрацию электрохимических данных с одновременной регистрацией спектров оптического поглощения. Задание и регистрацию электрохимических параметров синтеза осуществляли с помощью потенциостата/гальваностата НА-501G (Hokuto Denko Ltd.) и цифрового запоминающего осциллографа Nicolet 2090 (Nicolet Inc.). Спектры оптического поглощения в процессе синтеза ПП (350–950 нм), а также последующие спектроэлектрохимические исследования полученных пленок в водном растворе 0.1 М NaClO4 проводили с помощью скоростного сканирующего однолучевого спектрофотометра Avantes 2048. Электронные спектры поглощения в ближней УФ-видимой-ближней ИК-области спектра регистрировали на двулучевом спектрофотометре “Shimadzu UV-3101PC”.
Атомную силовую микроскопию (АСМ) пленок ПП, полученных в гальваностатическом режиме в присутствии различных электролитов, проводили на АСМ-микроскопе Enviroscope с контроллером Nanoscope V (Bruker) в полуконтактном режиме. Шероховатость пленок оценивали как среднеквадратическое отклонение неровностей профиля поверхности по кадру (5 × 5 мкм). Кадры снимали в разных частях пленки и усредняли.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Электрохимическая полимеризация
В литературе в основном представлены данные по электрополимеризации пиррола при достаточно высоких концентрациях как мономера, так и электролита, в том числе и для полиэлектролитов [5–10]. Для выявления роли полиэлектролита при синтезе ПП нами были проведены исследования влияния концентрации мономера на скорость и характер синтеза при постоянном мольном соотношении мономер/сульфокислотные группы (1/2). На рис. 1а видно, что при достаточно низких концентрациях пиррола (0.015 М) по сравнению с работами [5–10] (0.1–0.05 М) потенциостатический (ПС) синтез ПП в присутствии ПАМПСК идет с высокой скоростью и имеет автокаталитический характер. При уменьшении концентрации мономера до 0.005 М величина тока электрополимеризации значительно снижается. В случае гальваностатического (ГС) синтеза (рис. 1б) при концентрациях мономера 0.015 и 0.01 М полимеризация протекает при одинаковых потенциалах, а с уменьшением концентрации до 0.005 М потенциал синтеза со временем начинает расти, что приводит к переокислению получаемого полимера. Поэтому, дальнейшие исследования были проведены при концентрации пиррола 0.01 М.
Рис. 1.
Кинетические кривые заряда и тока (а) при ПС (0.65 В) и потенциала (б) при ГС-синтезах ПП в водных растворах ПАМПСК с различной концентрацией пиррола: 1 – 0.015 М, 2 – 0.01 М, 3 – 0.005 М, 4 – 0.0025 М. Молярное отношение пиррол/сульфокислотные группы поликислоты составляло 1 : 2 моль/г-экв.

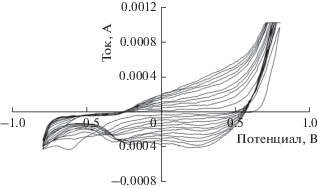
Для выявления влияния химического строения поликислоты и ее противоиона электрохимические синтезы ПП были проведены в водных растворах гибкоцепных поликислот ПССК и ПАМПСК и их Na+-солей, а также, для сравнения, в растворах H2SO4 и Na2SO4. На рис. 2 представлены циклические вольт-амперные (ЦВ) кривые, полученные при синтезе ПП в присутствии ПССК в потенциодинамическом (ПД) режиме в диапазоне от –0.8 до 0.8 В. Они имеют вид, который характерен для ЦВ-кривых ПД синтеза ПП в водной среде [3], и практически не зависит от химического строения поликислоты. Из табл. 1 видно, что потенциалы начала окисления мономера, определенные из 1 цикла ПД-синтеза, примерно одинаковы для синтеза в присутствии ПАМПСК и ПССК, но немного выше в присутствии их солей.
Рис. 2.
ЦВ-кривые, зарегистрированные при ПД синтезе ПП в водном растворе ПССК. Скорость развертки потенциала – 50 мВ/с.
В случае ПС-синтеза видно (рис. 3а), что продолжительность индукционного периода (рассчитанного из кинетических кривых изменения заряда) полимеризации пиррола (рис. 3а), отвечающего стадии димеризации катион-радикалов [12], зависит как от химического строения полиэлектролита, так и от природы его противоиона (табл. 1). При использовании полимерных кислот электрохимическая полимеризация пиррола начинается раньше (рис. 3б, кривые 1, 2) и протекает быстрее, чем в присутствии их солей (рис. 3б, кривые 1*, 2*). При этом в присутствии ПССК синтез ПП в ПС режиме идет при большем токе, чем в ПАМПСК (рис. 3б, кривые 1, 2). Наиболее длительный индукционный период наблюдается в присутствии натриевой соли ПАМПСК (рис. 3а). При ГС-синтезе (рис. 3в) полимеризация пиррола протекает при одинаковых потенциалах в присутствии как ПССК, так и ПАМПСК и их солей.
Рис. 3.
Кинетические кривые заряда (а), тока (б) при ПС синтезе (0.65 В) и потенциала (в) при ГС (100 мкА/см2) синтезе ПП в водных растворах ПССК (1), ПССNa (1*), ПАМПСК (2), ПАМПСNa (2*), H2SO4 (3), Na2SO4 (3*) и Na2SO4 (0.1 М) (4).

Рост потенциала окисления пиррола и длительности индукционного периода электросинтеза ПП в солевых формах полиэлектролитов, т.е. в средах с высокими pH, коррелирует с данными работы [13], где была показана роль ионов H+ и протонирования пиррола в процессе его электрополимеризации. Действительно при большей степени нейтрализации ПАМПСК (до pH 5.5) ПС-синтез идет еще медленнее, а ГС-синтез – при более высоком потенциале (табл. 1).
Необходимо отметить, что при проведении электрополимеризации ПП в серной кислоте при тех же условиях и концентрациях наблюдается значительное замедление синтеза (рис. 3а, 3б). При этом, потенциал окисления мономера остается примерно таким же, как и в полиэлектролитах, а индукционный период ПС-синтеза увеличивается в 2–6 раз. Потенциал ГС-синтеза в серной кислоте немного выше чем в полиэлектролитах. ПС-полимеризация пиррола в присутствии Na2SO4 при тех же концентрациях реагентов так же имеет автокаталитический характер, но протекает намного медленнее и с гораздо более длительным индукционными периодом, чем в полимерных электролитах и серной кислоте (табл. 1). В ГС-режиме наблюдается рост потенциала (рис. 3в), а потенциал окисления мономера в первом цикле ПД-синтеза в присутствии Na2SO4 (0.8 В) выше, чем потенциалы в присутствии полиэлектролитов (табл. 1). При увеличении концентрации мономера и Na2SO4 в 5 раз потенциал окисления мономера снижается и ПС-электросинтез ПП идет с высокой скоростью, а ГС-синтез – при более низком потенциале.
Таким образом, использование полимерных электролитов для полимеризации пиррола приводит к ускорению электрохимического синтеза и возможности использовать меньшие концентрации реагентов. Кроме того, получаемые пленки ПП высоко однородны и имеют хорошую адгезию к подложке. Необходимо отметить, что пленки ПП, полученные в H2SO4 и Na2SO4, менее однородны и после спектроэлектрохимических исследований, связанных с изменением объема пленки при изменении потенциала, отслаивались от электрода.
Обобщая наши данные по синтезу проводящих полимеров (полианилина и поли-3,4-этилендиокситиофена (ПЭДОТ)) в среде полиэлектролитов [14, 15], можно сказать, что в случае ПП использование полиэлектролитов при электрополимеризации пиррола также способствует локальному упорядочению положительно заряженных катион-радикалов вблизи отрицательно заряженных сульфокислотных групп, что приводит к ускорению полимеризации в этих условиях и образованию пленок лучшего качества.
На рис. 4 приведены спектры оптического поглощения, зарегистрированные в процессе ГС-синтеза пленок ПП. В случае солевых форм полиэлектролитов, H2SO4 и Na2SO4 (0.1 M) наблюдался монотонный рост оптического поглощения с достаточно выраженным максимумом на длине волны 730 нм, которое соответствует биполяронной форме ПП [16, 17] (рис. 4а). В случае кислотных форм полиэлектролитов наблюдается более широкая полоса поглощения, уходящая в ближнюю ИК-область (рис. 4б). Более того, для пленок ПП, синтезируемых в присутствии кислотных форм, более четко виден рост поглощения в области 500 нм, которое соответствует катион-радикалам ПП [17], а при полимеризации в солевой форме полиэлектролита это поглощение, по-видимому, смещается в коротковолновую область. Авторы [13] также наблюдали сдвиг поглощения катион-радикалов в область около 400 нм при увеличении pH раствора синтеза, при этом проводимость получаемых пленок ПП уменьшалась.
Рис. 4.
Электронные спектры поглощения пленок ПП, образующихся на рабочем электроде в процессе ГС-синтеза при 100 мкА/см2 в водных растворах ПАМПСNa (а), ПАМПСК (б).
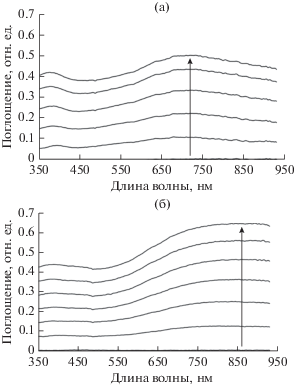
На рис. 5 представлены спектры оптического поглощения сухих пленок ПП, синтезированных в ГС-режиме. Необходимо отметить, что при одних и тех же условиях и заряде, затраченном на их получение (50 мКл/cм2), пленки полученные в серной кислоте имеют меньшую толщину и поглощение на спектре. Это может говорить о меньшем выходе ПП в данных условиях. Видно, что в случае использования кислотных форм полиэлектролитов наблюдается широкая полоса поглощения, уходящая в ближнюю ИК-область, что свидетельствует о том, что пленки находятся в полностью окисленной (биполяронной) форме [17]. На спектрах ПП, полученного в присутствии солевых форм полиэлектролитов и H2SO4, видно смещение поглощения в коротковолновую область и меньшее поглощение в ИК-области. Уменьшение содержания в ПП биполяронов может привести к снижению проводимости пленок.

Спектроэлектрохимия
Полученные таким образом гибридные пленки полипиррола были охарактеризованы в безмономерном водном растворе NaClO4. Перед спектроэлектрохимическими измерениями пленки ПП предварительно циклировали в водном растворе 0.1 М NaClO4 в диапазоне потенциалов от ‒0.6 до 0.6 В до получения стабильной ЦВ-кривой. Характерная для всех исследованных пленок ПП ЦВ-кривая представлена на рис. 6.
Рис. 6.
ЦВ-кривые пленки ПП, полученной в присутствии ПССК. Водный раствор 0.1 М NaClO4, скорость развертки потенциала – 50 мВ/с.

Влияние pH среды синтеза, т.е. природы противоиона полиэлектролита, отражается также и на спектроэлектрохимических свойствах полученных пленок ПП. На рис. 7 представлены спектры пленок ПП, синтезированных в присутствии кислотной и солевой форм полистиролсильфоната, при различных фиксированных потенциалах. Следует отметить, что спектры имеют форму, характерную для пленок ПП, полученных в неорганических электролитах [17]. При низких потенциалах наблюдается полоса около 400 нм, обусловленная π–π*-переходами, которая характеризует восстановленную форму ПП. При увеличении потенциала (окисление) интенсивность данной полосы снижается, при этом одновременно формируется слабая полоса поглощения около 500 нм, которую приписывают катион-радикалам (поляроннам), и происходит увеличение поглощения в области 800 нм (биполяронная форма) [16, 17]. Поглощение поляронов слабо выражено в случае ПП, полученного в кислотной форме полиэлектролита. В случае же использования полиэлектролита в солевой форме этой полосы не наблюдается, хотя при этом для всех пленок ПП видны изобестические точки, говорящие о переходе восстановленной формы в поляронную (и.т.1) и поляронной в биполяронную (и.т.2). Возможно, отсутствие видимого пика поглощения поляронов связано с его сдвигом в коротковолновую область спектра [17]. Действительно из табл. 2 видно, что для пленок ПП, полученных в присутствии натриевых форм, максимумы поглощения восстановленной формы сдвинуты в коротковолновую область на 9–10 нм, а положение изобестических точек на 20–30 нм. Аналогичные различия проявляются для пленок ПП, полученных в присутствии ПАМПСК и ПАМПСNa. Более выраженный пик катион-радикалов для ПП, синтезированного в присутствии кислотных форм полиэлектролитов, может быть связан с наличием в объеме пленки обособленных областей с различной степенью протонирования за счет присутствия в них захваченных молекул поликислоты, протоны которых не способны обмениваться с электролитом при циклах допирования/дедопирования. Однако данное предположение требует дополнительной экспериментальной проверки структурными методами (например, in situ спектроскопия комбинационного рассеяния). Кроме того, в литературе не раз упоминалось о гетерогенной структуре проводящих полимеров и, в частности, ПП. В работе [18] методом резистивной микроскопии (conductive-AFM) показано существование в пленках ПП областей с различной проводимостью, которая определяется различной степенью допирования этих областей.
Рис. 7.
Электронные спектры поглощения пленок ПП-ПССК (а) и ПП-ПССNa (б) при различных фиксированных потенциалах в водном растворе 0.1 М NaClO4.

Таблица 2.
Положения максимума полосы поглощения восстановленной формы ПП и изобестических точек, соответствующих переходам восстановленной формы в поляронную (и.т.1) и поляронной в биполяронную (и.т.2)
| Полиэлектролит | Восстановленная форма ПП, нм | и.т.1, нм | и.т.2, нм |
|---|---|---|---|
| ПССК | 404 | 474 | 590 |
| ПССNa | 397 | 467 | 555 |
| ПАМПСК | 404 | 495 | 582 |
| ПАМПСNa | 394 | 476 | 516 |
| H2SO4 | 375 | 466 | 480 |
| Na2SO4 (0.1 М) | 360 | 458 | 466 |
Морфология
Полученные пленки ПП были охарактеризованы методом атомно-силовой микроскопии (рис. 8). Однако, существенных отличий морфологии поверхности не было обнаружено. Поверхность пленок имела глобулярную регулярную структуру, характерную для ПП [6, 18]. Шероховатость пленок ПП, полученных в присутствии кислотных форм полиэлектролитов, немного выше, чем в солевых формах. Это может быть связано с более высокой скоростью синтеза в первом случае.

ЗАКЛЮЧЕНИЕ
Таким образом, нами проведено сравнительное исследование электрохимической полимеризации пиррола в присутствии солевых и кислотных форм гибкоцепных сульфокислотных полиэлектролитов. Электрохимическими методами в сочетании с мониторингом процесса методом in situ спектроскопии в УФ-видимой и ближней ИК-областях спектра показано, что использование полимерных электролитов при электрохимической полимеризации пиррола приводит к ускорению синтеза и возможности использовать меньшие концентрации реагентов по сравнению с электросинтезом в электролитах с неорганическими анионами. Установлено, что в присутствии кислотных форм полиэлектролитов формируются пленки ПП с более высокими скоростями, имеют большую шероховатость поверхности и имеют спектроэлектрохимические свойства, характерные для пленок ПП, полученных в электролитах с неорганическими анионами. Спектры пленок ПП, синтезированные в присутствии кислотных форм полиэлектролитов, демонстрируют более выраженный пик катион-радикалов в области около 500 нм, что может быть связано с наличием в объеме пленки обособленных областей, содержащих захваченные молекулы поликислоты, протоны которых не способны покидать пленку при циклах допирования/дедопирования. Однако, данное предположение требует дополнительной экспериментальной проверки, что будет предметом дальнейших исследований.
Список литературы
Handbook of Conducting Polymers. 3rd ed. Conjugated Polymers. Processing and Applications/Ed. by T.A. Skotheim, J.R. Reynolds. London, New York: Taylor & Francis Group, 2007.
Верницкая, Т.В., Ефимов, О.Н. Полипиррол как представитель класса проводящих полимеров (синтез, свойства, приложения). Успехи химии. 1997. Т. 66. № 5. С. 489. [Vernitskaya, T.V. and Efimov, O.N., Polypyrrole: a conducting polymer; its synthesis, properties and applications, Russ. Chem. Rev., 1997, vol. 66, no. 5, p. 443.]
Ko, J. M., Rhee, H. W., Park, S.-M., and Kim, C. Y., Morphology and Electrochemical Properties of Polypyrrole Films Prepared in Aqueous and Nonaqueous Solvents, J. Electrochem. Soc., 1990, vol. 137, no. 3, p. 905.
Lim, J.Y., Paik, W., and Yeo, I.-H., A Study on Ion Transports and Growth of Conducting Polypyrrole with Electrochemical Quartz Crystal Microbalance, Synth. Met., 1995. vol. 69, p. 451.
Kupila, E.-L. and Kankare, J., Electropolymerization of pyrrole: effects of pH and anions on the conductivity and growth kinetics of polypyrrole, Synth. Met., 1993, vol. 55, p. 1402.
Qu, L.-T., Shi, G.-Q., Liu, C., Yuan, J.-Y., and Qian, W.-B., Preparation, characterization and electrochemical properties of polypyrrole-polystyrene sulfonic acid composite film, Chinese J. Polymer Sci., 2005, vol. 23, no. 1, p. 37.
Shimidzu, T., Ohtani, A., Iyoda, T., and Honda, K., Charge-controllable polypyrrole/polyelectrolyte composite membranes Part II. Effect of incorporated anion size, J. Electroanal. Chem., 1987, vol. 224, p. 123.
Mamma, T., Ken, N., Osaka, T., Kondo, N., and Nakamura, S., Electrochemical Properties of a Polypyrrole/Polystyrenesulfonate Composite Film and Its Application to Rechargeable Lithium Battery Cathodes, J. Electrochem. Soc., 1994, vol. 141, no. 9, p. 2326.
Prezyna, L.A., Qiu, Y.-J., Reynolds, J.R., and Wnek, G.E., Interaction of Cationic Polypeptides with Electroactive Polypyrrole/Poly(styrenesulfonate) and Poly(N-methylpyrrole) / Poly(styrenesu1fonate) Films, Macromolecules, 1991, vol. 24, p. 5283.
Ignatova, M., Labaye, D., Lenoir, S., Strivay, D., Jerome, R., and Jerome, C., Immobilization of Silver in Polypyrrole/Polyanion Composite Coatings: Preparation, Characterization, and Antibacterial Activity, Langmuir, 2003, vol. 19, p. 8971.
Asavapiriyanont, S., Chandler, G.K., Gunawardena, G.A., and Pletcher, D., The electrodeposition of polypyrrole films from aqueous solutions, J. Electroanal. Chem., 1984, vol. 117, p. 229.
Qiu, Y.-J. and Reynolds, J.R., Electrochemically Initiated Chain Polymerization of Pyrrole in Aqueous Media, J. Polymer Sci.: Part A Polymer Chem., 1992, vol. 30, p. 1315.
Qian, R., Pei, Q., and Huang, Z., The role of H+ ions in the electrochemical polymerization of pyrrole, Makromol. Chem., 1991, vol. 192, p. 1263.
Gribkova, O.L., Nekrasov, A.A., Ivanov, V.F., Zolotorevsky, V.I., and Vannikov, A.V., Templating effect of polymeric sulfonic acids on electropolymerization of aniline, Electrochim. Acta, 2013, vol. 122, p. 150.
Якобсон, О.Д., Грибкова, О.Л., Некрасов, А.А., Ванников, А.В. Электрохимия. 2016. Т. 52. № 12. С. 1333. [Iakobson, O.D., Gribkova, O.L., Nekrasov, A.A., and Vannikov, A.V., The Effect of Counterion in Polymer Sulfonates on the Synthesis and Properties of Poly-3,4-ethylenedioxythiophene, Russ. J. Electrochem., 2016, vol. 52, no. 12, p. 1191.]
Breads, J.L., Scott, J.C., Yakushi, K., and Street, G.B., Polarons and bipolarons in polypyrrole: Evolution of the band structure and optical spectrum upon doping, Phys. Rev. B, 1983 vol. 30, p. 1023.
Arjomandi, J., Shah, A.-H. A., Bilal, S., Hoang, H.V., and Holze, R., In situ Raman and UV–vis spectroscopic studies of polypyrrole and poly(pyrrole-2,6-dimethyl-β-cyclodextrin), Spectrochim. Acta Part A, 2011, vol. 78, p. 1.
Lee, H.J. and Park, S.-M., Electrochemistry of Conductive Polymers. 30. Nanoscale Measurements of Doping Distributions and Current-Voltage Characteristics of Electrochemically Deposited Polypyrrole Films, J. Phys. Chem. B, 2004, vol. 108, p. 1590.
Дополнительные материалы отсутствуют.