

Координационная химия, 2023, T. 49, № 4, стр. 195-204
Синтез и кристаллическая структура двухъядерного комплекса ванадия(V) с лигандом на основе гидразида изоникотиновой кислоты и 1-фенил-1,3-бутандиона
П. Боурош 1, *, М. Коку 2, О. Данилеску 2, И. Булхак 2
1 Институт прикладной физики
Кишинев, Республика Молдова
2 Институт химии
Кишинев, Республика Молдова
* E-mail: pavlina.bourosh@ifa.md
Поступила в редакцию 16.05.2022
После доработки 26.07.2022
Принята к публикации 02.08.2022
- EDN: CUMNFL
- DOI: 10.31857/S0132344X23700196
Аннотация
Синтезирован новый молекулярный двухъядерный комплекc ванадия(V) [VO(L)(OCH3)]2 (I), в котором H2L был получен в результате взаимодействия гидразида изоникотиновой кислоты (изониазид) с 1-фенил-1,3-бутандионом. Лиганд H2L и комплекс I исследованы методами элементного анализа, ИК-спектроскопией, масс-спектрометрией и рентгеноструктурного анализа (CCDC № 2172124 и 2172125 соответственно), состав и строение лиганда в свободном состоянии дополнительно исследованы методом ЯМР-спектроскопии. Установлено, что бидепротонированный органический лиганд L2– координируется к атому металла тридентатно через набор донорных атомов ONO с образованием двух сочлененных металлоциклов. При этом в H2L и в соответственном координированном лиганде стабилизированы различные таутомерные формы.
Ванадий играет важную роль в биологических системах, являясь важным элементом для большинства живых существ, а также содержится в почве, нефти, воде и воздухе [1]. Роль ванадия в биологических системах проявляется как структурно, стабилизируя различные биологические формы соединений, так и функционально, обеспечивая ключевую активность различным центрам молекул белков, ферментов и коферментов [2]. Хотя биологическое значение ванадия было признано давно [3], и поскольку он является компонентом многих лекарств для лечения диабета, рака и заболеваний, вызываемых паразитами, этот элемент интересен также в области исследований координационной химии как металлический центр с различными степенями окисления, координационным числом и своими преференциями к донорным атомам.
Пиридинкарбонильные координирующие агенты, особенно основания Шиффа, представляют собой гибкие и универсальные лиганды, отличающиеся высокой и разнообразной способностью координировать к ионам металлов, образуя комплексы с различной молекулярной архитектурой [4]. Помимо этого, известно, что сам по себе гидразид изоникотиновой кислоты (изониазид, ГИНК) является противотуберкулезным лекарственным средством, поэтому часто используется для исследования процессов образования комплексов с биологическими свойствами. ГИНК является полидентатным лигандом, который может координировать как в кето-, так и в енольной форме, в зависимости от условий реакции. При взаимодействии с β-дикетонами ГИНК образует основания Шиффа, которые в присутствии ионов металлов образуют моно-, би- и полиядерные координационные соединения, в которых эти лиганды могут координироваться полидентатно как к одному атому металла, так и к двум в качестве мостиковых лигандов [5–12]. Ранее при взаимодействии гидразида изоникотиновой кислоты с 2,4-пентандионом в результате дополнительной конденсации одного концевого фрагмента нами был получен лиганд 5-гидрокси-3,5-диметил-4,5-дигидро-1H-пиразол-1-ил)(пириди-4-ил)метан, который в реакции с [Co(DfgH)2Br(H2O)] (DfgH2 = дифенилглиоксим) приводил к образованию комплекса, в котором этот лиганд замещал молекулу воды [13]. При этом выявлено, что при конденсации этих же органических молекул получен изоникотиноилгидразон 2,4-пентандиона, который в реакции с солями ванадия привел к образованию как моно-, так и биядерных комплексов [6, 7].
В настоящей работе представляем как новый комплекс ванадия(V) [VO(L)(OCH3)]2 (I) c лигандом, полученным при взаимодействии гидразида изоникотиновой кислоты с 1-фенил-1,3-бутандионом (Н2L), так и сам лиганд в свободном состоянии.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Для синтеза использовали коммерчески доступные реагенты (в том числе растворители) из каталогов фирм Sigma-Aldrich без предварительной очистки: гидразид изоникотиновой кислоты (98%) (HPLC), 1-фенил-1,3-бутандион (99%), ацетилацетонат оксованадия (98%), этиловый спирт (95%), метиловый спирт (99.8%).
Синтез изоникотиноилгидразона 1-фенил-1,3-бутандиона (H2L) [14]. К теплому раствору 1.37 г (10.0 ммоль) гидразида изоникотиновой кислоты в 7 мл этилового спирта добавляли 1.62 г (10.0 ммоль) 1-фенил-1,3-бутандиона. Полученный раствор нагревали при 75–78°С до появления коричневого цвета (~20 мин). После 12 ч образовывалось желтое кристаллическое вещество (1.45 г), которое отфильтровывали, промывали этанолом и диэтиловым эфиром и высушивали на воздухе. Кристаллы растворимы в хлороформе, диметилформамиде, диметилсульфоксиде, этиловом и метиловом спиртах и частично в воде. Выход ~50%.
ИК-спектр (ν, см–1) Н2L: 3220 ср, 3200 пл, 3058 сл, 3039 сл, 3026 сл, 2967 о.сл, 2927 сл, 2837 сл, 1664 с, 1596 с, 1574 ср, 1550 о.с, 1528 о.с, 1507 о.с, 1483 с, 1431 с, 1410 ср, 1378 ср, 1305 с, 1281 с, 1272 с, 1214 с, 1187 с, 1113 ср, 1086 ср, 1063 с, 1039 ср, 1027 ср, 1022 сл, 972 сл, 925 ср, 870 сл, 845 с, 833 с, 800 сл, 738 с, 714 с, 691 сл, 665 ср, 648 c, 620 cл, 613 ср, 565 ср, 540 сл, 529 сл, 490 сл, 485 сл, 432 ср, 413 сл, 406 сл.
ЯМР 1H (400 МГц, 25°С, CDCl3; δ, м.д. (J, Гц)): 2.10 (с., 3H, CH3), 3.01 (д., 1H, CH2, J = 18.6), 3.37 (д., 1H, CH2, J = 18.6), 5.32 (с., 1H, NH), 8.71 (д., 2H, Py, J = 3.7), 7.76 (д., 2H, Py, J = 4.5), 7.45 (д., 2H, Ph, J = 7.7), 7.38 (д., 2H, Ph, J = 7.4), 7.31 (т., 1H, Ph, J = 7.0). ЯМР 13С (400 МГц, 25°С, CDCl3; δ, м.д. (J, Гц)): 16.1 (1С, CH3), 53.8 (1C, CH2), 94.6 (1C, Ph), 123.5 (2C, Py), 123.9 (2C, Ph), 128.3 (1C, Ph), 128.8 (2C, Ph), 141.2 (1C, Py), 143.03 (1C, >C=N), 149.7 (2C, Py), 156.4 (1C, >C=О (бенз-ацет.)), 165.5 (1C, >C =O (гидр.)). ЯМР 15N (400 МГц, 25°С, CDCl3; δ, м.д. (J, Гц)): 213 (1N, =N–), 315 (1N, –NH–) (шкала Вruker).
Синтез [VO(L)(OCH3)]2 (I) [13]. К раствору 0.07 г (0.24 ммоль) H2L в 10 мл метилового спирта при нагревании (60–65°С) добавляли 0.06 г (0.24 ммоль) ацетилацетоната оксованадия (VO(Аcac)2) в 10 мл метилового спирта. Через 2–3 сут образовывалось черное кристаллическое вещество (0.07 г), которое промывали метиловым спиртом, затем диэтиловым эфиром. Кристаллы растворимы в хлороформе, диметилформамиде, диметилсульфоксиде, мало растворимы в этиловом и метиловом спиртах и нерастворимы в воде. Выход I ~ 55%.
ИК-спектр (ν, см–1) [VOL(OCH3)]2: 3070 cл, 3028 ср, 2964 сл, 2923 ср, 2817 ср, 1599 пл, 1589 ср, 1574 с, 1534 о.с, 1472 о.с, 1455 о.с, 1426 с, 1407 о.с, 1393 о.с, 1368 ср, 1337 с, 1314 ср, 1294 с, 1231 ср, 1210 о.сл, 1182 ср, 1146 ср, 1110 ср, 1076 сл, 1060 ср, 1015 о.с, 978 о.с, 889 ср, 881 ср, 837 с, 796 сл, 762 о.с, 744 с, 704 ср, 692 ср, 683 о.с, 647 ср, 611 о.с, 600 о.с, 569 ср, 504 ср, 454 ср, 425 ср, 403 о.сл.
Состав и строение лиганда устанавливали на основе элементного анализа, ИК-, ЯМР-спектроскопии и РСА, строение комплекса – на основе элементного анализа, массспектрометрии, ИК- спектроскопии и РСА.
ИК-спектры снимали на FT-IR Perkin-Elmer Spectrum 100 в вазелиновом масле в области 4000–400 см–1 и ATР в области 4000–650 cм–1. Спектры ЯМР 1H, 13С и 15N регистрировали на приборе Bruker Avance III (400 MHz, 25°С, CDCl3).
Масс-спектры измеряли на масс-спектрометре Agilent 6520 Series Accurate-Mass Quadrupole Time-of-Flight (Q-TOF) LC/MS. Растворы вводили в источник ионов электрораспыления (ESI) с помощью шприцевого насоса со скоростью потока 0.01 мл/мин. Данные собирали и обрабатывали с использованием ПО MassHunter Workstation Data Acquisition для серии 6200/6500, версия B.01.03.
РСА H2L и I проведен на дифрактометре Xcalibur Е (MoKα-излучение, λ = 0.71073 Å, графитовый монохроматор, ω-сканирование) при комнатной температуре. Экспериментальные данные для I переуточнены с учетом двойникового образца. Параметры элементарных ячеек уточнены c учетом полного массива экспериментальных данных. Кристаллические структуры решены прямыми методами и уточнены МНК в анизотропном полноматричном варианте для неводородных атомов (SHELX-97) [15]. Позиции атомов водорода частично рассчитаны геометрически, частично определены их Фурье-синтезы и уточнены изотропно в модели “жесткого тела”. Кристаллографические данные и характеристики эксперимента для H2L и I приведены в табл. 1, некоторые межатомные расстояния и валентные углы – в табл. 2, геометрические параметры водородных связей (ВС) – в табл. 3.
Таблица 1.
Кристаллографические данные и характеристики эксперимента для структуры H2L и I
| Соединение | H2L | I |
|---|---|---|
| Брутто-формула | C16H15N3O2 | C34H32N6O8V2 |
| М | 281.31 | 754.53 |
| Сингония | Триклинная | Триклинная |
| Пр. группа | $P\bar {1}$ | $P\bar {1}$ |
| а, Å | 9.0853(18) | 10.8850(8) |
| b, Å | 9.970(2) | 11.4822(8) |
| с, Å | 16.0900(18) | 15.9177(10) |
| α, град | 98.969(14) | 72.427(6) |
| β, град | 98.833(13) | 89.916(6) |
| γ, град | 90.033(18) | 62.183(7) |
| V, Å3 | 1422.2(5) | 1654.3(2) |
| Z | 4 | 2 |
| ρ(выч.), г/см3 | 1.314 | 1.515 |
| μ, мм–1 | 0.089 | 0.627 |
| F(000) | 592 | 776 |
| Размеры кристалла, мм | 0.20 × 0.10 × 0.06 | 0.35 × 0.35 × 0.05 |
| Область θ, град | 3.03–25.25 | 2.90–25.05 |
| Интервалы индексов отражений | –9 ≤ h ≤ 10, –7 ≤ k ≤ 11, –18 ≤ l ≤ 19 |
–9 ≤ h ≤ 12, –13 ≤ k ≤ 13, –18 ≤ l ≤ 18 |
| Число измеренных/независимых рефлексов (Rint) | 9234/5129 (0.0294) | 6204/6204 |
| Заполнение, % | 99.8 | 99.7 |
| Число рефлексов с I > 2σ(I) | 2714 | 3495 |
| Число уточняемых параметров | 379 | 456 |
| GOOF | 1.003 | 1.005 |
| R фактор (I > 2σ(I)) | R1 = 0.0422, wR2 = 0.0774 | R1 = 0.0819, wR2 = 0.1908 |
| R фактор (по всему массиву) | R1 = 0.0877, wR2 = 0.0815 | R1 = 0.1334, wR2 = 0.2103 |
| Δρmax/ρmin, e Å–3 | 0.191/–0.224 | 0.925/–0.736 |
Таблица 2.
Основные межатомные расстояния и валентные углы в соединениях H2L и I*
| Связь | d, Å | Связь | d, Å |
|---|---|---|---|
| H2L | |||
| O(1A)−C(6A) | 1.227(2) | O(1B)−C(6B) | 1.215(2) |
| C(6A)−N(2A) | 1.333(2) | C(6B)−N(2B) | 1.332(2) |
| N(2A)−N(3A) | 1.386(2) | N(2B)−N(3B) | 1.384(2) |
| N(3A)−C(7A) | 1.341(2) | N(3B)−C(7B) | 1.336(2) |
| C(7A)−C(8A) | 1.366(2) | C(7B)−C(8B) | 1.374(2) |
| C(8A)−C(9A) | 1.420(3) | C(8B)−C(9B) | 1.420(3) |
| C(9A)−O(2A) | 1.246(2) | C(9B)−O(2B) | 1.247(2) |
| I | |||
| V(1)−O(1A) | 1.912(5) | V(2)−O(1B) | 1.911(5) |
| V(1)−O(2A) | 1.841(5) | V(2)−O(2B) | 1.828(5) |
| V(1)−O(3A) | 1.574(5) | V(2)−O(3B) | 1.587(5) |
| V(1)−O(4A) | 1.811(4) | V(2)−O(4B) | 1.817(5) |
| V(1)−N(3A) | 2.106(6) | V(2)−N(3B) | 2.162(7) |
| V(1)−O(4A)#1 | 2.404(5) | V(2)−O(4B)#2 | 2.408(5) |
| O(1A)−C(6A) | 1.317(8) | O(1B)−C(6B) | 1.319(8) |
| C(6A)−N(2A) | 1.314(9) | C(6B)−N(2B) | 1.264(9) |
| N(2A)−N(3A) | 1.385(8) | N(2B)−N(3B) | 1.431(8) |
| N(3A)−C(7A) | 1.247(9) | N(3B)−C(7B) | 1.153(9) |
| C(7A)−C(8A) | 1.424(10) | C(7B)−C(8B) | 1.491(10) |
| C(8A)−C(9A) | 1.357(10) | C(8B)−C(9B) | 1.349(10) |
| C(9A)−O(2A) | 1.339(8) | C(9B)−O(2B) | 1.314(9) |
| Угол | ω, град | Угол | ω, град |
| H2L | |||
| O(1A)C(6A)N(2A) | 123.65(18) | O(1B)C(6B)N(2B) | 124.14(18) |
| C(6A)N(2A)N(3A) | 119.58(15) | C(6B)N(2B)N(3B) | 120.53(16) |
| N(2A)N(3A)C(7A) | 121.83(15) | N(2B)N(3B)C(7B) | 121.15(16) |
| N(3A)C(7A)C(8A) | 120.56(17) | N(3B)C(7B)C(8B) | 121.20(18) |
| C(7A)C(8A)C(9A) | 124.92(18) | C(7B)C(8B)C(9B) | 124.56(18) |
| C(8A)C(9A)O(2A) | 122.30(18) | C(8B)C(9B)O(2B) | 122.85(18) |
| I | |||
| O(1A)V(1)O(2A) | 153.2(2) | O(1B)V(2)O(2B) | 153.3(2) |
| O(1A)V(1)O(3A) | 98.1(3) | O(1B)V(2)O(3B) | 97.0(3) |
| O(1A)V(1)O(4A) | 94.7(2) | O(1B)V(2)O(4B) | 93.9(2) |
| O(1A)V(1)N(3A) | 75.0(2) | O(1B)V(2)N(3B) | 76.3(2) |
| O(1A)V(1)O(4A)#1 | 81.98(19) | O(1B)V(2)O(4B)#2 | 83.01(19) |
| O(2A)V(1)O(3A) | 100.4(3) | O(2B)V(2)O(3B) | 101.4(3) |
| O(2A)V(1)O(4A) | 99.9(2) | O(2B)V(2)O(4B) | 100.8(2) |
| O(2A)V(1)N(3A) | 83.5(2) | O(2B)V(2)N(3B) | 82.5(2) |
| O(2A)V(1)O(4A)#1 | 81.5(2) | O(2B)V(2)O(4B)#2 | 80.3(2) |
| O(3A)V(1)O(4A) | 102.6(2) | O(3B)V(2)O(4B) | 102.3(2) |
| O(3A)V(1)N(3A) | 97.4(3) | O(3B)V(2)N(3B) | 96.3(2) |
| O(3A)V(1)O(4A)#1 | 174.3(2) | O(3B)V(2)O(4B)#2 | 174.9(2) |
| O(4A)V(1)N(3A) | 158.6(2) | O(4B)V(2)N(3B) | 160.0(2) |
| O(4A)V(1)O(4A)#1 | 71.76(19) | O(4B)V(2)O(4B)#2 | 72.6(2) |
| N(3A)V(1)O(4A)#1 | 88.07(19) | N(3B)V(2)N(4B)#2 | 88.65(18) |
| O(1A)C(6A)N(2A) | 121.1(7) | O(1B)C(6B)N(2B) | 123.7(7) |
| C(6A)N(2A)N(3A) | 108.9(6) | C(6B)N(2B)N(3B) | 111.1(6) |
| N(2A)N(3A)C(7A) | 116.7(6) | N(2B)N(3B)C(7B) | 117.1(7) |
| N(3A)C(7A)C(8A) | 124.3(6) | N(3B)C(7B)C(8B) | 119.6(7) |
| C(7A)C(8A)C(9A) | 124.2(6) | C(7B)C(8B)C(9B) | 124.5(7) |
| C(8A)C(9A)O(2A) | 119.0(6) | C(8B)C(9B)O(2B) | 121.4(7) |
Таблица 3.
Геометрические параметры водородных связей в соединениях H2L и I
| Контакт D–H∙∙∙A | Расстояние, Å | Угол DHA, град | Координаты атомов А | ||
|---|---|---|---|---|---|
| D–H | H∙∙∙A | D∙∙∙A | |||
| H2L | |||||
| N(2A)–H(1)∙∙∙O(1B) | 0.86 | 2.05 | 2.820(2) | 148 | x, y, z |
| N(3A)–H(1)∙∙∙O(2A) | 0.86 | 1.99 | 2.650(2) | 132 | x, y, z |
| N(3A)–H(1)∙∙∙O(2A) | 0.86 | 2.31 | 2.963(2) | 133 | –x + 1, –y + 1, –z + 2 |
| N(2B)–H(1)∙∙∙O(1A) | 0.86 | 2.05 | 2.816(2) | 147 | x, y – 1, z |
| N(3B)–H(1)∙∙∙O(2B) | 0.86 | 2.02 | 2.668(2) | 132 | x, y, z |
| N(3B)–H(1)∙∙∙O(2B) | 0.86 | 2.28 | 2.926(2) | 132 | –x, –y, –z + 2 |
| C(16B)–H(16E)∙∙∙O(1A) | 0.96 | 2.66 | 3.482(2) | 145 | x, y – 1, z |
| I | |||||
| C(17A)–H(17B)∙∙∙N(1A) | 0.96 | 2.68 | 3.568(10) | 155 | x + 1, y – 1, z |
| C(17A)–H(17C)∙∙∙O(3A) | 0.96 | 2.53 | 3.036(9) | 1113 | x, y, z |
| C(17A)–H(17C)∙∙∙O(3B) | 0.96 | 2.65 | 3.213(8) | 156 | –x + 1, –y, –z + 1 |
| C(13A)–H(13A)∙∙∙O(3A) | 0.93 | 2.48 | 3.263(8) | 132 | –x + 1, –y, –z + 1 |
| C(17B)–H(17E)∙∙∙N(3B) | 0.96 | 2.68 | 3.349(9) | 127 | –x, –y, –z |
| C(17B)–H(17F)∙∙∙N(1A) | 0.96 | 2.63 | 3.553(11) | 161 | –x, –y + 1, –z + 1 |
Позиционные и тепловые параметры для H2L и I депонированы в Кембриджском банке структурных данных (КБСД) (№ 2172124 и 2172125 соответственно; deposit@ccdc.cam.ac.uk, http:// www.ccdc.cam.ac.uk).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
С целью получения новых комплексных соединений 3d-металлов с Шиффовыми основаниями на основе нуклеофильного агента ГИНК получен лиганд H2L с использованием 1-фенил-1,3-бутандиона (схема 1 ) и новый димерный комплекс [VO(L)(OCH3)]2, образованный в результате его взаимодействия с ацетилацетонатом оксованадия(IV). Таутомерные формы H2L, полученные в результате конденсации 1-фенил-1,3-бутандиона с ГИНК представлены на схеме 1 .
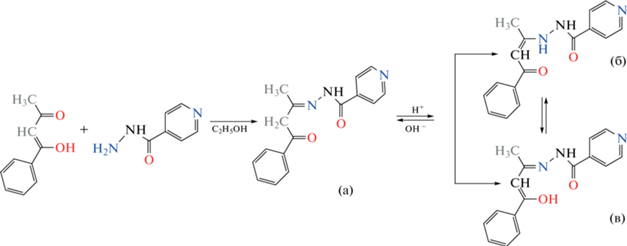
Схема 1 .
Спектральные характеристики ИК и ЯМР 1H для органического лиганда [16] подтверждают образование H2L в более стабильной таутомерной форме (а) [17].
В ИК-спектре H2L наблюдаются полосы поглощения средней интенсивности при 3200 см–1 с плечом на низкочастотной стороне, которая относится к ν(NH) [18] и серия полос поглощения более низкой интенсивности в интервале 3060–2800 см–1, которые относятся к ν(С–Н) разного происхождения: ароматического – 3058, 3038, 3026 см–1 и алифатического – 2967, 2927 и 2837 см–1 [19]. При 1664 см–1 проявляется интенсивная полоса поглощения, которую можно отнести к ассоциированной ν(С=О) [20]. Полосы поглощения при 1596, 1574, 1507 и 1483 см–1 относятся к плоскостным колебаниям скелета (С=С) в ароматических кольцах H2L [19]. Полоса поглощения при 1528 см–1 можно отнести к колебаниям амид II (δ(NH) + ν(C–N)) [19]. Метильные группы поглощают при 1431 см–1 – δаs(CH3) и 1378 см–1 – δs(CH3) [21].
Положение полос поглощения плоскостных колебаний δ(С–Н) в ароматических соединениях зависит от типа замещения и являются специфическими [21]. В спектре 1-замещенного цикла (пять незамещенных смежных атомов водорода) наблюдаются полосы поглощения при 1086 и 1063 см–1, а для 1,4-замещенного ароматического цикла (два незамещенных смежных атомов водорода) – при 1214 и две полосы при 1039 и 1027 см–1 cоответствено [21]. Неплоскостные колебания С–Н в ароматических кольцах также выражают тип замещения, но полосы в данном случае являются существенно более интенсивными: для 1-замещеного кольца (пять незамещенных смежных атомов водорода) – полосы при 714 и 664 см–1, тогда как для 1,4-замещеного кольца (два незамещенных смежных атомов водорода) – полоса при 833 см–1.
В протонном спектре ЯМР 1H раствора соединения H2L зарегистрировано восемь групп линий, которые были отнесены к протонам (δ, м.д.): одной группы CH3 (2.10 с.), одной группы CH2 (3.02–3.38 д.), одной группы NH (5.32 с.), пять сигналов атомов водорода фенильной группы (7.28–7.49) и четыре сигнала атомов водорода, принадлежащих пиридиновому кольцу (7.78–8.76).
Для подтверждения существования обеих таутомерных форм H2L (cхема 1а и 1б) к образцу, содержащему H2L растворенного в CDCl3, добавляли каплю трифторуксусной кислоты, после чего повторно регистрировали спектр ЯМР 1H. В спектре усилилась полоса протонов группы =СН– (δ = 6.30 м.д. (с.)), что свидетельствует о смещении равновесия в сторону образования таутомерной формы (б), концентрация которой увеличилась с уменьшением рН среды [17].
Данные спектра 13C ЯМР раствора соединения H2L указывают на присутствие 16 атомов углерода, так как присутствуют линии, принадлежащие одному атому углерода группы CH3 (16.1 м.д.), одному атому углерода группы CH2 (53.8 м.д.), пяти атомам углерода группы пиридил: два атома углерода в положениях 2 и 6 (149.72 м.д.), два атома углерода в положениях 3 и 5 (123.5 м.д.) и четвертичный атом углерода (141.2 м.д.), шести атомам углерода группы фенил: два орто- (123.9 м.д.), два мета- (128.8 м.д.), один пара-положение (128.3 м.д.) и четвертичный атом углерода (94.6 м.д.). В спектре ЯМР 13C также присутствуют линии, характерные одному азометинному атому углерода (143.03 м.д.) и двум атомам углерода карбонильных групп, принадлежащие 1-фенил-1,3-бутандиону (156.4 м.д.) и ГИНК (165.5 м.д.).
В спектре ЯМР 15N раствора соединения H2L зарегистрированы две линии, которые подтверждают присутствие двух атомов азота: =N– (213 м.д.) и –NH– (315 м.д.) (шкала Вruker) [22].
В процессе координации H2L к ионам ванадия, дважды депротонированный лиганд L2– стабилизируется в таутомерной форме (в). Этот факт подтверждается данными ИК-спектров и РСА.
В ИК-спектре [VO(L)(OCH3)]2, в отличие от спектра Н2L, не наблюдались полосы поглощения при 3220 см–1 ν(NH) и при 1664 см–1 ν(С=O). При этом появляется самая интенсивная полоса во всем спектре при 978 см–1, которая относится к ν(V=O) [23–25]. Полосы поглощения скелетных колебаний ν(С=С) и ν (C=N) связей ароматических колец проявляются при 1599, 1589, 1574 и 1456 см–1 [19]. Планарные деформационные колебания (δ(С–Н)) 1-замещенного кольца проявляются при 1146, 1076, а 1,4-замещенного кольца – при 1231 и 1060 см–1. Непланарные колебания δ(С–Н) 1-замещенных ароматических колец проявляются при 762 и 681 см–1 и для 1,4-замещенного бензольного кольца – при 837 см–1 [20, 21].
В комплексе [VO(L)(OCH3)]2 образуются металлоциклы с делокализацией электронов, поэтому в спектре наблюдаются полосы поглощения при 1574 и 1534 см–1 которые относятся к ν(С···С) + ν(С···О) и ν(С···О) + ν(С···С) соответственно [26, 27]. Поглощение при 921 см–1 можно отнести к ν[V–(μ-O)–V] [23], при 600 см–1 – к ν(V–N) [21], при 454 см–1 – к ν(V–О), смешанное с деформационным колебанием С–СН3, тогда как более низкочастотную полосу при 425 см–1 можно отнести практически к несмешанному валентному колебанию ν(V–О) [24, 26].
В масс-спектрах электронного удара в хлороформ-метанольном растворe [VO(L)(OCH3)]2 наблюдается интенсивный пик при m/z = 378.15, который соответствует дважды протонированному иону [M + 2H]2+ и два пика малой интенсивности при m/z = 400.13 и 777.22, соответствующие ионам [M + 2Na]2+ и [M + Na]+ (М = молекулярная масса C34H32N6O8V2).
Рентгеновское исследование H2L показало, что соединение кристаллизуется в триклинной пространственной группе $P\bar {1}$ (табл. 1), и его строение приведено на рис. 1. В независимой части элементарной ячейки находятся две кристаллографически независимые молекулы (А и В), которые стабилизированы в кристалле в одинаковой таутомерной форме (б). Этот факт подтверждается и расстояниями C(6)–O(1), C(6)–N(2), N(2)–N(3), C(7)–N(3), C(7)–C(8), C(8)–C(9), C(9)–O(2) между атомами в центральных фрагментах молекул А и В (табл. 2). Анализ КБСД [4] показал, что такое соединение известно, однако эта другая полиморфная форма, которая кристаллизуется в моноклинной пространственной группе Р21/c [22]. При этом как в последнем, так и в нашем случае для H2L стабилизирована та же таутомерная форма б). При этом конформация лиганда позволяет координироваться к атому металла как тридентатный хелатный лиганд, используя набор донорных атомов ONO (рис. 1).

В кристалле H2L в молекулах А и В выделяются сильные внутримолекулярные ВС N(3)–H∙∙∙O(2), в которых как донор вовлечена одна из двух групп NH (табл. 3, рис. 1), стабилизируя их конфигурацию формированными псевдогексациклами. Помимо этого, межмолекулярными ВС N(3)–H∙∙∙O(2) связаны между собой как молекулы А, так и В (табл. 3, рис. 2). Вторые группы NH молекул А и В участвуют в образовании межмолекулярных ВС N(2А)–H∙∙∙O(1В) и N(2В)–H∙∙∙O(1А), формируя цепочки, при этом эти молекулы объединены дополнительно слабыми межмолекулярными ВС С(16В)–H∙∙∙O(1А).

Молекулярное биядерное комплексное соединение [VO(L)(OCH3)]2 (I), полученное при взаимодействии H2L с VO(Аcac)2 (молярное соотношение 1 : 1) в среде метанола, кристаллизуется в триклинной пространственной группе $P\bar {1}$ (табл. 1). В независимой части элементарной ячейки содержатся две 1/2 кристаллографически независимых комплексов ванадия(V) A и B. На рис. 3 представлено строение этих центосиметричных комплексов.

В результате координирования к каждому атому металла по одному тридентатному бидепротонированному органическому лиганду L2– через донорные атомы ONO образуются два сочлененных металлоцикла: один – пятичленный VNNCO, другой шестичленный VOCCCN. Дополняет формирование идентичных координационных полиэдров металлов V(1) и V(2) один оксо-анион и два атома кислорода депротонированных лигандов метанола, которые объединяют два атома металла, a их мостиковая функция определяет образование димеров в кристалле (рис. 3а, 3б). Так как один метокси-анион расположен в экваториальной плоскости полиэдра, а другой – в одной из аксиальных позиций, а на второй аксиальной позиции расположен оксиатом, координационное число металлов 4 + 1 + 1 и их коор-динационные полиэдры в этих комплексах имеют форму квадратной бипирамиды. Длины связей V=O в координационных полиэдрах V(1) и V(2) равны 1.574(5) и 1.587(5) Å соответственно, а длины связи V–OСH3 в А и В имеют соответственно значения 1.811(4), 2.404(5) и 1.817(5), 2.408(5) Å (табл. 2). Расстояние V∙∙∙V между двумя атомами ванадия в центросимметричных димерах составляет 3.432 и 3.422 Å. Отметим, что эти комплексы стабилизированы дополнительно различными внутримолекулярными ВС С(17А)–H∙∙∙O(3А) и С(17В)–H∙∙∙N(3B) соответственно (табл. 3). Длины связей и валентные углы в координационных полиэдрах V(1) и V(2) близки к аналогичным величинам в моно- и биядерных комплексах этого металла с тридентатным изоникотиноилгидразоном 2,4-пентандиона [6, 7]. Длины связей в центральных частах молекул H2L и в органических лигандах L2– в I (табл. 2) чуть отличаются, а их бóльшие значения одних групп С=О указывают на стабилизацию координированного лиганда в таутомерной форме (в).
Анализ кристаллической структуры соединения [VO(L)(OCH3)]2 показывает, что супрамолекулярная архитектура формируется лишь за счет слабых межмолекулярных ВС, в которые вовлечены в качестве доноров СН-группы как метильных, так и фенильных групп. При этом комплексы А связаны между собой слабыми межмолекулярными ВС С−H···O в слой, а комплексы В объединены этими же слабыми связями с последними (табл. 3). В кристалле в слоях комплексов А можно выделить цепочки, между которыми расположены комплексы В, связанные с ними ВС С−H···O, образуя слой из чередующихся комплексов А и В (рис. 4).

Таким образом, при конденсации ГИНК с 1‑фенил-1,3-бутандионом нами получена другая полиморфная форма органического лиганда H2L, использованная для синтеза нового биядерного комплекса ванадия(V). В кристаллическом состоянии H2L и координированный L2− из комплексов ванадия(V) стабилизированы в различные таутомерные формы лиганда.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Gad S.C., Pham T. // Encyclopedia of Toxicology. Acad. Press, 2014. P. 909.
Krakowiak J., Lundberg D., Persson I. // Inorg. Chem. 2012. V. 51. № 18. P. 9598.
Rehder D. // Angew. Chem. Int. Ed. 1991. V. 30. P. 148.
Allen F.H. // Acta Crystallogr. B. 2002. V. 58. P. 380.
Nandy M., Shit S., Rizzoli C. et al. // Polyhedron. 2015. V. 88. P. 63.
Monfared H.H., Kheirabadi S., Lalami N.A., Mayer P. // Polyhedron. 2011. V. 30. P. 1375.
Monfared H.H., Farrokhi A., Alavi S., Mayer P. // Transition Met. Chem. 2013. V. 38. P. 267.
Yadav S., Yousuf I., Usman M. et al. // RSC Advances. 2015. V. 5. P. 50673.
Hosseini-Monfared H., Bikas R., Sanchiz J. et al. // Polyhedron. 2013. V. 61. P. 45.
Боурош П., Булхак И., Мырзак А. и др. // Коорд. химия. 2016. Т. 42. № 3. С. 137 (Bourosh P., Bulhac I., Mirzac A. et al. // Russ. J. Coord. Chem. 2016. V. 42. № 3. P. 157). https://doi.org/10.1134/S1070328416030015
Croitor L., Cocu M., Bulhac I. et al. // Polyhedron. 2021. V. 206. P. 115329.
Данилеску О., Булхак И., Шова С. и др. // Коорд. химия. 2020. Т. 46. № 12. С. 758 (Danilescu O., Bulhac I., Shova S. et al. // Russ. J. Coord. Chem. 2020. V. 46. № 12. P. 838). http://doi.org./10.1134/S1070328420090018
Cocu M., Bulhac I., Coropceanu E. и дp. // J. Molec. Struct. 2014. V. 1063. P. 274.
Rotaru M., Cocu M. // Acta et Comment. seria Științe Exacte și ale Naturii. 2017. № 1(3). P. 136.
Sheldrick G.M. // Acta Crystallogr. A. 2008. 64. № 1. P. 112.
Тарасевич Б.Н. ИК спектры основных классов органических соединений. Справочные материалы. М., 2012. 54 с.
Hernández-Molina R., Mederos A. // Comprehensive Coordination Chemistry, II. 2003. P. 411. https://doi.org/10.1016/B0-08-043748-6/01070-7
Беллами Л. Инфракрасные спектры сложных молекул. М.: ИЛ, 1963. 590 с.
Казицына Л.А., Куплетская Н.Б. Применение УФ-, ИК- и ЯМР-спектроскопии в органической химии. М.: Высш. школа, 1971. 264 с.
Наканиси К. Инфракрасные спектры и строение органических соединений. Практическое руководство. М.: Мир, 1965. 216 с.
Adams D.M. Metal-Ligand and Related Vibrations: A Critical Survey of the Infrared and Raman Spectra of Metallic and Organometallic Compounds. Ltd. London, 1967. 379 p.
Bikas R., Anarjan P.M., Aslekhademi S. et al. // Acta Crystallogr. E. 2012. V. 68. P. o412.
Kolawole G.A., Osowole A.A. // J. Coord. Chem. 2009. V. 62. № 9. P. 1437.
Monfared H.H., Kheirabadi S., Lalami N.A., Mayer P. // Polyhedron. 2011. V. 30. P. 1375.
Накамото K. ИК спектры и спектры неорганических и координационных соединений. М.: Мир, 1991. 536 с.
Кокшарова Т.В. // Вісник ОНУ. Хімія. 2014. Т. 19. В. 2. № 50. С. 27.
Bovey F.A. Nuclear Magnetic Resonance Spectroscopy, Acad. Press Inc., 1988. P. 461.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия