

Высокомолекулярные соединения (серия Б), 2020, T. 62, № 2, стр. 83-95
Синтез водорастворимых солей полиамидокислот и структурные особенности полученных из них волокон и пленок
В. М. Светличный a, *, Л. А. Мягкова a, Т. Е. Суханова a, b, Е. М. Иванькова a, Г. В. Ваганов a, А. Е. Чирятьева a, В. Ю. Елоховский a, М. Э. Вылегжанина a, Е. Н. Власова a, В. Е. Юдин a
a Институт высокомолекулярных соединений Российской академии наук
199004 Санкт-Петербург, Большой пр., 31, Россия
b Научно-исследовательский институт синтетического каучука имени академика С.В. Лебедева
198035 Санкт-Петербург, Гапсальская ул., 1, Россия
* E-mail: valsvet@hq.macro.ru
Поступила в редакцию 26.06.2019
После доработки 14.11.2019
Принята к публикации 28.11.2019
Аннотация
Осуществлен синтез трех различающихся по химическому строению водорастворимых солей поли-(4,4'-оксидифенилен)пиромеллитамидокислоты. Представлены данные об их стабильности во времени, реологических свойствах водных растворов, плотности и поверхностном натяжении, необходимые для проведения электроформования. Методами сканирующей электронной и атомно-силовой микроскопии изучены структурно-морфологические характеристики синтезированных водорастворимых солей полиамидокислот и субмикронных волокон, образующихся в ходе электроформования. На основе водорастворимых аммиачной и триэтиламмонийной солей поли-(4,4'-оксидифенилен)пиромеллитамидокислоты впервые получены волокнистые нетканые материалы. Для нетканого материала на основе триэтиламмонийной соли определены физико-механические свойства.
ВВЕДЕНИЕ
Технология электроформования нановолокон из растворов полимеров является перспективным направлением в области нанотехнологий для получения современных композиционных материалов, применяющихся в различных областях науки, техники и медицины [1–7]. Такие нановолокна отличаются сверхразвитой структурой, а высокие значения удельной поверхности и возможность ее функционализации [8] обусловливают их применение, например, в функциональных мембранах [9, 10], фотокатализаторах [6, 11], биосенсорах [12–14], а также в качестве полупроводников в наноэлектронике [15].
Среди различных полимеров, используемых при электроформовании, ароматические поли-имиды представляют большой интерес, так как материалы на их основе обладают высокой термостойкостью, высокими прочностными и упругими характеристиками, они устойчивы к действию кислот и радиации [16].
Первая работа по получению полиимидных нановолокон путем электроформования раствора полиамидокислоты (ПАК) в N-метил-2-пирролидоне (МП) на основе пиромеллитового диангидрида и 4,4'-оксидианилина с последующей термической имидизацией была опубликована в 2003 году [17]. В более поздних публикациях [18, 19] список используемых диангидридов и диаминов уже расширен. К настоящему времени накоплен богатый опыт получения полиимидных микро- и нановолокон методом электроформования растворов полиамидокислот в апротонных высококипящих растворителях, таких как МП, ДМАА, ДМФА с добавками бензола и толуола [20]. Для того чтобы избежать проблем, связанных с использованием токсичных органических растворителей, был предложен наиболее экологически чистый метод – через водорастворимые соли полиамидокислот (СПАК). Водорастворимость в данном случае достигается нейтрализацией карбоксильных групп полиамидокислоты подходящим основанием, солеобразующим компонентом здесь выступает аммиак или различные третичные амины [21–24]. Таким образом, использование в качестве прекурсора полиимида аминной соли ПАК на водной основе позволяет избежать применения токсичных органических растворителей.
В настоящее время интерес к водорастворимым солям полиамидокислот возрос в связи с возможностью получения на их основе волокнообразующих полиимидных материалов в процессе электроформования СПАК с последующей имидизацией [25, 26]. Общий подход заключается в следующем. Вначале ПАК синтезируют из мономеров – диангидрида и диамина в амидном растворителе, затем получают СПАК в водной среде при взаимодействии ПАК со стехиометрическим количеством (или небольшим избытком) основания (В), далее соль термически имидизуют, получая полиимид:
Цель настоящей работы – синтез и комплексное исследование структуры, морфологии и свойств водорастворимых солей поли-(4,4'-оксидифенилен)пиромеллитамидокислоты на основе различных солеобразующих компонентов, определение возможности их использования для получения волокнообразующих материалов в процессе электроформования их водных растворов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходными веществами служили соединения пиромеллитового диангидрида (ПМ) фирмы “Sigma–Aldrich” (CAS89-32-7, 97%, Тпл = 283–286°С) и 4,4’-диаминодифенилового эфира (ДАДФЭ) фирмы “Molekula” (CAS101-80-4, 99.5%, Тпл = 188°С), которые сушили под вакуумом: первое соединение при температуре 180–200°С в течение 4 ч, второе – при 150°С в течение 3 ч; диметилформамид квалификации х.ч. (“Вектон”) обезвоживали гидридом кальция и перегоняли под вакуумом.
Также использовали триэтиламин (ТЭА) квалификации х.ч. (“Компонент Реактив”), аммиак водный NH4OH (ч.д.а., “Вектон”), натрий углекислый безводный (х.ч., “Вектон”) и этанол (спирт этиловый ректификат, 96.3 об. %; Общество с ограниченной ответственностью “Гатчинский спиртовой завод”) без предварительной подготовки.
Методика синтеза водорастворимых солей ПАК(ПМ–ДАДФЭ)
Для получения волокнообразующих материалов методом электроформования синтезировали водорастворимые соли ПАК(ПМ–ДАДФЭ) в два этапа.
На первом этапе осуществляли синтез полиамидокислоты (ПМ–ДАДФЭ) – (I) в диметилформамиде. В качестве солеобразующих компонентов использовали триэтиламин (II), аммиак (III) или карбонат натрия (IV). Синтез ПАК проводили в растворе амидного растворителя: при интенсивном перемешивании в токе аргона растворяли 0.05 моля ДАДФЭ в небольшом количестве ДМФА, примешивали 0.05 моля ПМ при комнатной температуре, далее раствор доводили до полного растворения диангидрида, после чего добавляли ДМФА для получения раствора 20%-ной концентрации, затем раствор ПАК перемешивали 4 ч. Характеристическая вязкость синтезированной ПАК составляла [η] = 0.98 см3/г. Измерения вели вискозиметрическим методом (вискозиметр Уббелоде). Молекулярную массу полимера (M = 38 × 103) рассчитывали по уравнению Марка–Куна–Хаувинка: ${\eta } = K{{M}^{{\alpha }}}$, где К = = 1.1, α = 0.91 – константы, определяемые структурой полимера и параметрами растворителя [27].
На втором этапе синтезировали водорастворимые соли ПАК. Для получения СПАК необходимо было сначала удалить амидный растворитель при помощи раствора ПАК, осаждаемого в воду. Провели серию экспериментов, чтобы выявить оптимальную концентрацию разбавления исходных растворов ПАК. Оказалось, что повышенная концентрация раствора ПАК при выделении полимера в воду приводит к образованию больших агрегатов ПАК, которые не дают возможности быстрого подхода противоиона к карбоксильной группе и в несколько раз увеличивают время реакции образования солей ПАК. Напротив, в слишком разбавленных растворах при осаждении в воду образуется мелкодисперсный осадок, и выход продукта снижается до 30%.
Для дальнейшей работы использовали концентрированный раствор ПАК(I), разбавленный ДМФА до 3 мас. %. Полученный раствор медленно прикапывали в сосуд с дистиллированной водой при интенсивном перемешивании. В результате образовывалась белая волокнистая масса ПАК, которую отделяли и промывали дистиллированной водой при перемешивании. Эту операцию повторяли еще 4–5 раз. Затем волокнистую массу переносили на воронку Бюхнера и фильтровали с отжимом. Установлено, что после отжима и сушки на воздухе в навеске волокнистой массы содержится ∼11–13 мас. % сухой ПАК.
Далее к взвешенному выделенному волокнистому осадку добавляли расчетное количество дистиллированной воды и солеобразующий агент в соотношении 0.05 моля ПАК : 0.10 моля триэтиламина, либо 0.10 моля аммиака в виде 25%-ного раствора, или 0.05 моля Na2CО3 в виде 20%-ного водного раствора. Полученную в каждом опыте массу перемешивали при комнатной температуре до образования прозрачного водного раствора. Установлено, что для солей ПАК(ПМ–ДАДФЭ) концентрация раствора из расчета на ПАК не должна превышать 5 мас. %, иначе процесс солеобразования протекает очень медленно (до нескольких суток).
Для образования сухих солей ПАК удаление воды из раствора солей осуществляли на ротационном испарителе при 60–65°С. Затем соль сушили в вакуумном сушильном шкафу при температуре 50–60°С. Выход продуктов составлял 85–90%.
Спектры ЯМР 1Н растворов ПАК(ПМ–ДАДФЭ) и полученных солей ПАК при 25°С регистрировали на приборе “Bruker Avance 400” при 400 МГц, использовали растворители ДМСО-d6 и D2O.
ИК-спектры синтезированных образцов ПАК(ПМ–ДАДФЭ) и солей ПАК регистрировали на спектрометре “Vertex‑70” фирмы “Bruker” с применением микроприставки однократного нарушенного полного внутреннего отражения “Pike” (призма с углом 45° из материала ZnSe). Спектры записывали с разрешением 4 см–1 при тридцати сканированиях. При обработке спектров вводили поправку, учитывающую зависимость глубины проникновения излучения от длины волны.
Реологические свойства водных растворов солей ПАК определяли с помощью реометра “Physica MCR-301” (“Anton Paar”, Австрия) в сдвиговом режиме при 20°C. Коэффициент поверхностного натяжения σ оценивали с применением тензиометра “DSA-30” (“Kruss”, Германия).
Для исследования морфологии пленочных покрытий из растворов солей ПАК использовали сканирующий электронный микроскоп “Supra 55VP” (“Carl Zeiss”, Германия). Для получения проводящего покрытия перед исследованием на поверхность образцов напыляли слой золота толщиной ~25 нм при ионном токе 6 мА и межэлектродном напряжении 1.5 кВ на установке “Eiko-IB3”.
Изучение топографии поверхности пленочных покрытий, полученных из водных растворов синтезированных солей, осуществляли методом атомно-силовой микроскопии на приборе “Nanotop NT-206” (Общество с дополнительной ответственностью “Микротестмашины”, Беларусь). Измерения проводили в полуконтактном и контактном режимах в атмосферных условиях с использованием кремниевых кантилеверов NSC11/AlBS с коэффициентом жесткости k = = 1.5–5.0 Н/м и радиусом кривизны кончика острия менее 10 нм. Обработку экспериментальных данных осуществляли с помощью программы “Surface Explorer”.
Рентгеновские исследования выполняли на дифрактометре “Seifert XRD 3003 TT” (“GE”, Германия), снабженном первичным монохроматором при U = 40 кВ, I = 40 мА. Использовали CuKα-излучение с длиной волны λ = 1.5406 Å. Дифрактограммы получали в области углов рассеяния 2θ = 3°–40° с шагом 0.05° и временем сканирования 5 с в каждой точке. Толщина образцов, на которых проводили рентгеновские исследования, составляла в среднем 350 мкм.
Механические испытания нетканого материала соли ПАК осуществляли на универсальной разрывной машине “Instron 5943” (Великобритания) в режиме одноосного растяжения при комнатной температуре. Скорость растяжения составляла 10 мм/мин; длина образцов 30 мм, ширина – 5 мм. В процессе испытаний регистрировали диаграмму растяжения образца в режиме on-line, по результатам испытаний определяли модуль упругости Е, прочность при растяжении σр, разрывную деформацию εр.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
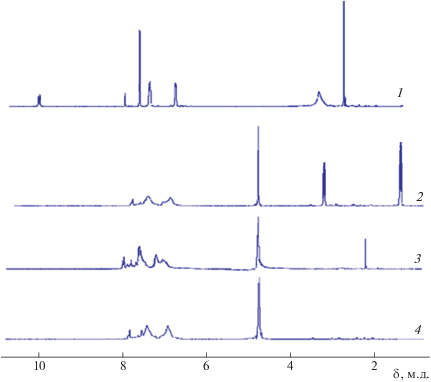
На рис. 1 представлены ЯМР 1Н-спектры ПАК(I) и синтезированных солей. В спектрах ЯМР 1Н раствора ПАК присутствуют сигналы протонов амидокислотной группы ПАК в области 10.5–11.0 м.д., сигналы ароматических фрагментов ПАК в области 7.5–8.5 м.д., как показано ранее в работе [28]. В солях ПАК сигналы амидокислотных групп отсутствуют (при сохранении сигналов ароматического звена), что указывает на практически полное взаимодействие этой функциональной группы с противоионами используемых солеобразующих реагентов Et3N (II), NH3 (III) и Na2CO3 (IV). В алифатической области спектра у образующихся солей регистрируются сигналы протонов, что особенно четко проявляется после реакции ПАК с ТЭА. Эти факты свидетельствуют об образовании солей ПАК с Et3N, NH3 и NA2CO3 при учете полной растворимости образцов в D2O.
Рис. 1.
Спектры ЯМР 1Н растворов ПАК(I) в ДМСО-d6 (1) и синтезированных солей (I)–II в D2O (2), (I)–III в D2O (3), (I)–IV в D2O (4).
Строение синтезированных солей ПАК(I) контролировали методом ИК-спектроскопии. На рис. 2 представлены ИК-спектры исходной полиамидокислоты и синтезированных СПАК. В спектре полиамидокислоты видны полосы, характерные для карбоксиамидных звеньев 3280, 1711, 1640 и 1540 см–1. Во всех спектрах солей исчезает полоса 1711 см–1 и появляются полосы в области 1590 и 1340 см–1, присущие группам СОО–. Также в спектрах сохраняются полосы 1640 и 1540 см–1, соответствующие амидному звену.

Проведенные реологические исследования показали высокую стабильность водных растворов СПАК. На рис. 3 представлено изменение динамической вязкости водных растворов триэтиламонийной соли ПАК(I) в сравнении с ПАК(I) в растворе ДМФ, а на рис. 4 – изменение вязкости во времени водных растворов СПАК.
Рис. 3.
Зависимость вязкости водных растворов триэтиламмонийных солей ПАК(I) от времени при концентрации 8 (1), 6 (2) и 3 мас. % (3) и изменение вязкости во времени ПАК(I) в растворе ДМФ (4).
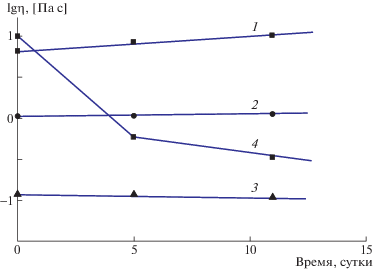
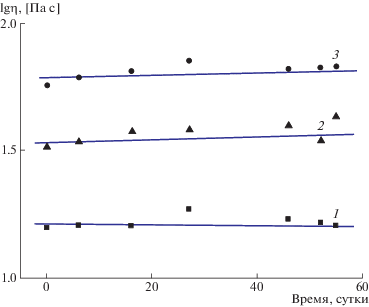
Рис. 4.
Изменение динамической вязкости водных растворов СПАК(I)–III (1), СПАК(I)–IV (2) и СПАК(I)–II (3) при концентрации 8 мас. % в течение 55 суток.
Перед процессом электроформования были определены значения плотности и поверхностного натяжения водных растворов СПАК. Так, водный 10%-ный раствор водорастворимых солей ПАК(I)–II имеет достаточно высокое поверхностное натяжение 61.5 мН/м. При таком высоком значении электроформование невозможно, поэтому в водный раствор был добавлен разбавитель с низким поверхностным натяжением – этиловый спирт. В процессе приготовления растворов для электроформования было установлено, что соли на основе Et3N растворимы в спирто‑водной смеси при соотношении 70 : 30 и концентрации 9–15 мас. %, а при использовании NH3 и Na2CO3 – в спирто-водной смеси 5% при соотношении 50 : 50 (таблица 1).
Таблица 1.
Значения плотности и поверхностного натяжения спирто-водных растворов солей ПАК
| Полимер | Солеобразующий компонент | Растворитель спирт : вода,% | Концентрация, мас. % | Плотность, г/см3 | Поверхностное натяжение, мН/м |
|---|---|---|---|---|---|
| ПАК(ПМ–ДАДФЭ) | Et3N | 70 : 30 | 9 | 0.9070 | 28.1 |
| 70 : 30 | 12 | 0.9113 | 28.1 | ||
| 70 : 30 | 15 | 0.9157 | 29.3 | ||
| 0 : 100 | 10 | 1.0121 | 61.5 | ||
| NH3 | 50 : 50 | 5 | 0.9376 | 28.5 | |
| Na2CO3 | 50 : 50 | 5 | 0.9362 | 28.6 |
При концентрации полимера 10–15 мас. % поверхностное натяжение раствора спирто‑водной смеси CПАК(I)–II составляет 28.1–29.3 мН/м, что оптимально для электроформования. При этом средняя электропроводность растворов, пригодных для формования, составляет 1685 мкСм/см.
Полученные спирто‑водные смеси (с соотношением 70 : 30) СПАК (I)–II, а также СПАК(I)–III и СПАК(I)–IV были использованы для электроформования.
Для более полной информации о структурно-морфологических особенностях синтезированных СПАК в работе проведено изучение тонких пленок, полученных из растворов СПАК(I)–II и СПАК(I)–IV с применением методов СЭМ (рис. 5–8, 10, 11) и АСМ (рис. 9–12).
Рис. 5.
СЭМ-микрофотография волокон, полученных методом электроформования из раствора CПАК(I)–II при концентрации 9 мас. %.

Рис. 6.
СЭМ-микрофотографии волокон, полученных методом электроформования из раствора CПАК(I)–II при концентрации 12 мас. % и увеличении 5000 (а), 100000 (б).

Рис. 7.
СЭМ-микрофотографии волокон, полученных методом электроформования из растворов CПАК(I)–II (а) и CПАК(I)–III (б) при концентрации 15 мас. %.


Рис. 9.
АСМ-изображения поверхности пленки CПАК(I)–II, полученной из раствора на стеклянной подложке: а, б – топография поверхности, в – 3D-изображение, г – профили выделенных участков поверхности.
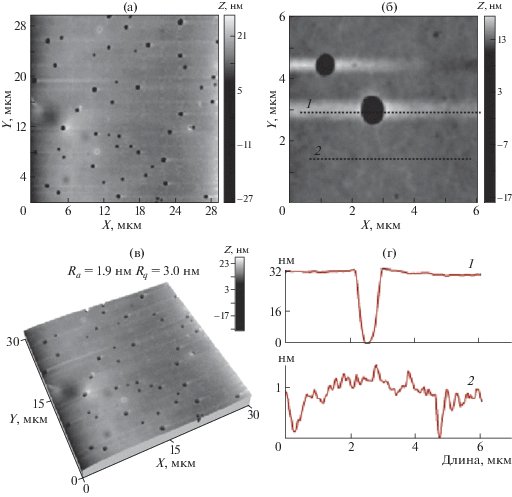
Рис. 10.
СЭМ-микрофотография поверхности пленки, полученной из раствора CПАК(I)–II при концентрации 12 мас. %.

Рис. 11.
СЭМ-микрофотография поверхности пленки, полученной из раствора CПАК(I)–IV при концентрации 5 мас. %.

Рис. 12.
АСМ-изображения поверхности пленки CПАК(I)–IV, полученной из раствора на стеклянной подложке: а, б – топография поверхности, в – 3D-изображение, г – профиль выделенного участка поверхности.
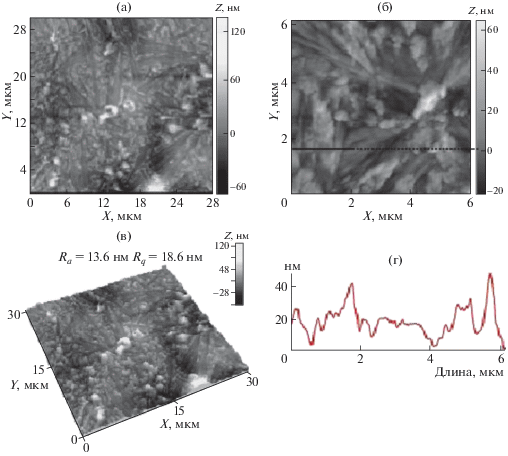
Рис. 13.
Оптическая фотография нетканого материала, полученного при электроформовании раствора CПАК(I)–II при концентрации 12 мас. %.

Методом СЭМ показано, что при увеличении концентрации спирто-водных (70 : 30) растворов CПАК(I)–II от 9 до 15 мас. % диаметр волокон (средние значения) увеличивается от ~0.47 мкм (рис. 5) до ~2.61 мкм (рис. 7а). При съемке на большом увеличении (рис. 6б) установлено, что волокна имеют гладкую на наноуровне поверхность без пор. При использовании аммиака в качестве солеобразующего компонента также проходило электроформование растворов соли и образовывались волокна (рис. 7б), однако в случае CПАК(I)–III формование осуществлялось менее стабильно, о чем свидетельствуют утолщения и наплывы на волокнах, видимые на микрофотографии. Следует отметить, что средняя толщина формуемых волокон из раствора CПАК(I)–III значительно меньше, чем из раствора CПАК(I)–II при одинаковой концентрации раствора.
При использовании раствора CПАК(I)–IV при электроформовании вместо волокон образовывались сферические частицы, которые слипались в крупные агрегаты (рис. 8).
На рис. 9 представлены АСМ-изображения поверхности пленки CПАК(I)–II, полученной из раствора c концентрацией 12 мас. % на стеклянной подложке. Методом АСМ на поверхности пленки соли CПАК(I)–II обнаружено незначительное количество отдельных крупных пор размерами 400–800 нм (рис. 9а–9в), глубиной до 35 нм (рис. 9г, профиль 1), вероятно, образовавшихся при испарении воды в процессе высушивания, при этом основная поверхность пленки выглядит очень гладкой, параметры шероховатости (для матрицы сканирования 30 × 30 мкм) составляют: среднеарифметическая шероховатость Ra = 1.9 нм и среднеквадратичная Rq = 3.0 нм (рис. 9в). При уменьшении размеров сканированного участка поверхности 6 × 6 мкм (рис. 9б) хорошо видна мелкопористая морфология гладких участков с размерами пор от 20 до 100 нм. Эти поры расположены в приповерхностном слое и имеют небольшую глубину до ~1 нм (рис. 9г, профиль 2).
На СЭМ-изображении (рис. 10) образца соли ПАК прослеживается такая же морфология с большим количеством дефектов в виде углублений на поверхности. Размеры лунок достигают 1.5 мкм. Такой характер морфологии может свидетельствовать об аморфной или мезоморфной надмолекулярной организации исследуемого образца.
Напротив, пленка CПАК(I)–IV – высоко кристаллическая, на СЭМ-микрофотографии (рис. 11) хорошо видны тонкие ламеллярные кристаллические пластинки, уложенные в рыхлые пачки. Известно, что на поверхности ламелей концентрируются нерастворимые примеси и образуются поры, что приводит к высокой хрупкости образца.
Анализ АСМ-изображений этого образца (рис. 12а–12в) показывает наличие пачек ламеллярных кристаллов в образце CПАК(I)–IV, кроме того (в дополнение к результатам СЭМ-анализа) обнаружено большое количество мелких глобулярных кристаллитов диаметром 200–300 нм в областях между ламеллярными кристаллами (рис. 12б).
Так, образец CПАК(I)–IV является высоко кристаллическим, имеет ламеллярную морфологию, ламели слабо связаны друг с другом и в процессе электроформования волокно не может образоваться, вероятно, из-за быстрой кристаллизации соли и последующего хрупкого разрушения.
Анализ морфологии формирующихся волокнистых материалов с помощью метода СЭМ показал, что создание волокон в ходе электроформования наблюдается как у образцов CПАК(I)–II (рис. 5–7а), так и CПАК(I)–III (рис. 7б), при этом в случае CПАК(I)–II для образования волокнистой структуры были использованы растворы разной концентрации, для того чтобы выбрать оптимальную концентрацию для проведения электроформования.
В связи с этим, для дальнейшего исследования физико-механических характеристик был изготовлен образец волокнистого нетканого материала большой площади толщиной ~30 мкм из раствора CПАК(I)–II при концентрации 12 мас. % (рис. 13).
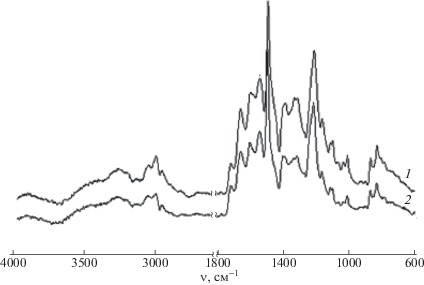
Сравнение ИК-спектров CПАК(I)–II и нетканого материала на ее основе (рис. 14) показывает, что они практически идентичны. В спектрах обоих образцов имеется полоса 1599 см–1, соответствующая группе СОО–, и отсутствует полоса СООН 1680 см–1, т.е. в процессе электроформования сохраняется структура исходной соли.
Рис. 14.
ИК-спектры образцов CПАК(I)–II (1) и нетканного материала, образующегося из этой соли в ходе электроформования (2).
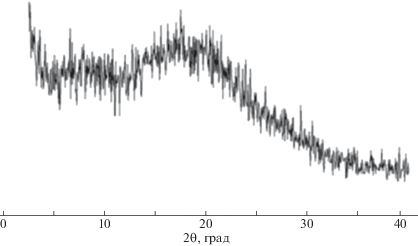
На рис. 15 представлены результаты рентгеновского исследования нетканого материала на основе CПАК(I)–II. На дифрактограмме можно выделить два диффузных рефлекса (гало) при 2θ ~ 6°–7° и 18°–20°. При исследовании образца в поляризованном свете на столике Боэтиуса (скрещенные николи) наблюдается слабое свечение, которое свидетельствует о том, что образец анизотропный. Сравнение данных, полученных методами РСА и оптической микроскопии, позволяет сделать вывод, что образец соли СПАК(I)–II мезоморфный. Однако это состояние не препятствует получению однородного по объему волокна. При этом следует отметить, что из мезоморфного состояния обычно получают волокна с более высокими физико-механическими характеристиками [29].
Оценка физико-механических характеристик полученного нетканого материала из CПАК(I)–II дает значения модуля упругости Е = 128 ± 12 МПа; прочности σ = 7.9 ± 0.6 МПа, и деформации при разрыве ε = 17 ± 2%.
ЗАКЛЮЧЕНИЕ
В работе осуществлен синтез водорастворимых солей поли-(4,4'-оксидифенилен)пиромеллитамидокислоты при использовании в качестве солеобразующих агентов триэтиламина, аммиака и углекислого натрия. Анализ результатов комплексного исследования стабильности во времени водных растворов синтезированных солей, их реологического поведения, плотности и поверхностного натяжения позволил определить условия, необходимые для проведения их электроформования. Впервые из спирто-водного (70 : 30) раствора соли ПАК(ПМ–ДАДФЭ)–Et3N 12 мас. % и соли ПАК(ПМ–ДАДФЭ)–NH3 15 мас. % получен волокнистый нетканый материал. Для волокнистого нетканого материала на основе соли ПАК(ПМ–ДАДФЭ)–Et3N определены физико-механические свойства. Показано, что в отличие от соли ПАК(ПМ–ДАДФЭ)–Et3N, образец соли ПАК(ПМ–ДАДФЭ)–Na2CO3 является высоко кристаллическим, что препятствует образованию волокон в процессе электроформования.
Авторы выражают благодарность А.Я. Волкову за техническую помощь в выполнении рентгеновских исследований.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (код проекта 18-03-00568_а).
Список литературы
Doshi J., Reneker D.H. // J. Electrostat. 1995. V. 35. P. 151.
Reneker D.H., Chun L. // Nanotechnology. 1996. V. 7. P. 216.
Филатов Ю.Н. Электроформование волокнистых материалов (ЭФВ-процесс). М.: Нефть и газ, 1997.
Dzenis Y . // Science. 2004. V. 304. P. 1917.
Li D., Xia Y.N. // Adv. Mater. 2004. V. 16. P. 1151.
Reneker D.H., Yarin A.L., Zussman E., Xu H. // Adv. Appl. Mechan. 2007. V. 41. P. 43.
Филатов И.Ю., Филатов Ю.Н., Якушкин М.С. // Вестн. МИТХТ. 2008. Т. 3. № 5. С. 3.
Greiner A., Wendorff J.H. // Angew. Chem. Int. Ed. 2007. V. 46. P. 5670.
Shin Y.M., Hohman M.M., Brenner M.P., Rutledge G.C. // Polymer. 2001. V. 42. P. 9955.
Megelski S., Stephens J.S., Chase D.B., Rabolt J.F. // Macromolecules. 2002. V. 35. P. 8456.
Li D., Wang Y., Xia Y.N. // Nano Lett. 2003. V. 3. P. 1167.
Acatay K., Simsek E., Ow-Yang C., Menceloglu Y.Z. // Angew. Chem. Int. Ed. 2004. V. 43. P. 5210.
Yuan X.Y., Zhang Y.Y., Dong C.H., Sheng J. // Polym. Int. 2004. V. 53. P. 1704.
Miyoshi T., Toyohara K., Minematsu H. // Polym. Int. 2005. V. 54. P. 1187.
Teo W.E., Ramakrishna S. // Nanotechnology. 2006. V. 17. P. 89.
Бессонов М.И., Котон М.М., Кудрявцев В.В., Лайус Л.А. Полиимиды – класс термостойких полимеров. Л.: Наука, 1983.
Changwoon N., Sang H.H., Myong H.L., Jong S.K., Dai S.L. // Polymer Int. 2003. V. 52. P. 429.
Chaobo H., Suqing W., Hean Z., Tingting L., Shu-iliang C., Chuilin L., Haoqing H. // Eur. Polym. J. 2006. V. 42. P. 1099.
Chen F., Peng X., Li T., Chen S., Wu X.-F., Reneker D.H., Hou H. // J. Phys. D. 2008. V. 41. 025308 (8 pp).
Matrenichev V.V., Popryadukhin P.V., Sklizkova V.P., Svetlichnyi V.M., Kryukov A.E., Smirnova N.V., Ivan’kova E.M., Popova E.N., Dobrovol’skaya I.P., Yudin V.E. // Polymer Science A. 2018. V. 60. № 4. P. 483.
Li Q., Yamashita T., Horie K., Yoshimoto H., Miwa T., Maekawa Y. // J. Polym. Sci., Polym. Chem. 1998. V. 36. P. 1329.
Ding Y., Bikson B., Nelson J.K. // Macromolecules. 2002. V. 35. № 3. P. 905.
Kreuz J.A., Endrey A.L., Gay F.P., Sroog C.E. // J. Polym. Sci., Polym. Chem. 1966. V. 4. P. 2607.
Reynolds R.J.W., Seddon J.D. // J. Polym. Sci., Polym. Symp. 1968. V. 23. P. 45.
Jiang S., Hou H., Agarwal S., Greiner A. // ACS Sustain. Chem. Eng. 2016. V. 4. № 9. P. 4797.
Xu H., Jiang S., Ding C., Zhu Y., Li J., Hou H. // Mater. Lett. 2017. V. 201. P. 82.
Котон М.М., Каллистов О.В., Кудрявцев В.В., Склизкова В.П., Силинская И.Г. // Высокомолек. соед. А. 1979. Т. 21. № 3. С. 532.
Jun Yang, Myong-Hoon Lee // Macromol. Res. 2004. V. 12. № 3. P. 263.
Sukhanova T.E., Baklagina Yu.G., Kudryavtsev V.V., Maricheva T.A., Lednicky F. // Polymer. 1999. V. 40. P. 6265.
Дополнительные материалы отсутствуют.
Инструменты
Высокомолекулярные соединения (серия Б)