НЕФТЕХИМИЯ, 2021, том 61, № 4, с. 504-519
УДК 544.47
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
ЗОЛОТОСОДЕРЖАЩИХ КАТАЛИЗАТОРОВ КОНВЕРСИИ ЭТАНОЛА
В БУТАНОЛ
© 2021 г. С. А. Николаев1,*, А. В. Чистяков2, П. А. Чистякова2, Д. И. Эзжеленко1,
Е. Ю. Либерман3, Т. В. Конькова3, М. В. Цодиков2
1Московский государственный университет имени М.В. Ломоносова, Москва, 119991 Россия
2Институт нефтехимического синтеза им. А.В. Топчиева РАН, Москва, 119991 Россия
3Российский химико-технологический университет им. Д.И. Менделеева, Москва, 125047 Россия
*Е-mail: serge2000@rambler.ru
Проступила в редакцию 24 февраля 2021 г.
После доработки 26 марта 2021 г.
Принята к публикации 12 апреля 2021 г.
C помощью физико-химических методов (ПЭМ, СЭМ, ЭДА, РФЭС, ТПД-NH3 и адсорбции N2) изуче-
на структура ряда носителей (Al2O3, SiO2, TiO2, ZrO2, C) и образцов катализатора Au/носитель (Au =
0.5%). Содержание в Au-катализаторах высокоактивных частиц золота размером 2-4 нм определяется
текстурой используемого носителя и изменяется в ряду: Au/TiO2 < Au/ZrO2 < Au/C < Au/SiO2 << Au/Al2O3.
Кислотность Au-катализаторов определяется природой носителя и изменяется в ряду: Al2O3 > TiO2 >
ZrO2 > SiO2 >> Au/C. Показано, что при 275°С углеродный носитель неактивен в конверсии этанола в
бутанол. На оксидных носителях целевая реакция протекает по механизму “бимолекулярной конденса-
ции” и с относительно низкой скоростью. На Au/Al2O3, Au/SiO2, Au/TiO2 и Au/ZrO2 реакция осущест-
вляется по механизму “альдольной конденсации” и с более высокой скоростью. При конверсии этанола
14-18% селективность по бутанолу изменяется в ряду: Au/C(0) << Au/SiO2 (0.4%) < Au/ZrO2 (1.5%) <
Au/TiO2 (2 %) << Au/Al2O3 (78%). Высокая эффективность Au/Al2O3 обусловлена сочетанием высокой
плотности центров Aln+-O2-, расположенных на поверхности носителя, и координационно-ненасыщен-
ных атомов Au0(KН), находящихся на поверхности частиц золота размером 2-4 нм.
Ключевые слова: носитель, Au, наночастицы, этанол, бутанол
DOI: 10.31857/S0028242121040067
Разработка каталитической конверсии биоэта-
кислотно-основных катализаторов, таких как
нола для получения синтетических углеводородов
Rb/NaX, K/Al2O3 MgO-Al2O3, CaO/Al2O3, MgO
является актуальной задачей, решение которой по-
[3-5, 6]. Включает в себя разрыв связи С-OH пер-
зволит снизить зависимость химической промыш-
вой молекулы этанола на кислотных центрах Mn+
ленности от использования нефти и перейти к бо-
и разрыв связи β-C-H второй молекулы этанола на
лее энергоэффективным технологиям [1, 2]. Одним
основных центрах O2-, с последующей рекомбина-
из перспективных процессов конверсии этанола в
цией фрагментов C2H5 и С2H4OH в бутанол, а OH
ценные продукты является реакция, приводящая к
и H в H2O. Необходимость использования высоких
образованию бутанола, который широко использу-
температур для разрыва связей этанола приводит к
ются в фармацевтике, нефтехимии и парфюмерии
высокой скорости побочных процессов. В резуль-
[3, 4].
тате кислотно-основные катализаторы проявляют
В настоящее время приняты два механизма
невысокую эффективность: при конверсии этано-
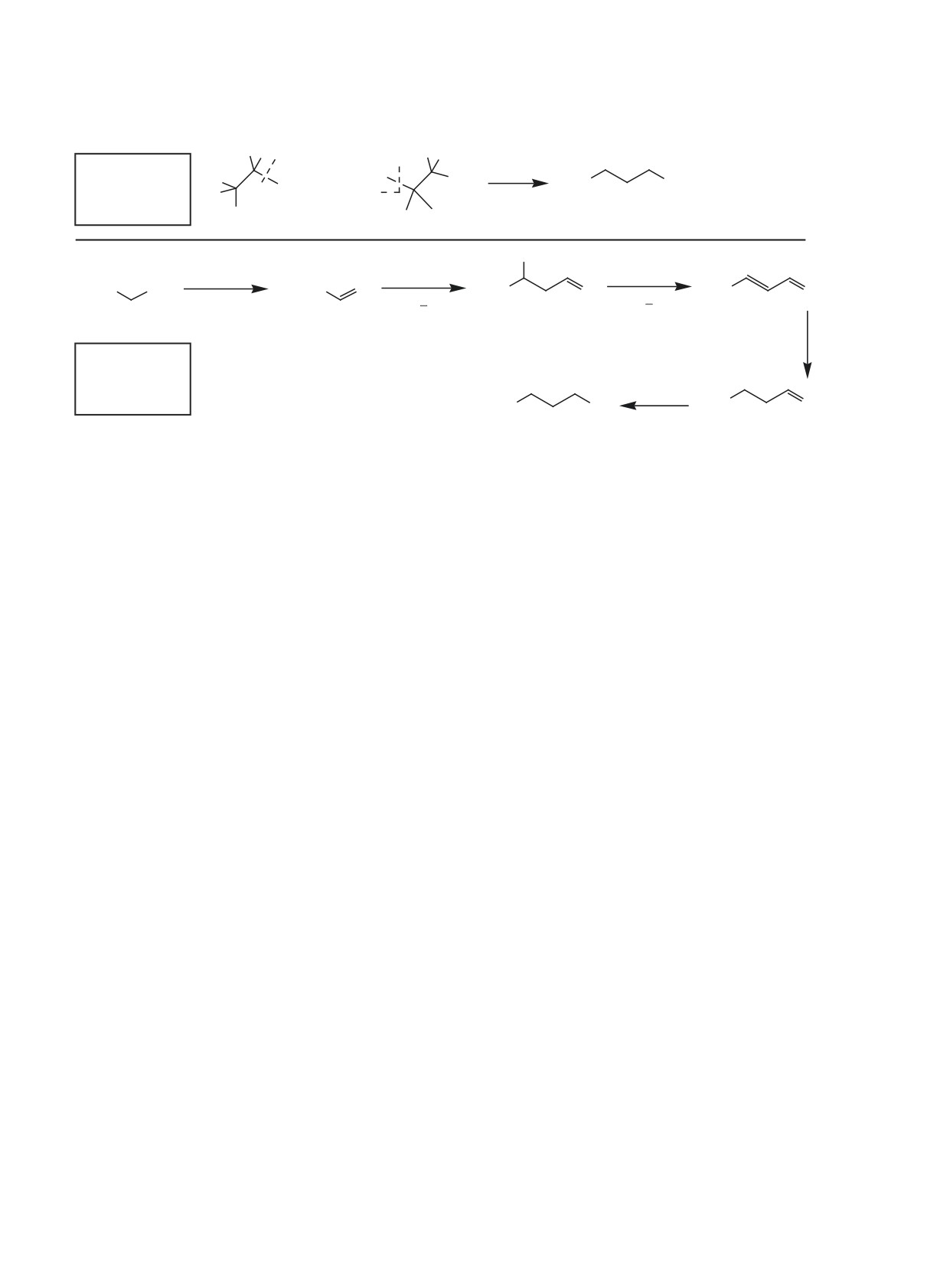
превращения этанола в бутанол (рис. 1). Меха-
ла 20-30% селективность по бутанолу составляет
низм (I) реализуется при 350-450°C в присутствии
18-37% [7, 8]. Так же есть сообщения о низкой
504
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
505
H H
H H
Механизм (I)
H
H
OH
Кат. = M1Ox
OH
+
2-
H3C
OH + H2O
H
O
T = 350-400°C
H M1n+
H
H
OH
−2H2
-H2O
H3C
OH
H3C
O
2
0
2
H3C
O
H3C
O
M
2
M1n+ - O2-
M1n+ - O2-
+H2
M0
Механизм (II)
Кат. = M2/M1Ox
+H2
T = 200-300°C
H3C
OH
H3C
O
0
M
2
Рис. 1. Механизмы конверсии этанола в присутствии кислотно-основных оксидов M1Ox (M1 = Al, Mg, Ca, Al-Si) и нане-
сенных катализаторов M2/M1Ox (M1 = Al, Mg, Ca, Al-Si; M2 = Pd, Ni, Pt, Rh). По данным [3-7].
стабильности работы оксидных систем, причина
Анализ приведенных выше данных позволяет
которой заключается в модификации кислотно-ос-
сделать вывод о том, что конверсия этанола в бу-
новных центров катализаторов побочными продук-
танол по механизму (II) протекает в более мягких
тами и водой [3, 5, 7-9].
условиях и с более высокой начальной селектив-
Механизм (II) реализуется при 200-300°C на би-
ностью. В то же время, для эффективного синтеза
бутанола из этанола требуется разработка новых
функциональных системах M0/носитель (M = Pd,
нанесенных катализаторов, устойчиво работаю-
Pt, Ni, и др.; носитель = Al2O3, MgO-Al2O3, и др.).
щих в присутствии CO и RH, образующихся в ходе
Механизм (II) представляет собой последователь-
побочных процессов. В этом отношении могут ока-
ность стадий: дегидрирование этанола в этаналь на
заться перспективными Au-содержащие системы.
центрах M0; конденсация этаналя в 2-бут-2-еналь
Известно, что в отличие от традиционных катали-
на кислотно-основных центрах носителя; гидри-
рования 2-бут-2-еналя в бутанол на металличе-
заторов-металлов, таких как палладий и никель,
золото в меньшей степени хемосорбирует CO и RH
ских центрах [3, 4, 10]. Наиболее активными ката-
[14], но при этом наночастицы золота проявляют
лизаторами конверсии этанола по механизму (II)
высокую активность в процессах гидрирования
являются композиты Pd/Al2O3 и Ni/Al2O3 [11, 12].
и дегидрирования RH [15, 16], которые являются
При 270°C, конверсия этанола на 0.1%Pd/Al2O3
ключевыми стадиями механизма (II) (см. рис. 1).
составляет 24%; селективность по бутанолу рав-
на 70% [13]. При 250°C, конверсия этанола на
В настоящее время изучено влияние размера
20%Ni/Al2O3 составляет 25%; селективность по
частиц Au [11, 17, 18], фазового состава частиц
бутанолу равна 70% [11]. Несмотря на высокую
[19, 20], температуры реакции [20] и агрегатного
начальную эффективность, и Pd- и Ni-катализато-
состояния этанола [19-21] на скорость образова-
ры обладают низкой стабильностью работы, кото-
ния бутанола из этанола. Показано, что иммоби-
рая выражается в снижении скорости образования
лизованные на оксиде алюминия частицы золота
бутанола на 80-95% спустя 12-20 ч эксплуатации
формируют активные центры Au0-Al2O3, которые
[11, 13]. Причиной дезактивации Pd/Al2O3 является
в оптимальных условиях (275°C, сверхкритическое
побочный процесс декарбонилирования ацетальде-
состояние этанола) позволяют проводить конвер-
гида с последующей хемосорбцией продукта (CO)
сию этанола с 70%-ной селективностью по целе-
на активных центрах Pd. Ni-катализатор провоци-
вому продукту; при этом катализатор не проявляет
рует разрыв связей -С-С- интермедиатов и отрав-
признаков дезактивации в течение 100 ч непрерыв-
ляется за счет коксования.
ной работы [20]. Таким образом, есть основания
НЕФТЕХИМИЯ том 61 № 4 2021
506
НИКОЛАЕВ и др.
полагать, что системы Au/оксид являются перспек-
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
тивными для разработки новых технологий кон-
Носители и катализаторы
версии этанола в бутанол.
В работе были использованы сертифицирован-
Известно, что носитель играет значительную
ные носители: Al2O3 (“АОК-63-11 (В)”, Ангар-
роль в формировании активных центров золотых
ский завод катализаторов и органического синтеза,
катализаторов дегидрирования, гидрирования и
1.0 мм гранулы; фазовый состав - γ-Al2O3 [13]);
окисления [14, 15]. При этом, влияние носителя
TiO2 (“Aerolyst®7710”, Evonik, 1 мм экструды,
структура TiO2 - анатаз); ZrO2 (“Aerolyst®6100”,
на активность Au-катализаторов конверсии этано-
Evonik, 2 мм экструды, структура ZrO2 - моно-
ла в бутанол изучено слабо. В ходе литературно-
клинная); углеродный носитель C
(“Сибунит”,
го поиска была найдена всего одна работа 2018 г.,
Институт катализа им. Г.К. Борескова СО РАН,
связанная с анализом активности Au/TiO2 и
1 мм гранулы, рентгеноаморфная модификация
Au/ZnO [17]. В [17] показано, что использование
[22]; SiO2 (№ 288616, Aldrich, 0.4 мм гранулы,
глобул TiO2 размером 20 нм позволяет получать
рентгеноаморфная модификация).
высокодисперсные частицы Au в системе Au/TiO2.
Образцы Au/носитель (Au = 0.5 мас. %) гото-
В результате Au/TiO2 проявляет высокую началь-
вили пропиткой по влагоемкости. В качестве пре-
ную активность, но быстро дезактивируется вслед-
курсора использовали водный раствор HAuCl4
ствие отложений кокса на кислых центрах TiO2.
(Sigma-Aldrich). В типовом синтезе 5 г носителя
Напротив, глобулы ZnO размером 8-40 нм позво-
прокаливали при 350°C в течение 3 ч и определяли
ляют получать менее дисперсные частицы Au, но
сорбционную емкость носителя по воде. Прокален-
при этом активные центры ZnO в меньшей степени
ный при 350°C носитель пропитывали раствором
дезактивируется в ходе реакции. В итоге Au/ZnO
HAuCl4 с известной концентрацией золота, суши-
проявляет умеренную, но стабильную активность.
ли при 25°C и прокаливали при 350°C в течение
Стоит также отметить, что для синтеза систем
3 ч. Фактическое содержание металла в образцах
Au/носитель авторы [17] использовали не гранулы
определяли с помощью атомной абсорбционной
носителей, а нанопыль, что затрудняет оценку эф-
спектрометрии (ААС) на приборе Thermo iCE 3000
фективности Au-катализаторов при использовании
[23]. Фактическое содержание Au в катализаторах
составило 0.5 ± 0.03 мас. %.
в промышленных реакторах, спроектированных,
как правило, в расчете на гранулированные или
Низкотемпературная адсорбция N2
прессованные катализаторы.
Измерения проводили на приборе NOVA 2000
(Quantachrome Instruments, США). Перед исследо-
Цель настоящей работы - установление зако-
ванием образцы дегазировали при 300°C в течение
номерностей формирования активных центров
2 ч, помещали в прибор и записывали изотермы ад-
конверсии этанола в системах Au/носитель (Au =
сорбции и десорбции азота.
0.5 мас. %; носитель = Al2O3, SiO2, TiO2, ZrO2, C) с
Расчет удельной поверхности (SBET) проводили
последующей оценкой начальной активности ком-
из изотерм адсорбции в интервале P/P0 = 0.05-0.3
позитов в конверсии этанола в бутанол при 275°C.
с помощью уравнения Брунауэра-Эммета-Теллера
Стоит подчеркнуть, что для синтеза катализаторов
(БЭТ). Относительная погрешность определения
были использованы гранулы промышленных носи-
SBET составляла 7-10%. Суммарный объем пор
телей и золото наносилось с использованием про-
(Vs) определяли по количеству адсорбированно-
питки. Такой подход при выборе носителя и метода
го азота при относительном давлении P/Ps = 0.99.
синтеза позволяет получать информацию, пред-
Объем микропор (Vм) определяли t-методом. Отно-
ставляющую интерес как для фундаментального
сительная погрешность определения величин Vм и
катализа, так и для химиков-технологов. Для более
Vs составляла 5-10%. Для построения дифферен-
точной оценки активности золотосодержащих ка-
циальных кривых распределения диаметра пор по
тализаторов были также изучены закономерности
объему (ДКР) использовали изотерму десорбции
превращения этанола на гранулах чистых носителей.
азота и уравнение Баррета-Джойнера-Халенды.
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
507
При расчетах использовали программное обеспе-
0.1 эВ. Для калибровки шкалы энергии использова-
чение, входящее в комплектацию прибора. Основ-
ли внешний стандарт [золотую фольгу с энергией
ной диаметр пор (D) определяли из ДКР. Относи-
связывания электронов Au 4f7/2 = (83.96 ± 0.03) эВ]
тельная погрешность определения D составляла
и внутренний стандарт (пик адсорбированного
10-14%.
углерода C 1s = 285.0 ± 0.05 эВ) [24]. Погрешность
определения экспериментальных величин энергии
Сканирующая и просвечивающая
связывания Au 4f в образцах Au/носитель состави-
электронная микроскопия
ла ±0.05 эВ.
Микрофотографии поверхности образцов полу-
Температурно-программированная
чали с помощью сканирующей электронной микро-
десорбция NH3
скопии (СЭМ) на приборе JCM-6000 с ускоряющим
напряжением электронов 5-15 кэВ и максимальной
Кислотность образцов измеряли с помощью
кратностью увеличения ×103 раз. Перед анализом
температурно-программированной десорбции NH3
гранулы образцов прокаливали при 350°C в тече-
(ТПД-NH3) на анализаторе хемосорбции УСГА-101
ние 3 ч, остужали до комнатной температуры и
[13]. Для этого 0.2 г образца помещали в кварцевый
фиксировали на обзорном столе с помощью прово-
реактор и прокаливали в токе He (скорость пода-
дящего скотча. Съемку проводили в режиме детек-
чи - 20 мл/мин) при температуре 400°С в течение
тирования вторичных электронов.
1 ч. Реактор охлаждали до 25°C и насыщали обра-
зец парами аммиака в течение 30 мин. Адсорбиро-
Исследование просвечивающей электронной
ванный физически аммиак удаляли прокаливанием
микроскопией (ПЭМ) проводили на приборе JEOL
JEM
2100F/UHR с ускоряющим напряжением
в токе He при 100°C в течение 1 ч. Затем проводили
линейный нагрев образца со скоростью 8°С/мин до
электронов 200 кЭВ и максимальной кратностью
750°С в потоке гелия (30 мл/мин). Выделяющий-
увеличения ×106 раз. Стандартная подготовка об-
ся аммиак регистрировался детектором по тепло-
разца для анализа ПЭМ приведена в [20]. Иденти-
проводности. Расчет кислотности образца [AS]
фикацию золотых частиц проводили с помощью
проводили отнесением количества выделившегося
локального энергодисперсионного анализа (ЭДА)
аммиака к массе навески образца. Погрешность
на приборе JED - 2300, входящего в комплекта-
цию электронного микроскопа. Диаметр частиц
определения [AS] составила ±7%.
Au определяли как максимальный линейный раз-
Каталитическая конверсия этанола
мер частицы. Погрешность определения размера
Каталитические тесты проводили на установ-
частиц составляла ± 3%. Средний размер (DAV) и
ке автоклавного типа Parr 5000 Series при подо-
долю активных частиц Au размером 2-4 нм [Au*]
бранной ранее оптимальной температуре 275°С
определяли из гистограмм распределения частиц
[13, 21]. В стандартном опыте в реактор помещали
по размерам, для чего проводили статистическую
30 мл этанола и 5 г катализатора. Реактор продува-
обработку выборки по 300 частицам [20].
ли Ar, нагревали до 275°С и проводили перемеши-
Рентгеновская фотоэлектронная
вание смеси при 1200 об/мин. Спустя 5 ч отклю-
спектроскопия
чали перемешивание и нагрев, вскрывали реактор
Рентгеновские фотоэлектронные спектры
и проводили отбор проб газовой смеси и жидкой
фракции продуктов.
(РФЭ-спектры) золота регистрировали на спектро-
метре Axis Ultra DLD (Kratos) с использованием
Продукты анализировали методом газовой хро-
монохроматического AlKα излучения (1486.6 эВ).
матографии: газообразные углеводороды С1-С4 -
Перед исследованием гранулы образцов прокали-
на хроматографе Кристалл-4000М (“Мета-хром”,
вали при 350°C в течение 3 ч, остужали до комнат-
Россия, ПИД, колонка HP-PLOT); СО, СО2 и Н2 -
ной температуры и фиксировали в приборе с помо-
на хроматографе Кристалл-4000
(“Мета-хром”,
щью проводящего скотча. Съемку производили с
Россия), детектор по теплопроводности, колонка
использованием электронной пушки для компенса-
СКТ). Качественный состав жидких органических
ции заряда на гранулах. Спектры регистрировали с
продуктов определяли на приборах MSD
6973
энергией пропускания анализатора 40 эВ с шагом
(“Agilent Technologies”, США, ПИД, колонка HP-
НЕФТЕХИМИЯ том 61 № 4 2021
508
НИКОЛАЕВ и др.
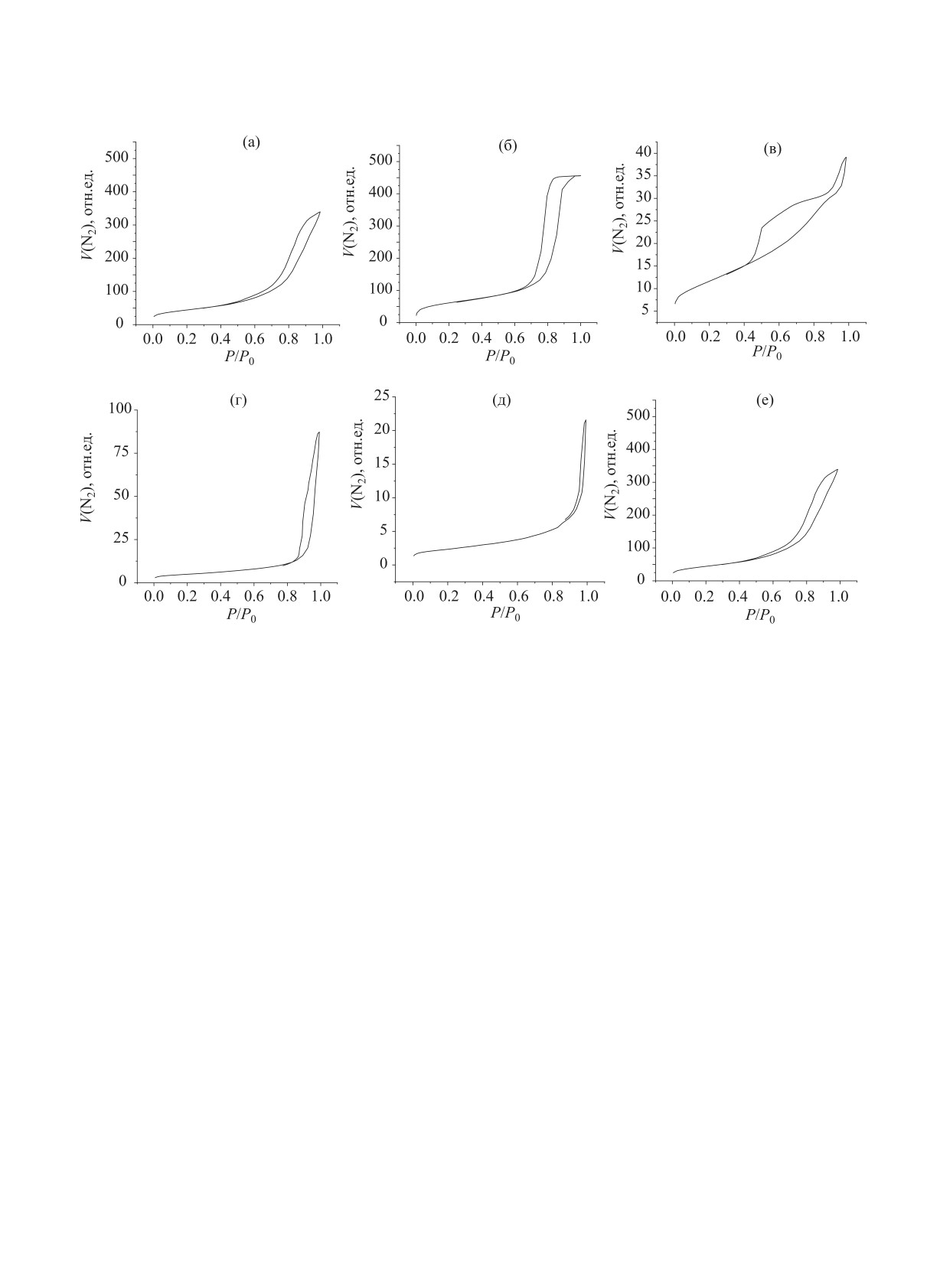
Рис. 2. Изотермы адсорбции и десорбции N2 на: (а) Al2O3; (б) SiO2; (в) C; (г) ZrO2; (д) TiO2; (е) Au/Al2O3.
5MS) и Automass-150 (“Delsi Nermag”, Франция,
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
ПИД, колонка CPSil-5) с EI = 70 эВ. Количествен-
Текстурные характеристики носителей
ное содержание жидких органических веществ
Из паспорта спецификации носителей следует,
определяли на приборе Varian
3600
(“Varian”,
что TiO2 имеет кристаллическую структуру аната-
США, ПИД, колонка Хроматэк SE-30, внутренний
за; ZrO2 представлен стабильной моноклинной мо-
стандарт - н-октан).
дификацией; SiO2 является рентгеноаморфным ма-
Конверсию этанола (α) определяли по фор-
териалом. Фазовый состав Al2O3 и С в паспортных
муле: α = (C2H5OH)кон·(C2H5OH)исх-1 × 100%, где
данных не указан, но рентгенофазовый анализ этих
(C2H5OH)кон - количество этанола в продуктах
носителей был проведен ранее в работах [13, 22].
реакции, моль; (C2H5OH)исх - количество исход-
Из данных [13, 22] следует, что С является рент-
ного этанола, моль. Селективность образования
геноаморфным материалом, а Al2O3 представлен
i-го компонента (Si) определяли по формуле: Si =
гамма модификацией (карточка JCPDS № 29-0063).
0.5I·ni·[(C2H5OH)исх - (C2H5OH)кон)]-1, где I - чис-
Изотермы адсорбции и десорбции N2 на исход-
ло атомов С в молекуле i-го компонента, шт.; ni -
ных носителях приведены на рис. 2a-д). Согласно
количество i-го компонента, моль; [(C2H5OH)исх -
IUPAC [25] полученные изотермы относятся к изо-
(C2H5OH)кон] - количество вступившего в реакцию
термам IV типа.
спирта, моль. Активность катализаторов рассчи-
Видно, что все изотермы содержат петлю капил-
тывали по формуле: A = ν(C4H9OH) × τ-1·m-1, где
лярно-конденсационного гистерезиса, что указыва-
ν(C4H9OH) - количество бутанола образованного в
ет на наличие у исследуемых материалов пористой
ходе реакции, моль; τ - время реакции, ч; m - масса
структуры. Полученные результаты согласуются с
навески катализатора, грамм.
известными данными по текстуре оксидных и угле-
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
509
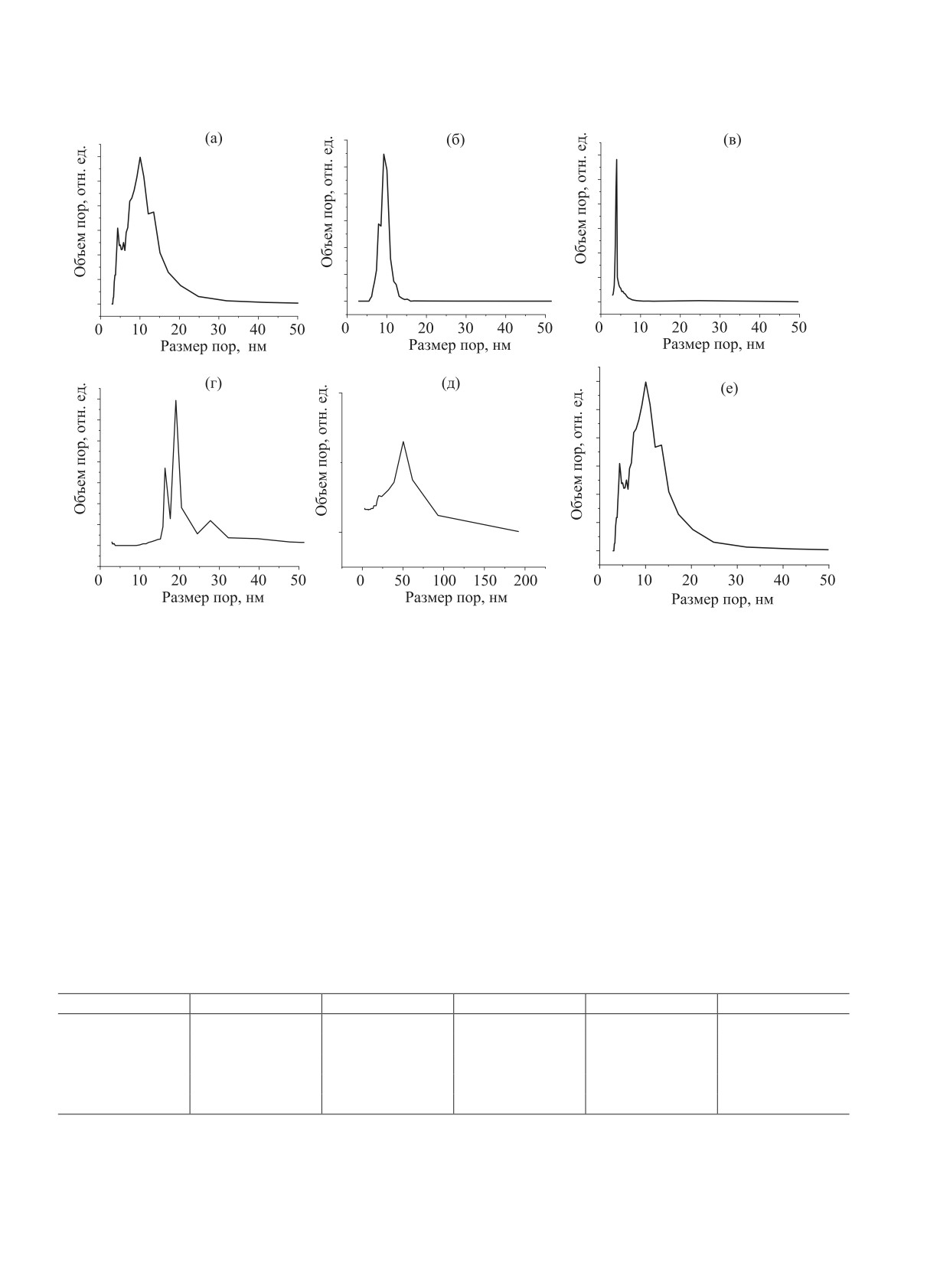
Рис. 3. Дифференциальные кривые распределения объема пор по размерам в образцах: (a) Al2O3; (б) SiO2; (в) C; (г) ZrO2;
(д) TiO2; (е) Au/Al2O3.
родных сорбентов [25]. По классификации Де Бура
Al2O3 обладает узким и бимодальным ДКР, при
[26], форма петли гистерезиса для Al2O3, SiO2 и С
этом большая часть пор имеет диаметр от 3 до
близка к типу А, который характерен для материа-
10 нм. ZrO2 характеризуется широким и полимо-
лов с цилиндрическими порами. Для TiO2 и ZrO2
дальным ДКР с максимумами в области 16, 19 и
форму петли гистерезиса можно отнести к типу В,
27 нм. TiO2 характеризуется широким ДКР с макси-
который характерен для материалов с щелевидны-
мумом в области 50 нм. Рассчитанные из экспери-
ми порами.
ментальных данных величины общего объема пор
Дифференциальные кривые распределения
(Vs) и объема микропор (Vм) носителей также при-
объема пор по размерам (ДКР) приведены на
ведены в табл. 1. Видно, что микропоры в образ-
рис. 3a-д. Видно, что SiO2 и С обладают узким и
цах практически отсутствуют, следовательно, их
мономодальным ДКР. Диаметр большей части пор
вкладом в пористую структуру можно пренебречь.
SiO2 и С равен 9 и 4 нм, соответственно (табл. 1).
Анализ диаметра пор носителей позволяет сделать
Таблица 1. Структурные параметры носителейа
Образец
D, нм
Vs, см3/г
Vм, см3/г
SBET, м2/г
[АS], мкмоль/г
Al2O3
3, 10
0.525
0.001
156
230
SiO2
9
0.706
0.018
212
20
С
4
0.061
0
41
0
ZrO2
16, 19, 27
0.135
0.001
18
65
TiO2
50
0.033
0
8
178
а D - основной размер пор, Vs - суммарный объем пор по азоту, Vм - объем микропор (t-метод), SBET - удельная поверхность по
БЭТ, [АS] - относительная кислотность по NH3
НЕФТЕХИМИЯ том 61 № 4 2021
510
НИКОЛАЕВ и др.
входящих в поверхностные структуры Mn+-O2-
[3-5, 12, 29]. Рассчитанные из профилей ТПД-NH3
значения кислотности [AS] см. в табл. 1. Видно,
что параметр [AS] составляет от 20 до 230 мкмоль/г
и увеличивается слева-направо в ряду: SiO2, ZrO2,
TiO2, Al2O3. Полученная зависимость согласуется
с известными данными по кислотности оксидных
носителей различной природы [30, 31].
Структура образцов Au/носитель
Пропитка носителей водным раствором пре-
курсора золота с последующим прокаливанием
приводит к формированию образцов Au/носитель.
В виду низкой концентрации модификатора (Au =
0.5 мас. %) пористая структура и кислотность
;
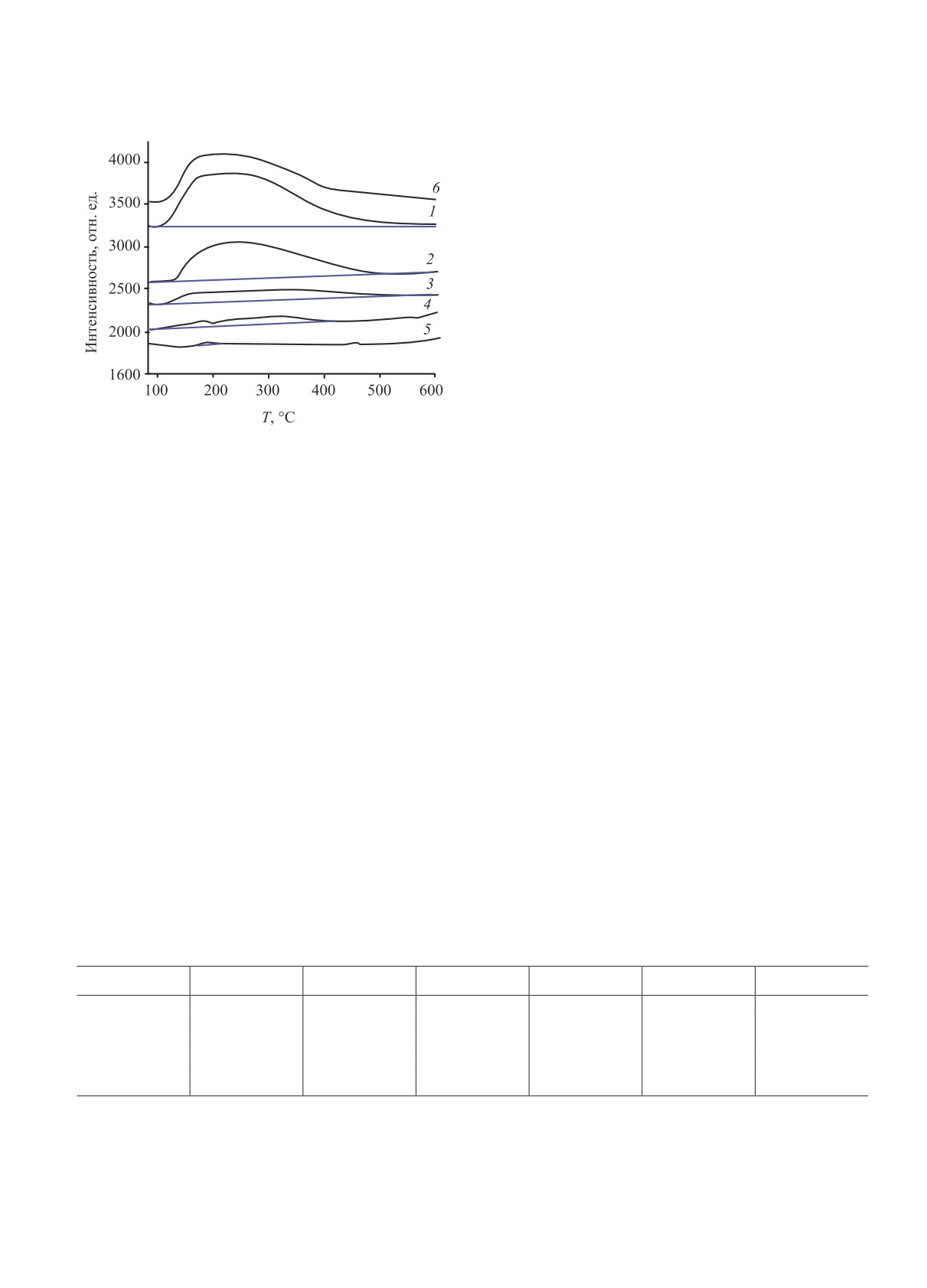
Рис. 4. Профили ТПД-NH3: (1) Al2O3; (2) TiO2; (3) ZrO2
носителя в составе образцов Au/носитель не пре-
(4) SiO2; (5) C; (6) - Au/Al2O3.
терпевают существенных изменений (табл. 1-2,
рис. 2-4). Ряды изменения величин SBET и [AS] об-
вывод о том, что носители содержат в основном
разцов Au/носитель соответствуют таковым для
мезопоры размером от 2 до 50 нм. Размер основной
носителей.
фракции пор увеличивается слева-направо в ряду:
Изображение внешней поверхности гранул об-
C, SiO2 ≈ Al2O3, ZrO2, TiO2 (см. табл. 1).
разцов Au/носитель приведено на микрофотогра-
Из рассчитанных по изотермам адсорбции
фиях СЭМ (рис. 5a-д). Видно, что поверхность
удельных поверхностей носителей SBET (табл. 1)
Au/Al2O3 не содержит визуально различимых
видно, что величина SBET составляет 8-212 м2/г и
фаз золота. Отсутствие крупных частиц золота в
увеличивается слева-направо в ряду: TiO2, ZrO2, C,
Au/Al2O3 можно объяснить высокой удельной по-
Al2O3, SiO2. Полученная зависимость согласуется с
верхностью Al2O3 в сочетании с относительно
известными данными по текстуре оксидных и угле-
малым размером пор (табл. 1). Эти особенности
родных носителей [27, 28].
текстуры препятствуют агрегации частиц на ста-
Профили ТПД-NH3 для носителей приведены
дии их формирования. Возможно и другое объяс-
на рис. 4а-д. Видно, что С не проявляет активно-
нение. Препятствовать агрегации частиц металлов,
сти в сорбции/десорбции аммиака, что обуслов-
может частичное растворение оксида алюминия в
лено отсутствием на его поверхности кислотных
ходе пропитки солянокислым раствором прекурсо-
центров. Профили оксидных носителей содержат
ра (HAuCl4*aq) с образованием аморфного Al2O3 в
широкий пик в области 100-450°C, который по-
порах после прокаливания предшественника ката-
является за счет десорбции аммиака со слабых и
лизатора. Предполагается, что в этом случае амор-
средних кислотных центров Бренстеда и Льюиса,
фный оксид алюминия сильно взаимодействует с
Таблица 2. Структурные параметры Au/носительа
Образец
[Au], мас. %
SBET, м2/г
[АS], мкмоль/г
Dср, нм
[Au*], %
Au 4f7/2, эВ
Au/Al2O3
0.5
154
238
5
25
84.0
Au/SiO2
0.5
215
18
10
3
83.9
Au/С
0.5
37
0
30
1
83.9
Au/ZrO2
0.5
16
64
100
0.6
83.9
Au/TiO2
0.5
8
183
120
0.5
83.9
а [Au] - содержание золота, SBET - удельная поверхность по БЭТ, [АS] - относительная кислотность по NH3; Dср - средний размер
частиц Au; [Au*] - относительное содержание активных частиц размером 2-4 нм; Au 4f7/2 - энергия связывания электронов
Au 4f7/2.
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
511
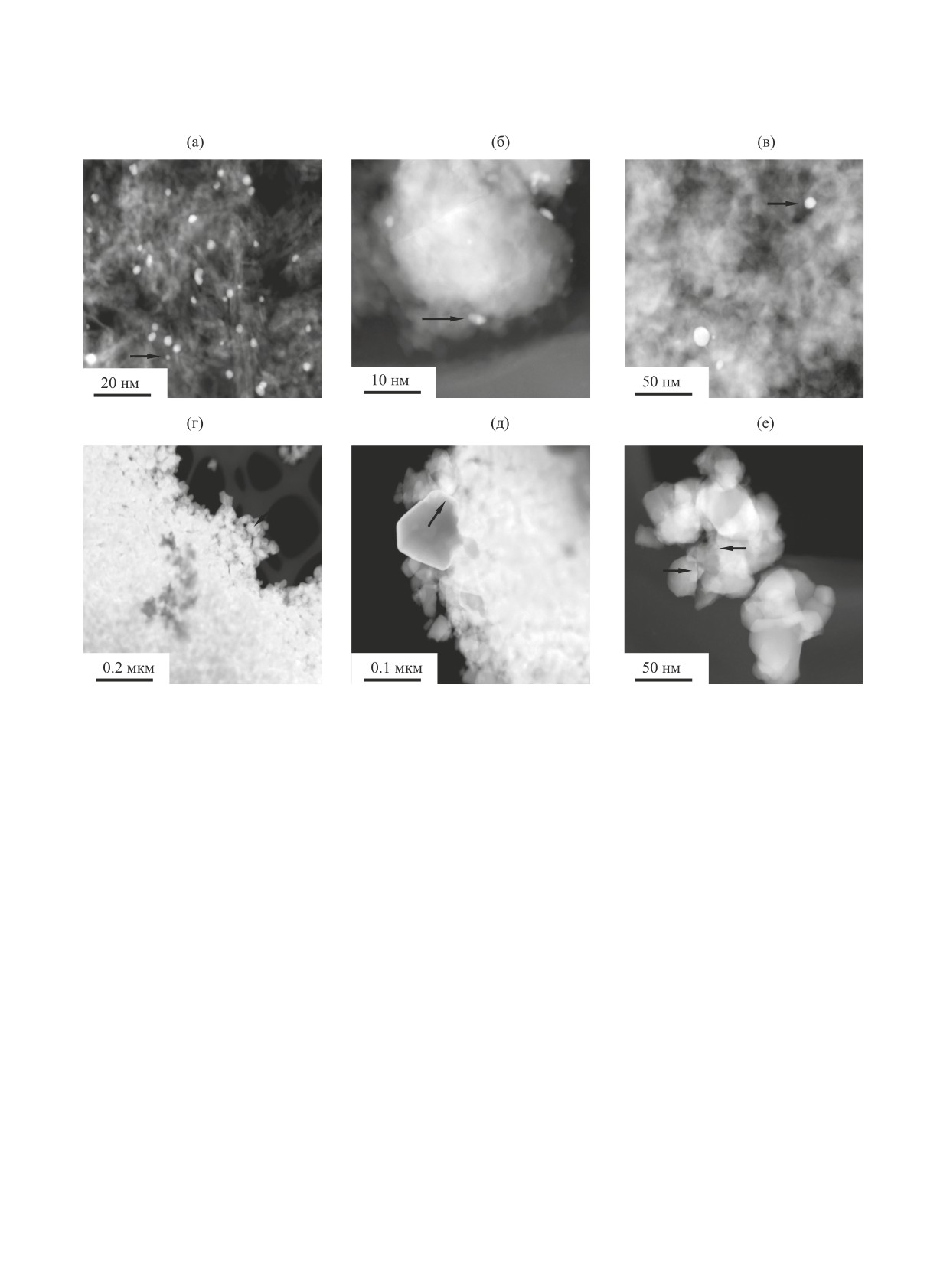
Рис. 5. Микрофотографии СЭМ образцов Au/носитель: (a) Au/Al2O3; (б) Au/SiO2; (в) Au/C; (г) Au/ZrO2; (д) Au/TiO2.
Скопления ультрадисперсных частиц золота отмечены кругами.
нанесенными частицами металлов и препятствует
работы [14] и объясняется слабым взаимодействи-
их спеканию [32].
ем частиц золота с поверхностью SiO2.
Внешняя поверхность гранул образцов Au/ZrO2,
В отличие от метода СЭМ, по методу ПЭМ ис-
Au/TiO2 и Au/C содержит ультрадисперсные ча-
следования осуществляются при при большей крат-
стицы Au. Наличие крупных частиц Au в составе
ности увеличения, что позволяет проводить анализ
катализаторов на основе ZrO2 и TiO2 можно объяс-
размера нанесенных частиц как в нано-, так и уль-
нить относительно низкой удельной поверхностью
традисперсном диапазоне. Типичные микрофото-
носителей (табл. 1), которая, в сочетании с боль-
графии ПЭМ образцов Au/носитель приведены на
шим размером пор, приводит к быстрому спеканию
рис. 6a-д. Видно, что поверхность образцов со-
малых кластеров золота, образующихся на стадии
держит темные частицы Au, контрастирующие
прокаливания предшественника Au/носитель. На-
с серым фоном носителя. Гистограммы распре-
личие крупных частиц в образце Au/C явление из-
деления частиц Au по размерам приведены на
вестное. В работе [14] сообщалось, что в отличие
рис. 7a-д. Рассчитанный из серии микрофотогра-
от оксидных носителей, углеродные сорбенты не
фий ПЭМ средний размер частиц Au приведен в
содержат катионов переходных металлов, которые
табл. 2. Видно, что средний размер изменяется в
могли бы взаимодействовать с нанесенной фазой
ряду: Au/TiO2 (120 нм) > Au/ZrO2 (100 нм) > Au/C
прекурсора, и таким образом препятствовать спе-
(30 нм) > Au/SiO2 (10 нм) Au/Al2O3 (5 нм). В целом
канию малых кластеров Au на стадии прокалива-
данные ПЭМ настоящего исследования хорошо со-
гласуются с обсужденными ранее данными СЭМ.
ния предшественника катализатора. Интересно
отметить, что Au/SiO2, полученный пропиткой си-
Известно, что высокая активность Au-содержа-
ликагеля с относительно высокой поверхностью,
щих композитов во многом определяется наличием
содержит крупные частицы золота, в то время как
в составе образцов атомов золота с низким коор-
пропитка его ближайшего аналога (Al2O3) не при-
динационным числом - Au0(KH) [14, 33]. Наибо-
водит к формированию крупных частиц золота
лее активные атомы Au0(KH) расположены на уг-
(рис. 5a, б). Этот результат согласуется с данными
лах и ребрах частиц золота и доля таких атомов на
НЕФТЕХИМИЯ том 61 № 4 2021
512
НИКОЛАЕВ и др.
Рис. 6. Микрофотографии ПЭМ образцов Au/носитель: (a) Au/Al2O3; (б) Au/SiO2; (в) Au/C; (г) Au/ZrO2; (д) и (е) Au/TiO2.
Индивидуальные частицы Au отмечены стрелками.
поверхности частиц зависит от размера. Так, при
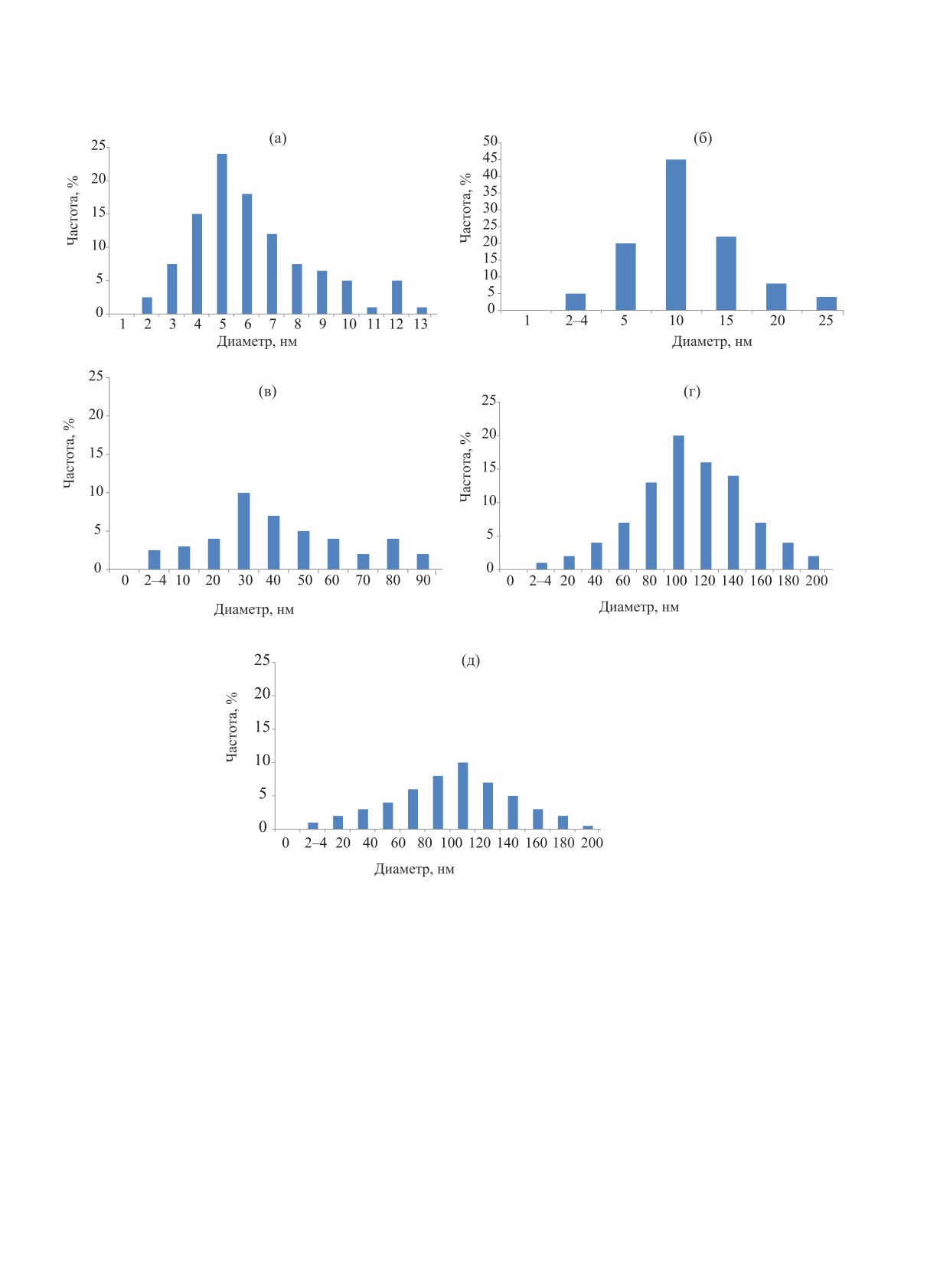
ния частиц по размерам (рис. 7). Значения [Au*]
15 нм относительное содержание Au0(KH) в части-
для систем Au/носитель приведены в табл.
2.
цах золота составляет 4%, а при размерах 2-4 нм
Видно, что параметр [Au*] изменяется антибатно
содержание Au0(KH) возрастает до 40-60% [33].
среднему размеру частиц в ряду: Au/TiO2 (0.5%) <
Из приведенных выше данных следует, что основ-
Au/ZrO2 (0.6%) < Au/C (1%) < Au/SiO2 (3%) <
ной вклад в активность нанесенных фаз Au должна
Au/Al2O3 (25%).
вносить фракция частиц Au размером 2-4 нм. Сто-
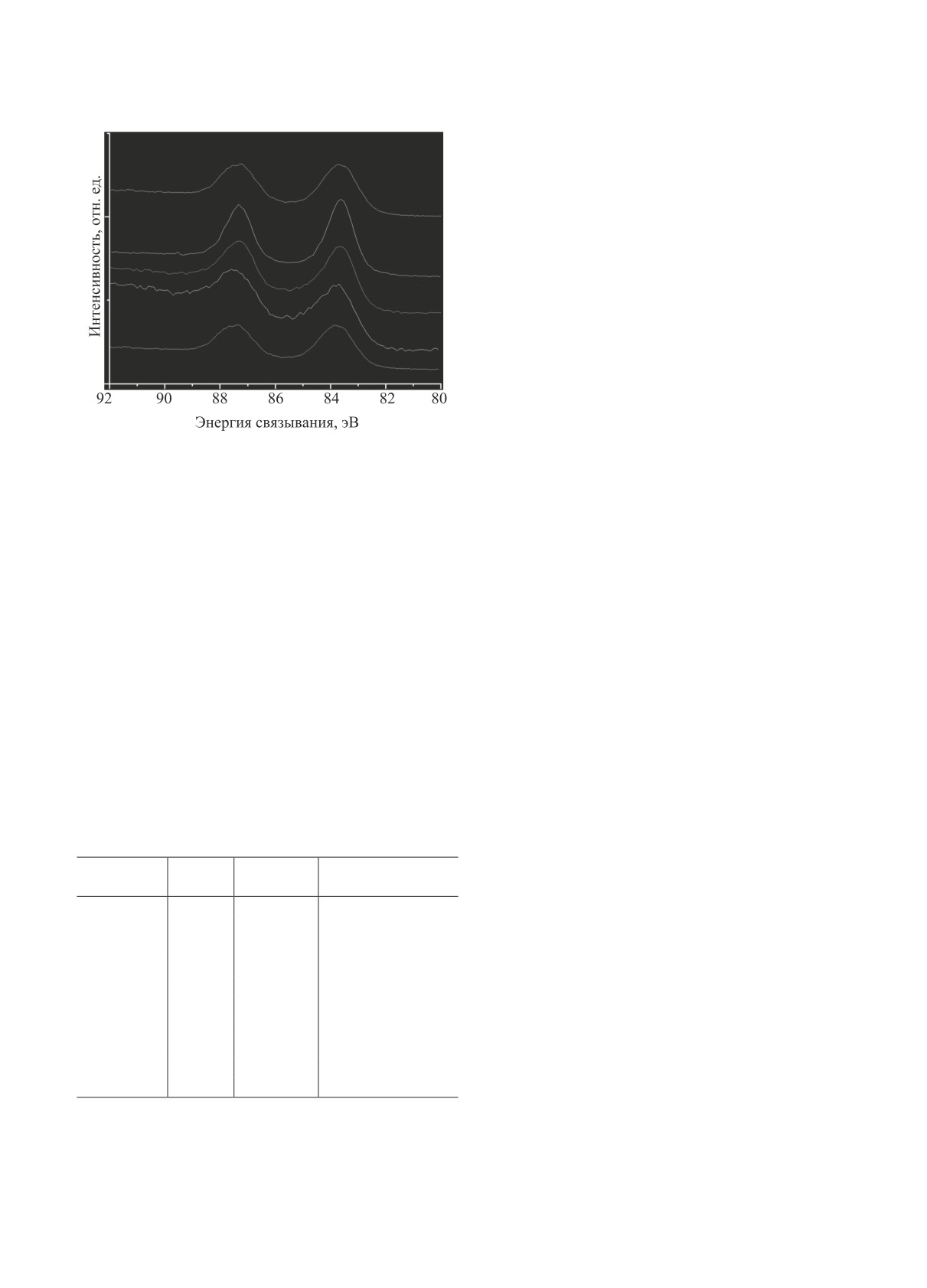
Химическое состояние нанесенных частиц зо-
ит отметить, что частицы размером 2-4 нм содер-
лота было изучено методом РФЭС. Спектры Au 4f
жат не только высокую плотность атомов с низким
золото-содержащих образцов приведены на рис. 8.
координационным числом. Их электронная струк-
Спектры содержат дублет пиков Au 4f7/2 и Au 4f5/2
тура занимает промежуточное положение между
с энергиями связывания электронов равными
структурой массивного металла и дискретными
83.9±0.1 и 88.7±0.1 эВ (табл. 2). Полученные зна-
уровнями отдельных атомов. Такая специфика
чения энергий связывания фотоэлектронов Au 4f
электронного строения приводится в качестве объ-
являются типичными для металлического золота
яснений высокой активности 2-4 нм частиц в раз-
[14, 17-20]. Таким образом, в отличие от разме-
личных размерно-чувствительных реакциях [14].
ра фаз Au, основное химическое состояние Au в
Содержание фракции частиц размером 2-4 нм
катализаторах не зависит от типа используемого
[Au*] было рассчитано из гистограмм распределе- носителя и определяется свойствами прекурсора
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
513
Рис. 7. Гистограммы распределения частиц золота по размерам в образцах Au/носитель: (a) Au/Al2O3; (б) Au/SiO2; (в) Au/C;
(г) Au/ZrO2; (д) Au/TiO2.
(HAuCl4), который при прокаливании предшествен-
Au/SiO2 было необходимо изучить специфику
ника катализатора до 350°C полностью разлагается
превращения этанола в присутствии чистых но-
по формуле: 2HAuCl4 → 2Au0 + 2HCl ↑+ 3Cl2 ↑.
сителей. В работах [13, 21] было установлено, что
Эффективность носителей в конверсии этанола
оптимальная температура для конверсии этанола в
в бутанол
бутанол в присутствии различных оксидных и на-
Известно, что оксиды могут проявлять актив-
несенных катализаторов равна 275°C, поэтому ана-
ность в конверсии этанола [3-10], поэтому пе-
лиз активности носителей проводили при той же
ред тестированием Au/Al2O3, Au/TiO2, Au/ZrO2 и
температуре.
НЕФТЕХИМИЯ том 61 № 4 2021
514
НИКОЛАЕВ и др.
таналь и 2-бутен-1-ол не обнаружено. Полученный
результат согласуется с данными работ Gabriëls и
др [3], Yang и др [6], Ndou и др [7]. Авторы работ со-
общали, что для высокой активности оксидных ка-
тализаторов необходимы температуры 420-450°C.
При этом катализ конверсии этанола в бутанол на
оксидах протекает по механизму (I).
Активность носителей в целевом процессе (A)
также приведена в табл. 3. Видно, что значение A
составляет (0.001-0.046)×10-4 моль·ч-1·г-1 и из-
меняется в ряду: Al2O3 >> TiO2 > ZrO2 > SiO2.
Углеродный носитель в превращениях этанола
неактивен. Наблюдаемый ряд активностей хоро-
шо согласуется с тем, что вероятный механизм
конверсии этанола на чистых носителях - это ме-
Рис. 8. РФЭ-спектры Au 4f в образцах Au/носитель:
ханизм (I). Поясним. При прочих равных условиях
(1) Au/Al2O3; (2) Au/SiO2; (3) Au/C; (4) Au/ZrO2; (5) Au/TiO2.
(температура, количество этанола, масса навески
катализатора), скорость образования бутанола по
механизму (I) должна быть пропорциональна чис-
В стандартном тесте (275°C, 5 ч) конверсия эта-
лу центров Mn+-O2- (рис. 1). Так как концентрация
нола на оксидах Al, Ti, Zr и Si составляет 0.1-5.3%,
центров Mn+ пропорциональна параметру кислот-
а селективность по бутанолу равна 0.04-0.5%
ности [AS], то при реализации механизма (I) долж-
(табл. 3). Основным продуктом реакции является
на наблюдаться прямая зависимость активности
этоксиэтан. Это вещество образуется в ходе деги-
носителя от параметра [AS]. Такая зависимость
дратации спирта на кислых центрах оксидов [34].
наблюдается: чем больше [AS], тем выше скорость
Селективность по этоксиэтану составляет 94-98%.
образования бутанола (табл. 1 и 3); а если параметр
Помимо этоксиэтана и бутанола в смеси присут-
[AS] равен 0 (углеродный носитель), то бутанол не
ствуют следовые количества бутанола, этоксибута-
образуется.
на и этилена, а так же углеводородов C4+ различ-
Особенности катализа конверсии этанола в бу-
ного строения. Интермедиатов конверсии этанола
танол на носителях можно обобщить следующим
в бутанол по механизму (II) таких, как этаналь, бу-
образом: (1) вероятный механизм конверсии эта-
нола в бутанол - это механизм (I); (2) углеродный
Таблица 3. Каталитическая эффективность образцов в
носитель неактивен в конверсии этанола; (3) ак-
стандартом тесте (275°С, 5 ч)a
тивность образования бутанола на оксидах равна
(0.001-0.046)×10-4 моль·ч-1·г-1 и изменяется в ряду
АBuOH×104,
Образец
αEtOH, %
SBuOH, %
моль·ч-1·г-1
Al2O3 >> TiO2 > ZrO2 > SiO2.
Al2O3
5.3
0.5
0.046
Эффективность образцов Au/носитель
TiO2
2
0.2
0.007
в конверсии этанола в бутанол
ZrO2
0.6
0.1
0.001
В стандартном тесте (275°C, 5 ч) конверсия
SiO2
0.1
0.04
< 0.001
этанола на образцах Au/Al2O3, Au/TiO2, Au/ZrO2
С
0
0
0
и Au/SiO2 составляет 16-18%; селективность по
Au/Al2O3
18
78
24.36
бутанолу зависит от природы оксида-носителя и
Au/TiO2
16
2
0.56
варьируется от 0.4 до 78% (табл. 3). Активность
Au/ZrO2
14
1.5
0.36
образцов Au/оксид в целевом процессе составляет
Au/SiO2
16
0.4
0.11
(0.11-24.3)×10-4 моль·ч-1·г-1 (табл. 3). Сравнение
Au/C
0.7б
0
0
a αEtOH - конверсия этанола, SBuOH - селективность по бутанолу,
активностей чистых и модифицированных золо-
ABuOH -активность образца в синтезе бутанола.
том носителей позволяет сделать вывод о том, что
б Основной продукт - этаналь.
добавки высокодисперсных фаз золота приводят
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
515
к резкому росту скорости целевого процесса. По-
Таблица 4. Состав органической смеси в стандартном
тесте (275°С, 5 ч, Au/Al2O3)
лученный результат согласуется с данными [18].
Quesada и др [18] сообщили, что TiO2 (P-25) про-
Вещество
%
являет низкую активность в конверсии этанола в
Этаналь
0.02
бутанол, а Au/TiO2 (P-25) проявляет более высокую
Бутаналь
0.10
активность.
2-Бутен-1-ол
0.02
Этанол
81.49
Ранее в настоящей работе была отмечена кор-
Бутанол
11.58
реляция между активностью оксидного носителя в
Этоксиэтан
1.33
конверсии этанола по механизму (I) и его кислот-
Этен
0.10
ностью. Поэтому рост скорости образования бута-
Бутан
0.46
нола после модификации носителя золотом мож-
1-Бутен
0.40
но было бы связать с ростом кислотности систем
Бутан-2-он
0.16
Au/оксид. Однако это не так. Из табл. 1 и 2 видно,
Этилацетат
0.22
что модификация носителя золотом не приводит к
Гексан-1-ол
1.50
увеличению параметра [AS], а значит, скорость об-
Гексан
0.30
разования бутанола по механизму (I) должна быть
Этоксибутан
0.12
одинаковой как для носителя, так и для Au-катали-
2-Этилбутан-1-ол
0.45
затора, сформированного на его основе. В чем же
Этоксигексан
0.50
тогда причина роста активности систем Au/оксид
2-Этилгексан-1-ол
0.07
по сравнению с оксидными носителями?
Прочие углеводороды
1.18
В работах [11-13] сообщалось, что на образцах
M0/M1Ox становится возможной конверсия этано-
Из рис. 1 видно, что механизм (II) состоит из
ла по механизму (II), отличительной особенностью
цепочки реакций: дегидрирование; конденсация;
которого является высокая скорость образования
дегидратация и гидрирование. Для катализа ста-
бутанола при 200-300°С. Так при 270°С, конверсия
дий конденсации этаналя и дегидратации альдоля
этанола на 0.1%Pd/Al2O3 составляет 24%; селек-
необходимы кислотно-основные центры Mn+-O2-
тивность по бутанолу равна 70% [13]. При 250°С,
[3, 4]. В системе Au/C такие центры отсутствуют,
конверсия этанола на
20%Ni/Al2O3 составляет
поэтому на качественном уровне можно сделать
25%; селективность по бутанолу равна 70% [11].
предположение, что целевой процесс на образце
Следовательно, наблюдаемый рост активности об-
Au/C идти не должен. Действительно, Au/C уско-
разования бутанола после модификации носите-
ряет только первую стадию дегидрирования этано-
ля золотом можно было бы связать с изменением
ла в этаналь (табл. 3). В отличие от Au/C, системы
механизма реакции. Для проверки этой гипотезы
Au/оксид обладают центрами Mn+-O2- и, следова-
был проведен хромато-масс-спектрометрический
тельно, могут принимать участие как в катализе
(ГХ-МС) анализ. Состав жидких органических
конденсации этаналя, так и дегидратации альдоля.
продуктов, полученных в ходе стандартного теста
Следовательно, на этих стадиях нет препятствий
на Au/Al2O3, приведен в табл. 4. Видно, что в про-
для конверсии этанола в бутанол. Поэтому наблю-
дуктах присутствуют интермедиаты механизма (II)
даемая активность Au/оксид больше нуля (табл. 3).
такие как этаналь, бутаналь и 2-бутен-1-ол. Таким
Если рассматривать скорости конденсации эта-
образом, есть основания полагать, что механизм
наля и дегидратации альдоля, то они определяются
реакции при катализе на системах Au/оксид - это
концентрацией центров Mn+-O2-. Так как концен-
механизм (II) и именно смена механизма приводит
трация центров Mn+ пропорциональна [AS], то
к росту скорости конверсии этанола в бутанол.
должна наблюдаться корреляция активность об-
Активность образцов Au/носитель в целевом
разца Au/оксид - величина [AS]. И действитель-
процессе изменяется в ряду: Au/Al2O3 >> Au/TiO2
но такая корреляция наблюдается. Для наиболее
> Au/ZrO2 > Au/SiO2 >> Au/C (табл. 3). Получен-
кислого образца (Au/Al2O3) активность образо-
ные результаты можно объяснить особенностями
вания бутанола высокая, для образцов со средней
структуры катализаторов и механизмом (II).
кислотностью (Au/TiO2 и Au/ZrO2) активность уже
НЕФТЕХИМИЯ том 61 № 4 2021
516
НИКОЛАЕВ и др.
меньше, а для наименее кислого образца (Au/SiO2)
ского золота. Средний размер частиц золота опре-
активность самая маленькая (табл. 2 и 3).
деляется текстурой носителя и изменяется в ряду:
Au/TiO2 (120 нм) > Au/ZrO2 (100 нм) > Au/C (30 нм) >
Из рис. 1 видно, что помимо стадий конденсации
Au/SiO2 (10 нм) Au/Al2O3 (5 нм). Содержание в Au-
и дегидратации в механизме (II) присутствуют ста-
катализаторах высокоактивных частиц золота раз-
дии дегидрирования и гидрирования. Для высокой
мером 2-4 нм определяется типом используемого
скорости дегидрирования и гидрирования необхо-
носителя и изменяется в ряду: Au/TiO2 < Au/ZrO2 <
димы активные центры металлов, например атомы
Au/C < Au/SiO2 << Au/Al2O3. Кислотность Au-ка-
на поверхности Pd, Ni, Pt [3, 4]. В случае золотых
тализаторов определяется природой носителя и из-
катализаторов центрами гидрирования-дегидриро-
меняется в ряду: Al2O3 > TiO2 > ZrO2 > SiO2 >> Au/C.
вания являются координационно-ненасыщенные
атомы Au0(KH), расположенные на поверхности
Углеродный носитель не проявляет активности
фаз золота [14]. Так, Fang и др. сообщали, что ато-
в конверсии этанола, что объясняется отсутстви-
мы Au0(KH) активны в дегидрировании фенилме-
ем в образце необходимых для катализа активных
танола при 100°C [16, 35], и этанола при 200°C
центров. Оксидные носители содержат центры
[36]. Следовательно, есть основания полагать, что
Mn+-O2-, что делает возможным протекание целе-
атомы Au0(KH) в образцах настоящей работы могут
вой реакции по механизму “бимолекулярной кон-
обеспечить высокую скорость дегидрирования эта-
денсации”. При 275°C, активность оксидных носи-
нола при температуре стандартного теста (275°C).
телей низкая: (0.001-0.046)×10-4 моль·ч-1·г-1.
Так же в работах [15, 33, 37] показано, что атомы
Модификация оксидных носителей золотом
Au0(KH) проявляют высокую активность в гидри-
приводит к формированию новых центров
ровании α,β-ненасыщенных альдегидов, алкенов и
Au0(KH)-Mn+-O2-. В результате становится воз-
алкинов при 100-300°C. Следовательно, есть осно-
можным протекание целевой реакции по механиз-
вания полагать, что атомы Au0(KH), в свою очередь
му “альдольной конденсации” и с более высокой,
в образцах настоящей работы могут обеспечить
чем на чистых носителях скоростью. При 275°C,
высокую скорость гидрирования интермедиатов
активность систем Au/оксид составляет
(0.11-
целевого процесса при температуре стандартного
24.3)×10-4 моль·ч-1·г-1. Модификация углеродного
теста (275°C).
носителя золотом не приводит к формированию
центров Au0(KH)-Mn+-O2-. В результате образец
Скорости дегидрирования этанола и гидриро-
Au/C не проявляет активности в целевом процессе.
вания интермедиатов целевого процесса определя-
ются концентрацией центров Au0(KH) в катализа-
Среди Au-катализаторов, наибольшую эф-
торе. Так как концентрация Au0(KH) определяется
фективность в конверсии этанола в бутанол про-
содержанием в катализаторе фракции частиц раз-
демонстрировал образец Au/Al2O3 (активность
мером 2-4 нм [Au*], то должна наблюдаться кор-
образования бутанола 24.3×10-4 моль·ч-1·г-1; селек-
реляция активность образца - величина параметра
тивность по бутанолу 78%). Высокая эффективность
[Au*]. Такая корреляция наблюдается (табл. 2 и 3).
Au/Al2O3 обусловлена наличием высокой плотно-
Для наиболее активного образца Au/Al2O3 пара-
сти необходимых для катализа бифункциональных
метр [Au*] равен 25% и активность Au/Al2O3 в об-
центров Au0(KH)--Аln+-O2-, сформированных на
разовании бутанола равна 24.36×10-4 моль·ч-1·г-1.
границе раздела фаз Au размером 2-4 нм и оксид-
Для остальных образцов Au/оксид параметр
ного носителя. На основании проведенной работы,
[Au*] составляет 0.5-3% и поэтому их активность
можно сделать вывод о том, что оптимальный но-
в образовании бутанола на порядок ниже
ситель для формирования золотых катализаторов
(0.56×10-4 моль·ч-1·г-1).
конверсии этанола в бутанол - оксид алюминия.
ЗАКЛЮЧЕНИЕ
ИНФОРМАЦИЯ О ВКЛАДЕ АВТОРОВ
Использование промышленных носителей
Николаев С.А. - концептуализация, проведение
(Al2O3, SiO2, TiO2, ZrO и C) позволяет получать
исследования методом ПЭМ, РФЭС, обсуждение
Au-катализаторы с фазой дисперсного металличе-
результатов, подготовка текста статьи.
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
517
Чистяков А.В. - проведение каталитических те-
“Нанохимия и наноматериалы” К.И. Маслакову,
стов, анализ состава продуктов методом ГЖХ, ГХ-
С.В. Максимову и С.В. Дворяку за помощь в
МС, обсуждение результатов.
изучении образцов с использованием оборудова-
Чистякова П.А.
- проведение исследования
ния, приобретенного в соответствии с Программой
развития МГУ.
ТПД-NH3, обсуждение результатов.
Цодиков М.В. - обсуждение результатов, подго-
товка текста статьи.
КОНФЛИКТ ИНТЕРЕСОВ
Либерман Е.Ю. - обсуждение результатов, под-
Авторы заявляют об отсутствии конфликта
готовка текста статьи.
интересов, требующего раскрытия в данной статье.
Конькова Т.В. - проведение исследования мето-
дом низкотемпературной сорбции N2.
СПИСОК ЛИТЕРАТУРЫ
Эзжеленко Д.И. - проведение исследования ме-
1.
Abdulrazzaq H.T., Schwartz T.J. Catalytic conversion of
тодом СЭМ, ЭДА, обсуждение результатов, подго-
ethanol to commodity and specialty chemicals // Etha-
товка иллюстративного материала статьи.
org/10.1016/B978-0-12-811458-2.00001-8
2.
Wu X., Fang G., Tong Y., Jiang D., Liang Z., Leng W.,
ИНФОРМАЦИЯ ОБ АВТОРАХ
Liu L., Tu P., Wang H., Ni J., Liet X. Catalytic upgrading of
Николаев Сергей Александрович, ORCID: http://
ethanol to n-butanol: Progress in catalyst development //
orcid.org/0000-0002-9091-3537
org/10.1002/cssc.201701590
Чистяков Андрей Валерьевич, ORCID: http://
3.
Gabriëls D., Hernández W.Y., Sels B., Voort P.V.D.,
orcid.org/0000-0002-4443-7998
Verberckmoes A. Review of catalytic systems and
Чистякова Полина Александровна, ORCID:
thermodynamics for the Guerbet condensation reaction
and challenges for biomass valorization // Catalysis
Science & Technology. 2015. V. 5. P. 3876-3902. https://
Цодиков Марк Вениаминович, ORCID: http://
doi.org/10.1039/C5CY00359H
orcid.org/0000-0002-8253-2945
4.
Kozlowski J.T., Davis R.J. Heterogeneous Catalysts for
the Guerbet Coupling of Alcohols // ACS Catalysis.
org/0000-0003-2218-8254
Конькова Татьяна Владимировна, ORCID: http://
cs400292f
orcid.org/0000-0002-7151-6317
5.
Dai J., Zhang H. Recent advances in selective C-C bond
coupling for ethanol upgrading over balanced Lewis
acid-base catalysts // Science China Materials. 2019.
ФИНАНСИРОВАНИЕ
Работа по развитию технологии превращения
019-9454-x
этанола в бутанол выполнена в рамках Государ-
6.
Yang C., Meng Z.Y. Bimolecular condensation of eth-
ственного задания ИНХС РАН и частично при под-
anol to 1-butanol catalyzed by alkali cation zeolites //
держке Российского Фонда Фундаментальных ис-
org/10.1006/jcat.1993.1187
следований (грант № 21-53-12006).
7.
Ndou A.S., Plint N., Coville N.J. Dimerization of ethanol
Структурные исследования и корреляции
to butanol over solid-base catalysts // Applied Catalysis
(структура-активность нанесенных образцов) вы-
полнены при финансовой поддержке Российского
org/10.1016/S0926-860X(03)00363-6
Фонда Фундаментальных исследований в рамках
8.
Carvalho D.L., de Avillez R.R., Rodrigues M.T.,
научного проекта № 20-33-90011.
Borges L.E.P., Appel L.G. Mg and Al mixed oxides
and the synthesis of n-butanol from ethanol // Applied
Catalysis A: General. 2012. V. 415-416. P. 96-100.
БЛАГОДАРНОСТИ
Авторы выражают благодарность сотрудни-
9.
Cosimo J.I. Di, Dıez V.K., Xu M., Iglesia E., Apeste-
кам Центра Коллективного Пользования МГУ
guıa C.R. Structure and Surface and Catalytic Properties
НЕФТЕХИМИЯ том 61 № 4 2021
518
НИКОЛАЕВ и др.
of Mg-Al Basic Oxides // J. of Catalysis. 1998. V. 178.
apcata.2017.12.004
10.
Sun Z., Vasconcelos A.C., Bottari G., Stuart M.C.A.,
19.
Chistyakov A.V., Zharova P.A., Nikolaev S.A., Tsodi-
Bonura G., Cannilla C., Frusteri F., Barta K. Efficient
kov M.V. Direct Au-Ni/Al2O3 catalysed cross-
catalytic conversion of ethanol to 1-butanol via the
condensation of ethanol with isopropanol into pentanol-2 //
Guerbet reaction over copper- and nickel-doped porous //
ACS Sustainable Chemistry & Engineering. 2017.
org/10.1016/j.cattod.2016.06.016
20.
Nikolaev S.A., Tsodikov M.V., Chistyakov A.V., Zharo-
acssuschemeng.6b02494
va P.A., Ezzgelenko D.I. The activity of mono- and
11.
Riittonen T., Toukoniitty E., Madnani D.K., Leino A.-R.,
bimetallic gold catalysts in the conversion of sub-
Kordas K., Szabo M., Sapi A., Arve K., Wärnå J., Mik-
and supercritical ethanol to butanol // J. of Catalysis.
kola J.-P. One-pot liquid-phase catalytic conversion of
ethanol to 1-butanol over aluminium oxide — the effect
jcat.2018.11.017
of the active metal on the selectivity // Catalysts. 2012.
21.
Chistyakov A.V., Nikolaev S.A., Zharova P.A., Tsodi-
kov M.V., Manenti F. Linear α-alcohols production from
12.
Marcu I.-C., Tanchoux N., Fajula F., Tichit D. Catalytic
supercritical ethanol over Cu/Al2O3 catalyst // Energy.
conversion of ethanol into butanol over M-Mg-Al
mixed oxide catalysts (M = Pd, Ag, Mn, Fe, Cu, Sm,
energy.2018.10.071
Yb) obtained from LDH precursors // Catalysis Letters.
22.
Taran O.P., Descorme C., Polyanskaya E.M., Ayushe-
ev A.B., Besson M., Parmon V.N. Sibunit-based catalytic
012-0935-9
materials for the deep oxidation of organic ecotoxicants
13.
Nikolaev S.A., Tsodikov M.V., Chistyakov A.V.,
in aqueous solutions. III: Wet air oxidation of phenol
Chistyakova P.A., Ezzhelenko D.I., Shilina M.I.
over oxidized carbon and Rr/C catalysts // Catalysis in
PdCu nanoalloy supported on alumina: A stable and
selective catalyst for the conversion of bioethanol to
S2070050413020104
23.
Nikolaev S.A., Golubina E.V., Krotova I.N., Shilina M.I.,
org/10.1016/j.cattod.2020.06.061
Chistyakov A.V., Kriventsov V.V. The effect of metal
14.
Takei T., Akita T., Nakamura I., Fujitani T., Okumura M.,
deposition order on the synergistic activity of Au-Cu
Okazaki K., Huang J., Ishida T., Haruta M.
and Au-Ce metal oxide catalysts for CO oxidation //
Heterogeneous Catalysis by Gold // Advances in
Applied Catalysis B: Environmental. 2015. V. 168-169.
B978-0-12-385516-9.00001-6
24.
Наумкин А.В., Васильков А.Ю., Волков И.О., Смир-
15.
McEwan L, Julius M, Roberts S, Fletcher J.C.Q.
нов В.В., Николаев С.А. Фотоэлектронные спектры и
A review of the use of gold catalysts in selective
строение композитов, полученных иммобилизацией
hydrogenation reactions // Gold Bulletin. 2010. V. 43.
наночастиц Au, Ni и Au+Ni из коллоидных растворов
в триэтиламине на SiO2 // Неорганические материа-
16.
Fang W., Chen J., Zhang Q., Deng W., Wang Y.
лы. 2007. Т. 43. № 4. С. 445 49 [Naumkin A.V., Vasil’-
Hydrotalcite-supported gold catalyst for the oxidant-
kov A.Yu., Volkov I.O., Smirnov V.V., Nikolaev S.A. X-ray
free dehydrogenation of benzyl alcohol: studies on
photoelectron spectra and structure of composites pre-
support and gold size effects // Chemistry A European
pared via deposition of Au, Ni, and Au+Ni nanoparticles
on SiO2 from colloidal solutions in trimethylamine //
chem.201002469
Inorganic Materials. 2007. V. 43. № 4. P. 381-385].
17.
Morales M.V., Asedegbega-Nieto E., Castillejos-López
E., Bachiller-Baezab B., Guerrero-Ruiz A. Difference
25.
Sing K.S.W., Everett D.H., Haul R.A.W., Moscow L.,
in the deactivation of Au catalysts during ethanol
Pierotti R.A., Rouquérol J., Siemieniewska T. Reporting
transformation when supported on ZnO and on TiO2 //
physisorption data for gas/solid systems with special
reference to the determination of surface area and
org/10.1039/C8RA00314A
porosity // Pure and Applied Chemistry. 1985. V. 57.
18.
Quesada J., Arreola-Sánchez R., Faba L., Díaz E.,
Rentería-Tapia V. M., Ordóñez S. Effect of Au
26.
De Boer J.H. The structure and properties of porous
nanoparticles on the activity of TiO2 for ethanol upgra-
materials / Eds. Everett D.H., Stone F.S. London: But-
ding reactions // Applied Catalysis B: Environmental.
terworths, 1958. 68 р.
НЕФТЕХИМИЯ том 61 № 4 2021
ВЛИЯНИЕ НОСИТЕЛЯ НА ФОРМИРОВАНИЕ И АКТИВНОСТЬ
519
27. Kanda Y., Nakata K., Temma C., Sugioka M., Uemichi Y.
32. Слептерев А.А., Иост К.Н., Темерев В.Л., Талзи В.П.,
Effects of support on formation of active sites and
Леонтьева Н.Н., Цырульников П.Г. Модифицирова-
hydrodesulfurization activity of rhodium phosphide
ние дефектной структуры алюмооксидного носителя
catalyst // J. of the Japan Petroleum Institute. 2012.
кислотной обработкой // Омский научный вестник.
2013. Т. 117. C. 55-58.
28. Liu D., Zhu H. Zhao J., Pan L., Dai P., Gu X., Li L.,
33. Nikolaev S.A., Smirnov V.V. Synergistic and size
effects in selective hydrogenation of alkynes on gold
Liu Y., Zhao X. Synthesis of mesoporous g-Al2O3 with
nanocomposites // Catalysis Today. 2009. V. 147S.
spongy structure: in-situ conversion of metal-organic
frameworks and improved performance as catalyst
34. Lee J., Szanyi J., Kwak J.H. Ethanol dehydration on
support in hydrodesulfurization // Materials. 2018.
γ-Al2O3: Effects of partial pressure and temperature //
29. Hu Z., Li W.-Z., Sun K.-Q., Xu B.-Q. Effects of support
org/10.1016/j.mcat.2016.12.013
pre-calcination on the NOx storage and reduction
35. Fang W., Zhang Q., Chen J., Deng W., Wang Y. Gold
performance of Pt-BaO/Al2O3 catalysts // Catalysis
nanoparticles on hydrotalcites as efficient catalysts for
Science and Technology. 2013. V. 3. P. 2062-2071.
oxidant-free dehydrogenation of alcohols // Chemical
30. Shamanaev I.V., Deliy I.V., Gerasimov E.Yu.,
org/10.1039/B923047E
Pakharukova V.P., Kodenev E.G., Aleksandrov P.V.,
36. Wang C., Garbarino G., Allard L.F., Wilson F., Busca G.,
Bukhtiyarova G.A. Synergetic effect of Ni2P/SiO2 and
Flytzani-Stephanopoulos M. Low-temperature dehy-
γ-Al2O3 physical mixture in hydrodeoxygenation of
drogenation of ethanol on atomically dispersed gold
methyl palmitate // Catalysts. 2017. V. 7. Article № 329.
supported on ZnZrOx // ACS Catalysis. 2016. V. 6.
31. Qin B., Shen Y., Xu B., Zhu S., Li P., Liu Y. Mesoporous
37. Bus E., Prins R., van Bokhoven J.A. Origin of the cluster-
TiO2-SiO2 adsorbent for ultra-deep desulfurization of or-
size effect in the hydrogenation of cinnamaldehyde over
ganic-S at room temperature and atmospheric pressure //
supported Au catalysts // Catalysis Communications.
org/10.1039/C8RA00112J
catcom.2006.11.040
НЕФТЕХИМИЯ том 61 № 4 2021