ЖУРНАЛ ОБЩЕЙ ХИМИИ, 2023, том 93, № 5, с. 684-694
УДК 542.06:547.779.1
РАЗРАБОТКА ВОСПРОИЗВОДИМОГО И
МАСШТАБИРУЕМОГО МЕТОДА СИНТЕЗА
БИОЛОГИЧЕСКИ АКТИВНЫХ ПРОИЗВОДНЫХ
ПИРАЗОЛО[1,5-a]ПИРИМИДИНА
© 2023 г. Д. С. Новикова1,*, Ф. Дарвиш1, Т. А. Григорьева1, В. Г. Трибулович1
1 Санкт-Петербургский государственный технологический институт (технический университет),
Московский пр. 24-26/49, Санкт-Петербург, 190013 Россия
*e-mail: dc.novikova@gmail.com
Поступило в редакцию 24 апреля 2023 г.
После доработки 24 апреля 2023 г.
Принято к печати 25 апреля 2023 г.
Разработан воспроизводимый и масштабируемый метод синтеза, получена и описана серия
3,6-замещенных производных пиразоло[1,5-a]пиримидина, которые являются основой для рациональ-
ного дизайна селективных ингибиторов АМФ-активируемой протеинкиназы. При формировании новых
типов углеродного скелета показана возможность применения кросс-сочетания по Сузуки-Мияуре с
использованием лигандов Бухвальда для образования С-С связи в стерически затрудненном положении
6 5,7-диметилзамещенного пиразоло[1,5-a]пиримидина.
Ключевые слова: пиразоло[1,5-a]пиримидин, пиридин-1H-пиразол-5-амин, ингибитор АМФК,
Compound C, кросс-сочетание
DOI: 10.31857/S0044460X23050049, EDN: DBOQGZ
АМФ-Активируемая протеинкиназа (АМФК)
мые последние годы, в частности, за авторством
является одним из ключевых белков-регуляторов,
наиболее авторитетного специалиста в этой обла-
устойчиво привлекающих внимание исследова-
сти профессора Харди [4]. Однако, если прямая
телей. Это обусловлено высокой вовлеченностью
активация АМФК возможна при действии низко-
АМФК в энергетический гомеостаз как отдельной
молекулярных соединений, аффинных к одному из
клетки, так и организма в целом [1]. Несмотря на
трех сайтов активации, которые имеют достаточно
сложную структуру киназы, представляющую со-
уникальное строение, допускающее разработку
бой мультисубъединичный комплекс, к настояще-
селективных активаторов [5, 6], ингибиторы мо-
му моменту разработан ряд подходов к прямой и
гут быть обнаружены только среди соединений,
косвенной активации данного фермента, в частно-
способных к непосредственному взаимодействию
сти через киназа-киназные процессы [2]. В связи
с АТФ-связывающим карманом за счет образова-
с этим основные усилия исследователей направле-
ния водородных связей аналогично взаимодей-
ны на поиск и разработку новых низкомолекуляр-
ствию аденина, в рамках современных парадигм.
ных активаторов АМФК [3].
В настоящий момент выбор низкомолекулярных
Очевидно, что для подобного многофункцио-
агентов, позволяющих с определенной степенью
нального белка-регулятора представляет интерес
селективности ингибировать активность АМФК,
не только активация, но и ингибирование. Это
осуществляется из двух структур - дорсоморфина
подтверждается работами, опубликованными в са-
(Compound C) и SBI-0206965 [7, 8]. Принимая во
684
Р
АЗРАБОТКА ВОСПРОИЗВОДИМОГО И МАСШТАБИРУЕМОГО МЕТОДА СИНТЕЗА
685
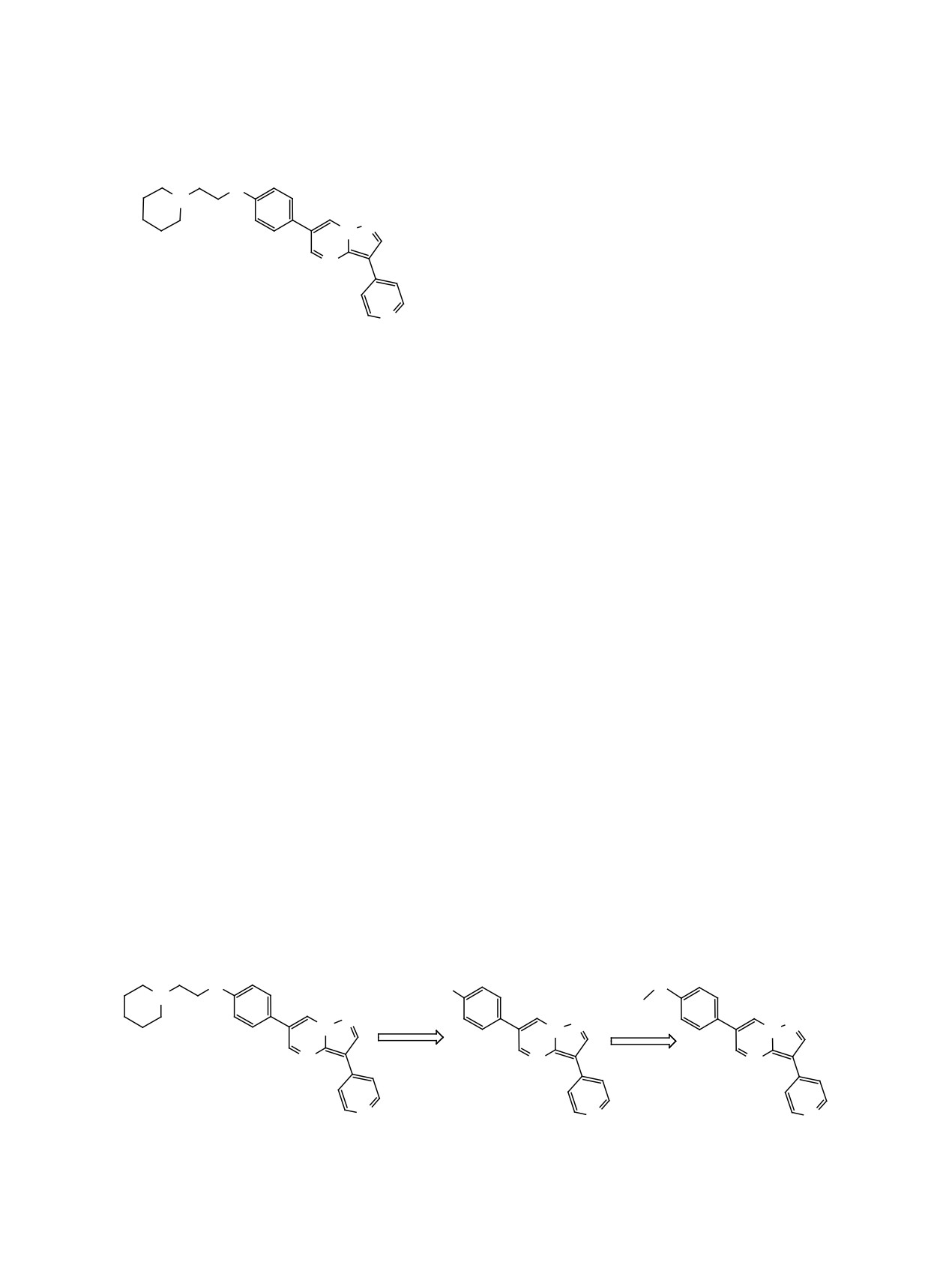
в отношении KDR (Kinase insert domain receptor;
Схема 1.
тирозинкиназа, рецептор фактора роста сосуди-
O
стого эндотелия, тип 2) [10] и киназ, регулирую-
N
щих BMP-зависимые (Bone morphogenetic protein;
N
N
костный морфогенетический белок) сигнальные
пути [11].
N
Начальные этапы рационального дизайна тре-
буют создания библиотеки соединений из несколь-
N
ких десятков соединений для первичного исследо-
Дорсоморфин (Compound C)
вания зависимости структура-активность, причем
варьирование структуры должно носить систем-
ный характер и обеспечивать объяснимые с точки
внимание предельную консервативность АТФ-свя-
зрения компьютерного моделирования различия
зывающего кармана киназ, становится понятной
в связывании низкомолекулярного соединения с
сложность разработки ингибиторов, обладающих
белком-мишенью. Простейший анализ позволяет
высокой селективностью.
установить ключевую для построения библиотеки
структуру 11а (схема 2), которая позволит иссле-
Исторически первым ингибитором АМФК стал
довать влияние на биологическую активность трех
дорсоморфин (схема 1). Он был идентифицирован
молекулярных фрагментов:
в результате высокопроизводительного скринин-
га. В дальнейших исследованиях дорсоморфин не
- изменение положения метоксигруппы в фе-
нильном фрагменте позволяет направленно из-
проявил значительного ингибирования ряда струк-
менять расположение в пространстве концевой
турно родственных по отношению к АМФК киназ
алкиламиновой цепочки конечного аналога дорсо-
и был зафиксирован в качестве первого селектив-
морфина;
ного ингибитора АМФК [9].
- наличие заместителей в пиримидиновом
Несмотря на высокий интерес к модулирова-
кольце позволяет обеспечить поворот фенильно-
нию активности АМФК, систематических иссле-
го кольца вокруг С-С связи относительно гетеро-
дований связи структура-активность для дорсо-
циклического ядра;
морфина, как и попыток рационального дизайна
высокоселективных ингибиторов АМФК на ос-
- изменение положения в пиридиновом фраг-
нове данной молекулы не проводилось. В то же
менте С-С связи с гетероциклическим ядром по-
время способность дорсоморфина ингибировать
зволяет исследовать особенности взаимодействия
различные киназы все же привлекла внимание ис-
низкомолекулярного соединения с АТФ-связываю-
следователей. Были осуществлены попытки смены
щей полостью АМФК.
мишени и исследования связи структура-актив-
Гетероциклический фрагмент рассматривае-
ность производных пиразоло[1,5-a]пиримидина
мых соединений представляет собой жесткую и
Схема 2.
O
HO
O
N
N
N
N
N
N
N
N
N
N
N
N
N
11a
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
686
НОВИКОВА и др.
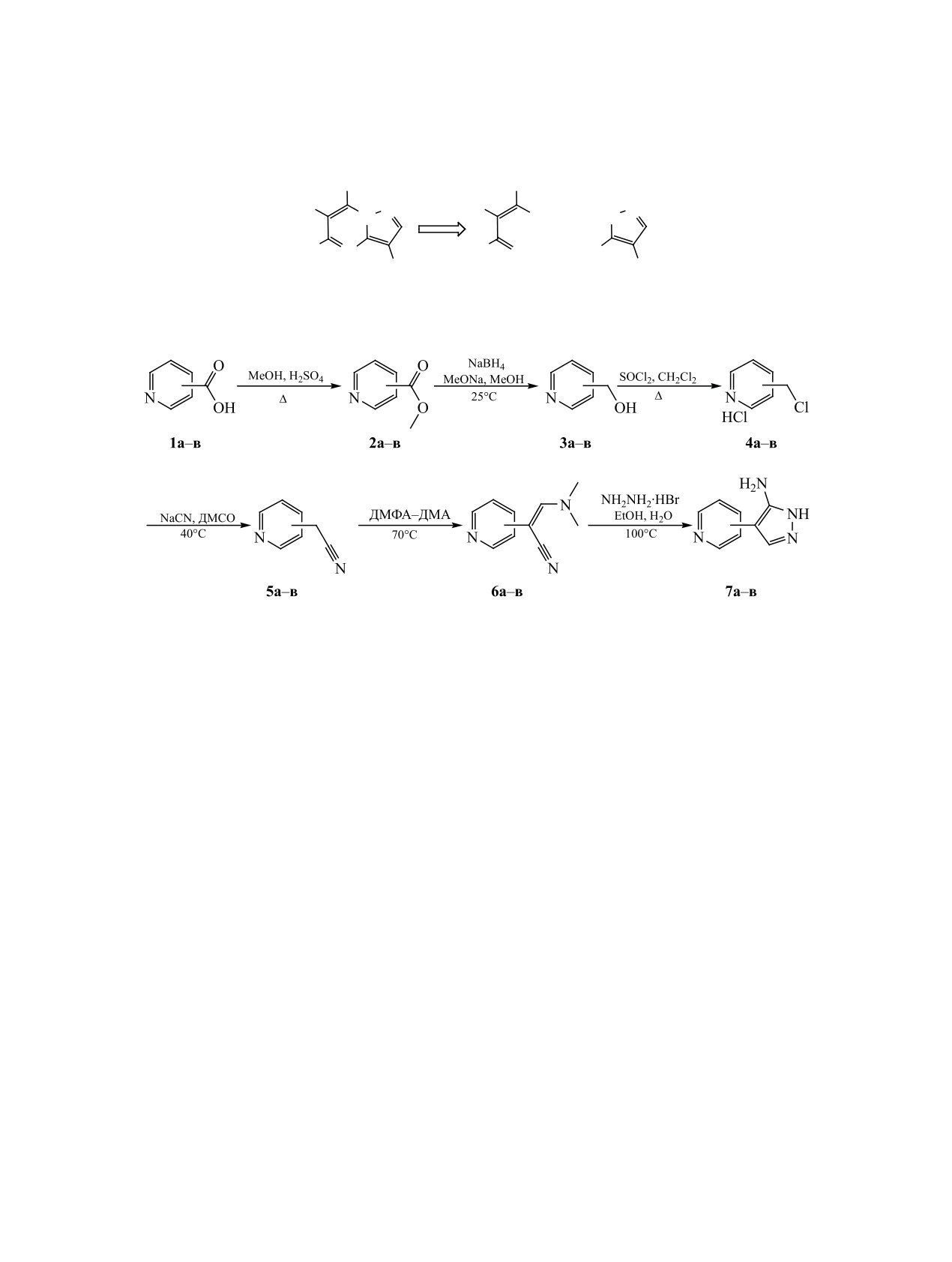
Схема 3.
R7
R7
R6
N
R6
N
N
OH
HN
+
R5
N
R5
O
H2N
R3
R3
Схема 4.
4-пиридил (а), 3-пиридил (б), 2-пиридил (в).
плоскую молекулу пиразоло[1,5-a]пиримидина,
синтетической процедуры позволила увеличить
который является одной из привилегированных
выход до 84-92%. Восстановление соединений
структур для дизайна комбинаторных библиотек,
2а-в до пиридинметанолов 3а-в осуществляли
направленных на поиск лекарственных препа-
боргидридом натрия в сравнительно новой моди-
ратов [12, 13]. Это, в частности, обусловлено на-
фикации с катализом метилатом натрия [15]. При
личием пяти положений для введения связываю-
проведении данной реакции следует обратить вни-
щих фрагментов по всей периферии скаффолда.
мание на разложение весьма устойчивого боратно-
Наиболее доступным методом формирования пи-
го комплекса целевого продукта с помощью HCl.
разоло[1,5-a]пиримидинового ядра является ци-
Получение хлорметилпиридинов 4а-в проводили
клоконденсация 5-аминопиразола с различными
по методу [16] с дополнительной кристаллизаци-
1,3-бисэлектрофилами, в частности, с замещенны-
ей продукта из изопропанола в связи с необходи-
ми β-дикарбонильными соединениями (схема 3).
мостью наиболее полного удаления кислотных
Ключевым гетероциклическим элементом
загрязнений для успешного осуществления следу-
данной схемы является 4-замещенный 1H-пира-
ющей стадии синтеза. Привлекательная возмож-
зол-5-амин. Для синтеза данного соединения нами
ность использования хлорметилпиридинов в виде
была выбрана линейная стратегия, исходя из изо-
оснований [17] оказалась неосуществимой ввиду
никотиновой (1а), никотиновой (1б) и пиколино-
их высокой склонности к самоконденсации.
вой (1в) кислот (схема 4).
Получение 2-пиридилацетонитрилов 5а-в осу-
Стадию этерификации для получения метило-
ществляли путем нуклефильного замещения ци-
вых эфиров 2а-в проводили по модифицирован-
анистым натрием в апротонном биполярном рас-
ной методике на основе работы [14], оптимизация
творителе (ДМСО). Методика синтеза с выходом
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
Р
АЗРАБОТКА ВОСПРОИЗВОДИМОГО И МАСШТАБИРУЕМОГО МЕТОДА СИНТЕЗА
687
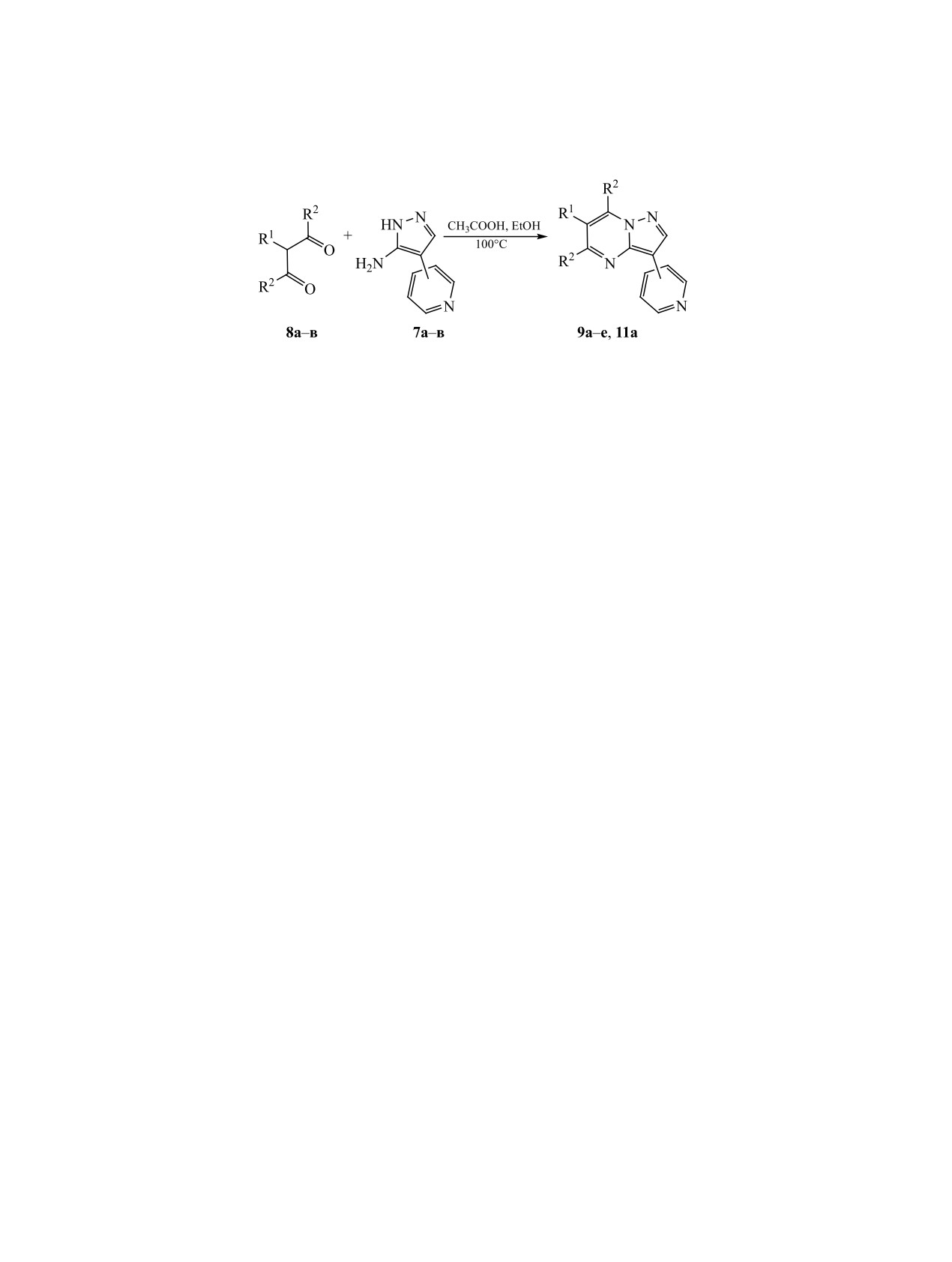
Схема 5.
R1 = Br, R2 = H (8а), CH3 (8б); R1 =4-OCH3-C6H4, R2 = H (8в); R1 = Br, R2 = H, 4-пиридил (9а), 3-пиридил (9б), 2-пи-
ридил (9в); R1 = Br, R2 = CH3, 4-пиридил (9г), 3-пиридил (9д), 2-пиридил (9е); R1 =4-OCH3-C6H4, R2 = H, 4-пиридил
(11а).
продукта 34% была впервые предложена в работе
разработанному методу очистки, оценить истин-
[18]. Более поздние модификации [16, 19] позво-
ный цвет кристаллических продуктов 6а-в как
лили увеличить выход, однако при этом исполь-
желтоватый.
зовался достаточно большой избыток цианистого
Стадия циклизации
3-диметиламино-2-пири-
натрия, так как один эквивалент нуклеофила ухо-
дилакрилонитрилов 6а-в в аминопиразолы 7а-в
дил на нейтрализацию исходного хлорметилпи-
проводилась нами как с основанием (гидразином),
ридина, взятого в виде гидрохлорида. Альтерна-
так и с различными его солями. Наиболее высо-
тивное эквимолярное добавление триэтиламина
кие выходы были получены при применении ре-
в реакционную смесь для получения основания
комендованного в [11] гидробромида гидразина в
хлорметилпиридина in situ приводит к заметному
среде этанол-вода. Таким образом, шестистадий-
снижению выхода. Применение минимального из-
ный синтез 4-пиридил-1H-пиразол-5-аминов 7а-в
бытка цианистого натрия (2.2 экв.), в отличие от
по результатам неоднократных повторов осущест-
ранее предложенного трехкратного [19], позволи-
влен нами со средним выходом 41% в пересчете на
ло получить 2-пиридилацетонитрилы с устойчи-
исходные кислоты 1а-в.
выми выходами, превышающими 80%. Выделение
Следующий этап синтеза приводит к формиро-
продукта реакции возможно с помощью вакуум-
ванию непосредственно пиразоло[1,5-a]пирими-
ной дистилляции при остаточном давлении 0.7-
динового фрагмента. В зависимости от применяе-
2 кПа, при этом отсутствует необходимость при-
мого 1,3-биэлектрофила мы можем получить либо
менения установки Кюгельрор [16]. Полученные
функционализированный полупродукт при R1 = Br
2-пиридилацетонитрилы достаточно стабильны в
(схема 5), либо целевой продукт при R1 = 4-OМе-
виде основания, хранятся при 4°С и для дальней-
С6Н4. С точки зрения стратегии химического син-
шего синтеза не требуют превращения в гидрохло-
теза, второй вариант имеет некоторые преимуще-
риды [11].
ства за счет использования параллельного синтеза
Литературная методика синтеза 3-диметилами-
замещенного 1,3-биэлектрофила, однако, с точки
но-2-пиридилакрилонитрилов 6а-в [11] требовала
зрения комбинаторной стратегии, вариант постро-
применение двадцатикратного избытка диметила-
ения функционализированной структуры пиразо-
цеталя диметилформамида и приводила к преиму-
ло[1,5-a]пиримидина выглядит более предпочти-
щественному смолообразованию. Оптимизация
тельным. В данной работе нами были опробованы
оба варианта.
процесса позволила снизить избыток диметилаце-
таля диметилформамида до 1.3, провести синтез в
Броммалоновый альдегид 8а синтезировали по
существенно более мягких условиях и, благодаря
методике [20, 21], показавшей высокую воспроиз-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
688
НОВИКОВА и др.
Схема 6.
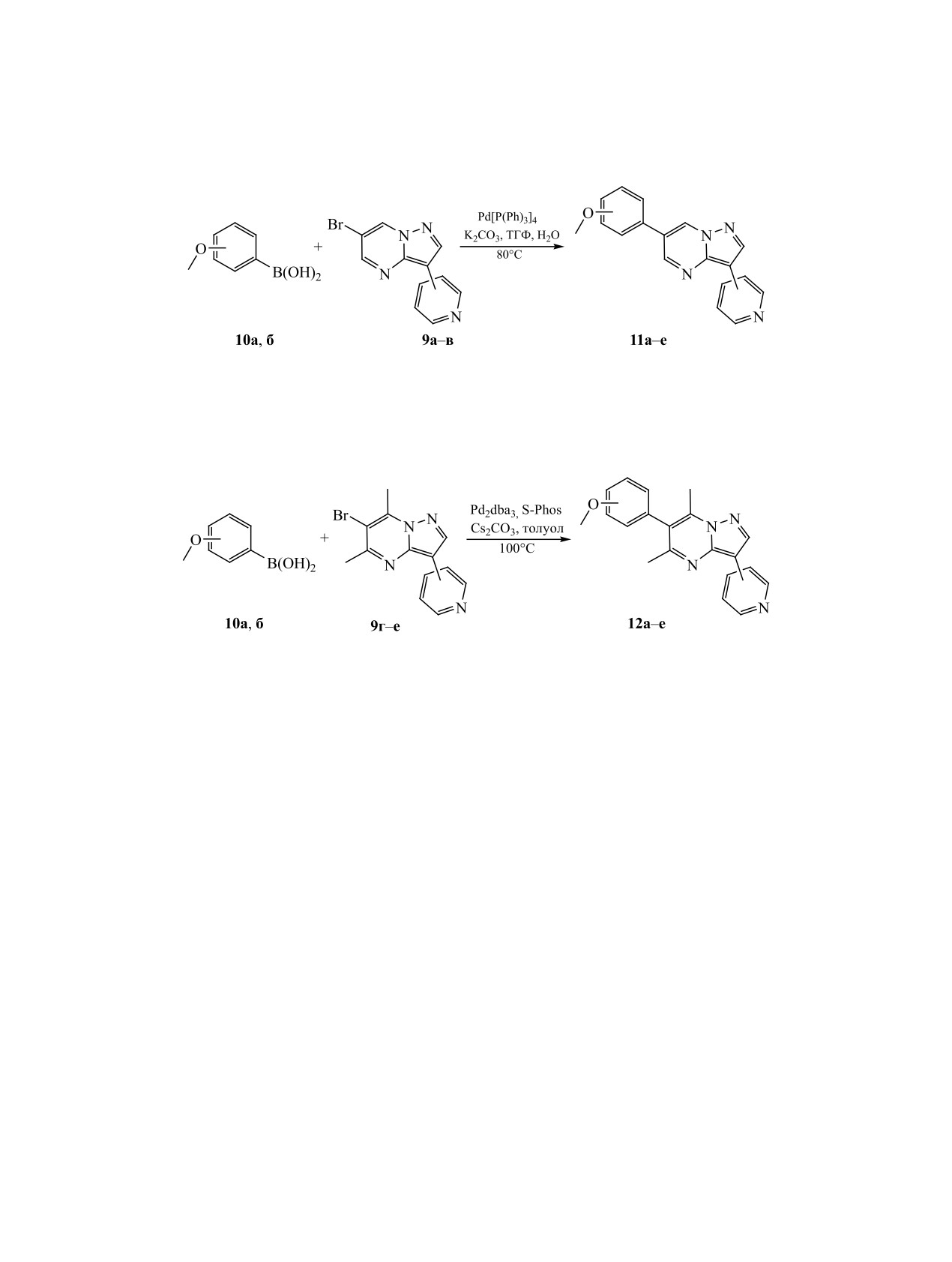
4-OCH3, 4-пиридил (11а), 3-пиридил (11б), 2-пиридил (11в); 3-OCH3, 4-пиридил (11г), 3-пиридил (11д), 2-пиридил
(11е).
Схема 7.
4-OCH3, 4-пиридил (12а), 3-пиридил (12б), 2-пиридил (12в); 3-OCH3, 4-пиридил (12г), 3-пиридил (12д), 2-пиридил
(12е).
водимость по выходу и чистоте продукта. Ацети-
ными выходами (82-87%) и позволила получить
лацетон бромировали до 3-бромацетилацетона 8б,
продукты 11а-е высокой чистоты.
исходя из общих принципов получения α-галоген-
В то же время использование в качестве катали-
карбонильных соединений. 4-Метоксифенилмало-
затора стандартного Pd[P(Ph)3]4 для образования
новый альдегид 8в синтезировали по методу [22],
С-С связи в случае 6-бром-5,7-диметилзамещен-
исходя из 4-метоксифенилуксусной кислоты. Все
ных 3-пиридилпиразоло[1,5-a]пиримидинов 9г-е
реакции циклоконденсации с образованием сое-
не привело к образованию детектируемых коли-
динений 9а-е и 11а проводили в среде этанол-ук-
честв целевого продукта. При этом применение
сусная кислота. Как правило, продукты реакции
каталитической системы Pd2dba3/S-Phos, предло-
не требовали специфической очистки, однако вы-
женной Бухвальдом для стерически затрудненных
деление соединений 9г-е требовало большего вре-
случаев кросс-сочетания [24], показало удовлетво-
мени.
рительные результаты и позволило получить сое-
Образование связи С-С между 6-бром-3-пири-
динения 12а-е с выходом 67-78% (схема 7).
дилпиразоло[1,5-a]пиримидинами и 3-, 4-метокси
Таким образом, нами предложен воспроизво-
замещенным фенилом проводилось с использова-
димый и масштабируемый лабораторный метод
нием реакции Сузуки-Мияуры (схема 6). Для это-
синтеза производных пиразоло[1,5-a]пиримиди-
го по методике, описанной в [23], были синтезиро-
на для создания библиотеки потенциально актив-
ваны 3-метоксифенил- и 4-метоксифенилборные
ных ингибиторов киназной активности АМФК.
кислоты. Реакция проходила с удовлетворитель-
В ходе работы были охарактеризованы ранее не
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
Р
АЗРАБОТКА ВОСПРОИЗВОДИМОГО И МАСШТАБИРУЕМОГО МЕТОДА СИНТЕЗА
689
описанные соединения и синтезированы новые
перемешивали 3 ч при комнатной температуре,
производные пиразоло[1,5-a]пиримидина. Пока-
после чего добавляли 150 мл метанола для уда-
зана высокая эффективность кросс-сочетания по
ления избытка NaBH4. Метанол отгоняли досу-
Сузуки-Мияуре для создания углеродного скелета
ха при пониженном давлении, затем прибавляли
АМФК-направленных пиразоло[1,5-a]пиримиди-
100 мл 20%-ного раствора HCl и нагревали при
нов.
перемешивании в течение 1 ч, после чего охлаж-
дали и нейтрализовали сухим K2CO3 до pH 8-9.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Полученную смесь солей, воды и отслоившегося
пиридинметанола экстрагировали этилацетатом
Спектры ЯМР на ядрах 1Н и 13С регистрирова-
(3×100 мл). Органический слой сушили Na2SO4,
ли на спектрометре Bruker Avance III HD 400 на ча-
растворитель отгоняли при пониженном давлении.
стотах 400 и 100 МГц соответственно в ДМСО-d6
и CDCl3. Масс-спектры снимали на моноквадру-
Пиридинметанол очищали перегонкой в вакууме
при остаточном давлении 0.13 кПа. Выход 12.4-
польном хромато-масс-спектрометре LCМS-2020
13.0 г (78-82%). Описание полученных соедине-
(Shimadzu) c ионизацией электрораспылением
ний приведено в Дополнительных материалах.
(ESI) в положительном режиме. Очистку продук-
тов для идентификации проводили на флеш-хро-
Общая методика получения гидрохлоридов
матографе Isolera Four (Biotage) с использованием
хлорметилпиридинов 4а-в. К раствору 15 г пи-
картриджей SNAP KP-Sil 100 g (Biotage).
ридинметанола 3а-в (0.14 моль) в 200 мл дихлор-
метана при перемешивании прибавляли по каплям
Общая методика получения метилового
эфира пиридинкарбоновых кислот 2а-в. 50 г
20 мл тионилхлорида (32.8 г, 0.28 моль) таким
пиридинкарбоновой кислоты 1а-в (0.41 моль) су-
образом, чтобы температура смеси не превышала
спендировали в 300 мл безводного метанола. При
30°С. По окончании прибавления реакционную
перемешивании 26 мл серной кислоты (0.49 моль)
смесь перемешивали 3 ч, после чего растворитель
прибавляли в течение 30 мин, при этом реакци-
отгоняли досуха при пониженном давлении. К
онная смесь гомогенизировалась. Полученный
остатку прибавляли 200 мл изопропанола, переме-
раствор кипятили 12 ч. По окончании реакции ме-
шивали суспензию в течение 30 мин, после чего
танол отгоняли при пониженном давлении, оста-
отгоняли растворитель досуха при пониженном
ток выливали на 300 г льда и добавлением сухого
давлении. Гидрохлорид хлорметилпиридина пере-
Na2CO3 доводили pH до 8-9. Продукт экстраги-
кристаллизовывали из изопропанола с применени-
ровали этилацетатом (3×150 мл). Объединенные
ем активированного угля. Полученные таким об-
органические слои промывали водой (2×100 мл),
разом продукты были стабильны и не содержали
сушили Na2SO4, растворитель отгоняли при пони-
кислотных примесей. Выход 17.8-19.8 г (79-88%).
женном давлении. Метиловый эфир пиридинкар-
Описание полученных соединений приведено в
боновой кислоты очищали перегонкой в вакууме
Дополнительных материалах.
при остаточном давлении 2.7 кПа. Выход 47-51 г
Общая методика получения 2-пиридилаце-
(84-92%). Описание полученных соединений при-
тонитрилов 5а-в. 22 г NaCN (0.45 моль) помеща-
ведено в Дополнительных материалах.
ли в фарфоровую ступку, заливали 200 мл безвод-
Общая методика получения пиридинмета-
ного ДМСО и перетирали образовавшийся сольват
нолов 3а-в. К 20 мл безводного метанола добав-
до получения равномерной суспензии, после чего
ляли 0.2 г металлического натрия. После полного
переносили в колбу. При перемешивании неболь-
растворения натрия при охлаждении добавляли
шими порциями добавляли 30.8 г сухого гидрохло-
20 г метилового эфира пиридинкарбоновой кис-
рида хлорметилпиридина 4а-в (0.19 моль) таким
лоты 2а-в (0.15 моль), затем при интенсивном
образом, чтобы температура не превышала 40°С.
перемешивании добавляли 11.4 г свежерастертого
По окончании прибавления смесь перемешива-
NaBH4 (0.3 моль). Реакция восстановления имеет
ли 2 ч при 40°С, после чего выливали в раствор
индукционный период и может протекать весьма
100 г K2CO3 и 14 г KOH в 500 мл воды. Полу-
энергично, при резком повышении температуры
ченную смесь экстрагировали этилацетатом
следует поместить колбу в ледяную баню. Смесь
(4×200 мл), при этом наблюдалось образование
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
690
НОВИКОВА и др.
трех слоев. Объединенные органические слои про-
приведено в Дополнительных материалах.
мывали 100 мл раствора NaCl и сушили Na2SO4,
Броммалоновый альдегид (8а). 25 г 1,1,3,3-
растворитель отгоняли при пониженном давлении.
тетраметоксипропана (0.152 моль), 25 мл воды и
2-Пиридилацетонитрил выделяли двукратной пе-
1.1 мл концентрированной HCl перемешивали до
регонкой при пониженном давлении. Первую пе-
получения гомогенного раствора. Смесь охлажда-
регонку проводили при остаточном давлении 0.5-
ли на ледяной бане и при перемешивании прибав-
0.6 кПа без разделения фракций. Вторая перегонка
ляли по каплям 7.8 мл брома (24.3 г, 0.152 моль) с
начиналась при 2.7 кПа, при этом перегонялся
такой скоростью, чтобы реакционная смесь успе-
захваченный экстракцией ДМСО, затем давление
вала обесцвечиваться, и ее температура не пре-
снижали до 1.3 кПа и перегоняли 2-пиридилацето-
вышала 15°С. По окончании прибавления смесь
нитрил. Выход 18.2-19.3 г (81-86%). Полученные
упаривали при 2.7 кПа и температуре бани 40°С.
2-пиридилацетонитрилы достаточно стабильны в
Полученную влажную кристаллическую массу
виде основания, хранятся при 4°С и для дальней-
промыли 50 мл ледяной воды и 20 мл холодного
шего синтеза не требуют превращения в гидро-
CH2Cl2. Продукт досушивали при пониженном
хлориды [11]. Описание полученных соединений
давлении. Выход 17-20 г (73-86%).
приведено в Дополнительных материалах.
3-Бромацетилацетон (8б). К смеси 15 г аце-
Общая методика получения 3-диметилами-
тилацетона (0.15 моль), 5 мл концентрированной
но-2-пиридилакрилонитрилов 6а-в. Смесь 20 г
HCl и 100 мл воды при охлаждении и при переме-
2-пиридилацетонитрила 5а-в (0.17 моль), 26 г
диметилацеталя диметилфрмамида (0.22 моль) и
шивании прибавляли по каплям 7.7 мл брома (24 г,
150 мл ДМФА перемешивали при 70°С в течение
0.15 моль) с такой скоростью, чтобы реакционная
4 ч. По окончании реакции смесь отгоняли досуха
смесь успевала обесцвечиваться, и ее температура
при пониженном давлении и затем максимально
не превышала 15°С. По окончании реакции к сме-
досушивали при остаточном давлении 70 Па. По-
си добавляли 200 мл этилацетата, перемешивали
лученную массу перекристаллизовывали из ми-
5 мин, органический слой отделяли и промывали
нимального количества этилацетата. Выпавший
его до нейтральной реакции 10%-ным раствором
осадок растворяли в 800 мл этилацетата, отфиль-
NaHCO3. Растворитель отгоняли при пониженном
тровали на бумажном фильтре, затем пропускали
давлении, полученный 3-бромацетилацетон при-
через 10 см слой силикагеля. Отгонка растворите-
меняли для дальнейшего синтеза без дополнитель-
ля позволила получить бледно-желтую кристалли-
ной очистки.
ческую массу 3-диметиламино-2-пиридилакрило-
Общая методика получения 6-замещенных
нитрила. Выход 23.8-25.0 г (80-85%). Описание
3-пиридилпиразоло[1,5-a]пиримидинов
9а-е,
полученных соединений приведено в Дополни-
11а.
16 г
4-пиридил-1H-пиразол-5-амина
7а-в
тельных материалах.
(0.1 моль) и 0.1 моля дикарбонильного соединения
Общая методика получения 4-пиридил-1H-
8а-в растворяли в смеси 150 мл уксусной кислоты
пиразол-5-аминов 7а-в. Смесь 20 г 3-диметил-
и 150 мл этанола. Полученный раствор кипятили
амино-2-пиридилакрилонитрила 6а-в (0.12 моль),
при перемешивании в течение 6 ч. Реакционную
26 г гидробромида гидразина (0.23 моль), 200 мл
смесь отгоняли досуха, затем добавляли 400 мл
этанола и 30 мл воды кипятили при перемешива-
воды, 20 г K2CO3 и перемешивали полученную
нии в течение 6 ч. По окончании реакции из реак-
суспензию 10 мин. Осадок отфильтровывали, про-
ционной смеси отгоняли этанол при пониженном
мывали небольшим количеством этанола. При не-
давлении, добавляли 300 мл воды. рН смеси дово-
обходимости продукт перекристаллизовывали из
дили до 8-9 добавлением сухого K2CO3, после чего
толуола. Выход 23.7-25.3 г (86-92%) для соеди-
интенсивно перемешивали в течение 30 мин. Из
нений 9а-в, 24.2-25.3 г (75-82%) для соединений
смеси отгоняли 200 мл воды при пониженном дав-
9г-е и 24.2 г (80%) для соединения 11а.
лении, остаток отфильтровывали, осадок сушили
на воздухе. Сырой 4-пиридил-1H-пиразол-5-амин
Общая методика получения 6-арилзамещен-
перекристаллизовывали из этанола. Выход 16.3-
ных
3-пиридилпиразоло[1,5-a]пиримидинов
17 г (88-92%). Описание полученных соединений
11а-е. В 80 мл ТГФ при перемешивании раство-
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
Р
АЗРАБОТКА ВОСПРОИЗВОДИМОГО И МАСШТАБИРУЕМОГО МЕТОДА СИНТЕЗА
691
ряли 2 г 6-галогензамещенного 3-пиридилпира-
(CArOCH3). Масс-спектр, m/z (Iотн, %): 303.1 (100)
золо[1,5-a]пиримидина 9а-в (7.3 ммоль), 0.05 г
[M + H]+.
Pd[P(Ph)3]4 (0.043 ммоль, 0.5 мол%), 1.66 г меток-
6-(4-Метоксифенил)-3-(пиридин-2-ил)пира-
сифенилборной кислоты 10а, б (11 ммоль). По-
золо[1,5-a]пиримидин (11в). Выход 1.81 г (82%),
лученную смесь перемешивали 30 мин в слабом
желтое кристаллическое вещество, т. пл. 184-
токе аргона, затем прибавляли раствор 5 г K2CO3
185°C. Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.: 3.82
(36 ммоль) в 50 мл воды. Реакцию проводили при
с (3H, OCH3), 7.10 д (2H, CHAr, 3JHH 8.8 Гц), 7.23
кипении с интенсивным перемешиванием в слабом
д. д. д (1H, CHAr, 3JHH 7.5, 4.8, 5JHH 1.2 Гц), 7.83 д
токе аргона в течение 4 ч. По окончании реакции
(2H, CHAr, 3JHH 8.8 Гц), 7.87 т. д (1H, CHAr, 3JHH
растворитель отгоняли досуха при пониженном
7.8, 4JHH 1.9 Гц), 8.46 д. т (1H, CHAr, 3JHH 7.9, 5JHH
давлении. Остаток перемешивали в 200 мл кипя-
1.1 Гц), 8.60 д. д. д (1H, CHAr, 3JHH 4.9, 4JHH 1.9,
щего толуола, после чего отфильтровали горячим.
5JHH 1.0 Гц), 8.81 с (1H, CHAr), 9.09 д (1H, CHAr,
Фильтрат осветляли 1 г силикагеля при кипении и
4JHH 2.3 Гц), 9.49 д (1H, CHAr, 4JHH 2.3 Гц). Спектр
перемешивании, вновь отфильтровали и доводили
ЯМР 13С (ДМСО-d6), δC, м. д.: 55.31 (OCH3), 109.66
объем раствора до 15 мл. Через сутки отфильтро-
(CAr), 114.69 (CHAr), 120.30 (CHAr), 121.11 (CHAr),
вали выпавший осадок и сушили. Выход 1.80-1.92
121.79 (CAr), 125.47 (CAr), 128.23 (CHAr), 132.54
г (82-87%).
(CHAr), 136.74 (CHAr), 143.57 (CAr), 144.07 (CHAr),
6-(4-Метоксифенил)-3-(пиридин-4-ил)пира-
149.39 (CHAr), 150.44 (CHAr), 150.93 (CAr), 159.63
золо[1,5-a]пиримидин (11а). Выход 1.92 г (87%),
(CArOCH3). Масс-спектр, m/z (Iотн, %): 303.1 (100)
желтоватое кристаллическое вещество, т. пл.
[M + H]+.
215°C. Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.: 3.83
6-(3-Метоксифенил)-3-(пиридин-4-ил)пира-
с (3H, OCH3), 7.10 д (2H, CHAr, 3JHH 8.8 Гц), 7.84
золо[1,5-a]пиримидин (11г). Выход 1.9 г (86%),
д (2H, CHAr, 3JHH 8.8 Гц), 8.15 д (2H, CHAr, 3JHH
желтоватое кристаллическое вещество, т. пл. 169-
6.2 Гц), 8.59 д (2H, CHAr, 3JHH 6.2 Гц), 8.96 с (1H,
170°C. Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.: 3.86
CHAr), 9.12 д (1H, CHAr, 4JHH 2.3 Гц), 9.52 д (1H,
с (3H, OCH3), 7.01-7.05 м (1H, CHAr), 7.44-7.47
CHAr, 4JHH 2.3 Гц). Спектр ЯМР 13С (ДМСО-d6),
м (3H, CHAr), 8.16 д (2H, CHAr, 3JHH 6.2 Гц), 8.59
δC, м. д.: 55.33 (OCH3), 106.08 (CAr), 114.72 (CHAr),
д (2H, CHAr, 3JHH 6.2 Гц), 8.99 с (1H, CHAr), 9.16
119.62 (CHAr), 122.16 (CAr), 125.34 (CAr), 128.28
д (1H, CHAr, 4JHH 2.3 Гц), 9.62 д (1H, CHAr, 4JHH
(CHAr), 132.61 (CHAr), 139.28 (CAr), 143.80 (CHAr),
2.3 Гц). Спектр ЯМР 13С (ДМСО-d6), δC, м. д.:
143.84 (CAr), 149.98 (CHAr), 150.79 (CHAr), 159.70
55.33 (OCH3), 106.19 (CAr), 112.35 (CHAr), 114.34
(CArOCH3). Масс-спектр, m/z (Iотн, %): 303.1 (100)
(CAr), 119.17 (CHAr), 119.66 (CHAr), 122.15 (CAr),
[M + H]+.
130.34 (CHAr), 133.66 (CHAr), 134.54 (CAr), 139.20
6-(4-Метоксифенил)-3-(пиридин-3-ил)пира-
(CAr), 144.13 (CHAr), 149.99 (CHAr), 150.93 (CHAr),
золо[1,5-a]пиримидин (11б). Выход 1.85 г (84%),
159.95 (CArOCH3). Масс-спектр, m/z (Iотн, %): 303.1
желтоватое кристаллическое вещество, т. пл. 165-
(100) [M + H]+.
166°C. Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.: 3.83 с
6-(3-Метоксифенил)-3-(пиридин-3-ил)пира-
(3H, OCH3), 7.10 д (2H, CHAr, 3JHH 8.9 Гц), 7.48 д.
золо[1,5-a]пиримидин (11д). Выход 1.91 г (87%),
д. д (1H, CHAr, 3JHH 8.0, 4.8, 5JHH 0.9 Гц), 7.83 д (2H,
желтоватое кристаллическое вещество, т. пл.
CHAr, 3JHH 8.9 Гц), 8.45 д. д (1H, CHAr, 3JHH 4.7, 4JHH
173-174°C. Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.:
1.6 Гц), 8.52 д. д. д (1H, CHAr, 3JHH 8.0, 4JHH 2.3,
3.86 с (3H, OCH3), 7.00-7.05 м (1H, CHAr), 7.43-
1.7 Гц), 8.88 с (1H, CHAr), 9.07 д (1H, CHAr, 4JHH
7.47 м (3H, CHAr), 7.48 д. д. д (1H, CHAr, 3JHH 8.0,
2.3 Гц), 9.36 д. д (1H, CHAr, 4JHH 2.3, 5JHH 0.8 Гц),
4.8, 5JHH 0.9 Гц), 8.46 д. д (1H, CHAr, 3JHH 4.7, 4JHH
9.48 д (1H, CHAr, 4JHH 2.3 Гц). Спектр ЯМР 13С
1.6 Гц), 8.52 д. д. д (1H, CHAr, 3JHH 8.0, 4JHH 2.3,
(ДМСО-d6), δC, м. д.: 55.32 (OCH3), 105.80 (CAr),
1.7 Гц), 8.91 с (1H, CHAr), 9.11 д (1H, CHAr, 4JHH
114.70 (CHAr), 121.81 (CAr), 123.78 (CHAr), 125.48
2.3 Гц), 9.37 д. д (1H, CHAr, 4JHH 2.3, 5JHH 0.9 Гц),
(CAr), 128.03 (CAr), 128.21 (CHAr), 132.30 (CHAr),
9.59 д (1H, CHAr, 4JHH 2.3 Гц). Спектр ЯМР 13С
132.39 (CHAr), 142.95 (CHAr), 143.29 (CAr), 146.68
(ДМСО-d6), δC, м. д.: 55.32 (OCH3), 105.92 (CAr),
(CHAr),
146.89 (CHAr),
150.23 (CHAr),
159.63
112.27 (CHAr), 114.27 (CHAr), 119.11 (CHAr), 121.80
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
692
НОВИКОВА и др.
(CAr), 123.79 (CHAr), 127.95 (CAr), 130.33 (CHAr),
(ДМСО-d6), δ, м. д.: 2.35 с (3H, CH3), 2.48 с (3H,
132.44 (CHAr), 133.36 (CHAr), 134.67 (CAr), 143.31
CH3), 3.84 с (3H, OCH3), 7.10 д (2H, CHAr, 3JHH
(CHAr), 143.58 (CAr), 146.72 (CHAr), 146.96 (CHAr),
8.7 Гц), 7.33 д (2H, CHAr, 3JHH 8.7 Гц), 8.18 д (2H,
150.36 (CHAr), 159.95 (CArOCH3). Масс-спектр, m/z
CHAr, 3JHH 6.2 Гц), 8.56 д (2H, CHAr, 3JHH 6.2 Гц),
(Iотн, %): 303.1 (100) [M + H]+.
8.90 с (1H, CHAr). Спектр ЯМР 13С (ДМСО-d6), δC,
6-(3-Метоксифенил)-3-(пиридин-2-ил)пира-
м. д.: 14.89 (CH3), 24.87 (CH3), 55.17 (OCH3), 105.26
золо[1,5-a]пиримидин (11е). Выход 1.83 г (82%),
(CAr), 114.26 (CHAr), 119.40 (CHAr), 122.21 (CAr),
желтое кристаллическое вещество, т. пл. 171°C.
126.91 (CAr), 131.24 (CHAr), 139.78 (CAr), 142.81
Спектр ЯМР 1Н (ДМСО-d6), δ, м. д.: 3.86 с (3H,
(CHAr), 143.84 (CAr), 144.14 (CAr), 149.86 (CHAr),
OCH3), 7.00-7.05 м (1H, CHAr), 7.24 д. д. д (1H,
159.04 (CAr), 159.51 (CArOCH3). Масс-спектр, m/z
CHAr, 3JHH 7.5, 4.8, 5JHH 1.2 Гц), 7.43-7.46 м (3H,
(Iотн, %): 331.2 (100) [M + H]+.
CHAr), 7.87 т. д (1H, CHAr, 3JHH 7.7, 4JHH 1.9 Гц),
6-(4-Метоксифенил)-5,7-диметил-3-(пи-
8.47 д. т (1H, CHAr, 3JHH 7.9, 5JHH 1.1 Гц), 8.60
ридин-3-ил)пиразоло[1,5-a]пиримидин
(12б).
д. д. д (1H, CHAr, 3JHH 4.8, 4JHH 1.9, 5JHH 0.9 Гц),
Выход 1.52 г (70%), бесцветное кристалличе-
8.84 с (1H, CHAr), 9.12 д (1H, CHAr, 4JHH 2.3 Гц),
ское вещество, т. пл. 156-158°C. Спектр ЯМР 1Н
9.59 д (1H, CHAr, 4JHH 2.3 Гц). Спектр ЯМР 13С
(ДМСО-d6), δ, м. д.: 2.35 с (3H, CH3), 2.48 с (3H,
(ДМСО-d6), δC, м. д.: 55.32 (OCH3), 109.76 (CAr),
CH3), 3.84 с (3H, OCH3), 7.10 д (2H, CHAr, 3JHH
112.33 (CHAr), 114.24 (CHAr), 119.13 (CHAr), 120.35
8.8 Гц), 7.33 д (2H, CHAr, 3JHH 8.8 Гц), 7.51 д. д. д
(CHAr), 121.17 (CHAr), 121.78 (CAr), 130.31 (CHAr),
(1H, CHAr, 3JHH 8.0, 4.8, 5JHH 0.9 Гц), 8.47 д. д (1H,
133.59 (CHAr), 134.66 (CAr), 136.75 (CHAr), 143.85
CHAr, 3JHH 4.7, 4JHH 1.6 Гц), 8.50 д. д. д (1H, CHAr,
(CAr), 144.41 (CHAr), 149.40 (CHAr), 150.57 (CHAr),
3JHH 8.0, 4JHH 2.3, 1.7 Гц), 8.78 с (1H, CHAr), 9.33
150.86 (CAr), 159.94 (CArOCH3). Масс-спектр, m/z
д. д (1H, CHAr, 4JHH 2.3, 5JHH 0.8 Гц). Спектр ЯМР
(Iотн, %): 303.1 (100) [M + H]+.
13С (ДМСО-d6), δC, м. д.: 14.88 (CH3), 24.86 (CH3),
Общая методика получения 6-арил-5,7-ди-
55.16 (OCH3), 104.70 (CAr), 114.30 (CHAr), 121.69
метилзамещенных
3-пиридилпиразоло[1,5-a]-
(CAr), 123.68 (CHAr), 125.33 (CAr), 127.95 (CAr),
пиримидинов
12а-е. В
120 мл толуола при
131.30 (CHAr), 136.25 (CHAr), 139.40 (CAr), 141.55
перемешивании растворяли
0.04 г Pd2(dba)3
(CAr), 143.99 (CAr), 146.47 (CHAr), 150.23 (CHAr),
(0.044 ммоль, 0.66 мол%) и 0.08 г 2-дициклогек-
158.97 (CAr), 159.40 (CArOCH3). Масс-спектр, m/z
силфосфино-2′,6′-диметоксибифенила
(Sphos,
(Iотн, %): 331.2 (100) [M + H]+.
0.195 ммоль), полученный раствор перемешивали
6-(4-Метоксифенил)-5,7-диметил-3-(пи-
в течение 30 мин в небольшом токе аргона. К ре-
акционной смеси прибавляли 2 г 6-галоген-5,7-ди-
ридин-2-ил)пиразоло[1,5-a]пиримидин
(12в).
Выход 1.58 г (72%), бесцветное кристалличе-
метилзамещенного
3-пиридилпиразоло[1,5-a]-
пиримидина 9г-е (6.6 ммоль), 1.66 г метоксифе-
ское вещество, т. пл. 170-172°C. Спектр ЯМР 1Н
нилборной кислоты 10а, б (11 ммоль) и 6 г Cs2CO3
(ДМСО-d6), δ, м. д.: 2.35 с (3H, CH3), 2.48 с (3H,
(18.4 ммоль). Реакцию проводили при 100°С с ин-
CH3), 3.84 с (3H, OCH3), 7.10 д (2H, CHAr, 3JHH
тенсивным перемешиванием в слабом токе аргона
8.8 Гц), 7.26 д. д. д (1H, CHAr, 3JHH 7.5, 4.8, 5JHH
в течение 16 ч. По окончании реакции объем то-
1.2 Гц), 7.53 д (2H, CHAr, 3JHH 8.8 Гц), 7.90 т. д (1H,
луола доводили до 200 мл, доводили до кипения
CHAr, 3JHH 7.8, 4JHH 1.9 Гц), 8.43 д. т (1H, CHAr, 3JHH
и фильтровали горячим. Фильтрат осветляли 1 г
7.9, 5JHH 1.1 Гц), 8.57 д. д. д (1H, CHAr, 3JHH 4.9, 4JHH
силикагеля при кипении и перемешивании, вновь
1.9, 5JHH 1.0 Гц), 8.76 с (1H, CHAr). Спектр ЯМР
фильтровали и доводили объем раствора до 15 мл.
13С (ДМСО-d6), δC, м. д.: 14.89 (CH3), 24.87 (CH3),
Через сутки отфильтровывали выпавший осадок и
55.17 (OCH3), 108.92 (CAr), 114.26 (CHAr), 120.55
сушили. Выход 1.46-1.7 г (67-78%).
(CHAr), 121.63 (CHAr), 122.02 (CAr), 126.75 (CAr),
6-(4-Метоксифенил)-5,7-диметил-3-(пи-
131.43 (CHAr), 136.53 (CHAr), 139.07 (CHAr), 143.28
ридин-4-ил)пиразоло[1,5-a]пиримидин
(12а).
(CAr), 144.79 (CHAr), 149.44 (CHAr), 150.48 (CHAr),
Выход
1.7 г
(78%), бесцветное кристалличе-
159.01 (CAr), 159.50 (CArOCH3). Масс-спектр, m/z
ское вещество, т. пл. 175-177°C. Спектр ЯМР 1Н
(Iотн, %): 331.2 (100) [M + H]+.
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
Р
АЗРАБОТКА ВОСПРОИЗВОДИМОГО И МАСШТАБИРУЕМОГО МЕТОДА СИНТЕЗА
693
6-(3-Метоксифенил)-5,7-диметил-3-(пи-
(CHAr), 121.07 (CHAr), 121.77 (CAr), 127.44 (CHAr),
ридин-4-ил)пиразоло[1,5-a]пиримидин
(12г).
130.88 (CHAr), 136.39 (CAr), 137.93 (CAr), 142.55
Выход 1.64 г (75%), бесцветное кристалличе-
(CAr), 144.62 (CHAr), 149.20 (CHAr), 150.24 (CAr),
ское вещество, т. пл. 169-170°C. Спектр ЯМР 1Н
159.06 (CAr), 159.64 (CArOCH3). Масс-спектр, m/z
(ДМСО-d6), δ, м. д.: 2.35 с (3H, CH3), 2.48 с (3H,
(Iотн, %): 331.2 (100) [M + H]+.
CH3), 3.87 с (3H, OCH3), 7.00-7.05 м (1H, CHAr),
ИНФОРМАЦИЯ ОБ АВТОРАХ
7.24-7.26 м (2H, CHAr), 7.43-7.45 м (1H, CHAr),
8.18 д (2H, CHAr, 3JHH 6.2 Гц), 8.56 д (2H, CHAr,
Новикова Дарья Сергеевна, ORCID: https://
3JHH 6.2 Гц), 8.93 с (1H, CHAr). Спектр ЯМР 13С
orcid.org/0000-0002-5310-4570
(ДМСО-d6), δC, м. д.: 14.89 (CH3), 24.87 (CH3),
Григорьева Татьяна Алексеевна, ORCID: https://
55.17 (OCH3), 105.77 (CAr), 112.12 (CHAr), 114.22
orcid.org/0000-0003-1271-0328
(CAr), 118.98 (CHAr), 119.25 (CHAr), 122.10 (CAr),
Трибулович Вячеслав Генрихович, ORCID:
128.42 (CHAr), 129.98 (CHAr), 134.86 (CAr), 139.49
(CAr), 144.15 (CAr), 149.86 (CHAr), 159.03 (CAr),
159.69 (CArOCH3). Масс-спектр, m/z (Iотн, %): 331.2
БЛАГОДАРНОСТЬ
(100) [M + H]+.
Работа выполнена с использованием оборудо-
6-(3-Метоксифенил)-5,7-диметил-3-(пи-
вания Инжинирингового центра Санкт-Петербург-
ридин-3-ил)пиразоло[1,5-a]пиримидин
(12д).
ского государственного технологического инсти-
Выход 1.46 г (67%), бесцветное кристалличе-
тута.
ское вещество, т. пл. 165-167°C. Спектр ЯМР 1Н
(ДМСО-d6), δ, м. д.: 2.35 с (3H, CH3), 2.48 с (3H,
ФИНАНСОВАЯ ПОДДЕРЖКА
CH3), 3.87 с (3H, OCH3), 7.00-7.05 м (1H, CHAr),
7.24-7.27 м (2H, CHAr), 7.44-7.46 м (1H, CHAr),
Работа выполнена при финансовой поддержке
7.50 д. д. д (1H, CHAr, 3JHH 3JHH 8.0, 4.8, 5JHH
Российского научного фонда (проект № 21-73-
0.9 Гц), 8.48 д. д (1H, CHAr, 3JHH 4.7, 4JHH 1.6 Гц),
00296).
8.51 д. д. д (1H, CHAr, 3JHH 8.0, 4JHH 2.3, 1.7 Гц),
КОНФЛИКТ ИНТЕРЕСОВ
8.85 с (1H, CHAr), 9.35 д. д (1H, CHAr, 4JHH 2.3, 5JHH
0.9 Гц). Спектр ЯМР 13С (ДМСО-d6), δC, м. д.: 14.89
Авторы заявляют об отсутствии конфликта ин-
(CH3), 24.87 (CH3), 55.17 (OCH3), 105.69 (CAr),
тересов.
112.03 (CHAr), 114.01 (CHAr), 118.94 (CHAr), 121.40
(CAr), 123.54 (CHAr), 127.33 (CAr), 127.93 (CHAr),
ДОПОЛНИТЕЛЬНЫЕ МАТЕРИАЛЫ
130.36 (CHAr), 134.37 (CAr), 138.24 (CHAr), 143.01
Дополнительные материалы для этой статьи
(CHAr), 143.79 (CAr), 146.93 (CAr), 150.54 (CHAr),
доступны по doi
10.31857/S0044460X23050049
159.04 (CAr), 159.65 (CArOCH3). Масс-спектр, m/z
для авторизованных пользователей.
(Iотн, %): 331.2 (100) [M + H]+.
6-(3-Метоксифенил)-5,7-диметил-3-(пи-
СПИСОК ЛИТЕРАТУРЫ
ридин-2-ил)пиразоло[1,5-a]пиримидин
(12е).
1. Trefts E., Shaw R.J. // Mol. Cell. 2021. Vol. 81. N 18.
Выход 1.54 г (71%), бесцветное кристалличе-
P. 3677. doi 10.1016/j.molcel.2021.08.015
ское вещество, т. пл. 160-162°C. Спектр ЯМР 1Н
2. Tarasiuk O., Miceli M., Di Domizio A., Nicolini G. //
(ДМСО-d6), δ, м. д.: 2.35 с (3H, CH3), 2.48 с (3H,
Biology. 2022. Vol. 11. N 7. P. 1041. doi 10.3390/
CH3), 3.87 с (3H, OCH3), 7.00-7.05 м (1H, CHAr),
biology11071041
7.24-7.27 м (3H, CHAr), 7.43-7.45 м (1H, CHAr),
3. Новикова Д.С., Гарабаджиу А.В., Мелино Дж.,
Барлев Н.А., Трибулович В.Г. // Изв. АН. Сер. хим.
7.90 т. д (1H, CHAr, 3JHH 7.7, 4JHH 1.9 Гц), 8.44 д.
2015. № 7. С. 1497; Novikova D.S., Garabadzhiu A.V.,
т (1H, CHAr, 3JHH 7.9, 5JHH 1.1 Гц), 8.57 д. д. д (1H,
Melino G., Barlev N.A., Tribulovich V.G. // Russ. Chem.
CHAr, 3JHH 4.8, 4JHH 1.9, 5JHH 0.9 Гц), 8.78 с (1H,
Bull. 2015. Vol. 64. N 7. P. 1497. doi 10.1007/s11172-
CHAr). Спектр ЯМР 13С (ДМСО-d6), δC, м. д.: 14.89
015-1036-x
(CH3), 24.87 (CH3), 55.17 (OCH3), 109.53 (CAr),
4. Russell F.M., Hardie D.G. // Int. J. Mol. Sci. 2021.
112.04 (CHAr), 114.13 (CHAr), 118.96 (CHAr), 120.15
Vol. 22. N 1. P. 186. doi 10.3390/ijms22010186
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023
694
НОВИКОВА и др.
5.
Novikova D.S., Grigoreva T.A., Ivanov G.S.,
Smuga D., Bazydło K., Dubiel K., Wieczorek M. // Eur.
Barlev N.A., Tribulovich V.G. // ChemMedChem. 2020.
J. Med. Chem. 2018. Vol. 155. P. 96. doi 10.1016/j.
Vol. 15. N 24. P. 2521. doi 10.1002/cmdc.202000579
ejmech.2018.05.043
6.
Novikova D.S., Grigoreva T.A., Zolotarev A.A.,
15.
Prasanth C.P., Ebbin J., Abhijith A., Nair D.S., Ibnusaud I.,
Garabadzhiu, A.V., Tribulovich, V.G. // RSC Adv. 2018.
Raskatov J., Singaram B. // J. Org. Chem. 2018. Vol. 83.
Vol. 8. N 60., P. 34543. doi 10.1039/C8RA07576J
N 3. P. 1431. doi 10.1021/acs.joc.7b02993
7.
Dasgupta B., Seibel W. // Methods Mol. Biol. 2018.
16.
Rüger N., Roatsch M., Emmrich T., Franz H., Schüle R.,
Vol. 1732. P. 195. doi 10.1007/978-1-4939-7598-3_12
Jung M., Link A. // ChemMedChem. 2015. Vol. 10.
8.
Dite T.A., Langendorf C.G., Hoque A., Galic S.,
N 11. P. 1875. doi 10.1002/cmdc.201500335
Rebello R.J., Ovens A.J., Lindqvist L.M., Ngoei K.R.W.,
17.
Kaushik M.P., Vaidyanathaswamy R. // Phosphorus,
Ling N.X.Y., Furic L., Kemp B.E., Scott J.W.,
Oakhill J.S. // J. Biol. Chem. 2018. Vol. 293. N 23.
Sulfur, Silicon, Relat. Elem. 1995. Vol. 102. N 1-4.
P. 8874. doi 10.1074/jbc.RA118.003547
P. 45. doi 10.1080/10426509508042541
9.
Zhou G., Myers R., Li Y., Chen Y., Shen X., Fenyk-
18.
Borne R.F., Aboul-Enein H.Y. // J. Heterocycl.
Melody J., Wu M., Ventre J., Doebber T., Fujii N.,
Chem. 1980. Vol. 17. N 7. P. 1609. doi 10.1002/
Musi N., Hirshman M.F., Goodyear L.J., Moller D.E. //
jhet.5570170753
J. Clin. Invest. 2001. Vol. 108. N 8. P. 1167. doi 10.1172/
19.
Demchuk O.P., Hryshchuk O.V., Vashchenko B.V.,
JCI13505
Radchenko D.S., Kovtunenko V.O., Komarov I.V.,
10.
Fraley M.E., Rubino R.S., Hoffman W.F.,
Grygorenko O.O. // Eur. J. Org. Chem. 2019. Vol. 2019.
Hambaugh S.R., Arrington K.L., Hungate R.W.,
N 34. P. 5937. doi 10.1002/ejoc.201901001
Bilodeau M.T., Tebben A.J., Rutledge R.Z., Kendall R.L.,
20.
Trofimenko S. // J. Org. Chem. 1963. Vol. 28. N 11.
McFall R.C., Huckle W.R., Coll K.E., Thomas K.A. //
P. 3243. doi 10.1021/jo01046a526
Bioorg. Med. Chem. Lett. 2002. Vol. 12. N 24. P. 3537.
doi 10.1016/s0960-894x(02)00827-2
21.
Tomsho J.W., McGuireb J.J., Coward J.K. // Org.
11.
Cuny G.D., Yu P.B., Laha J.K., Xing X., Liu J.F.,
Biomol. Chem. 2005. Vol. 3. N 18. P. 3388. doi 10.1039/
Lai C.S., Deng D.Y., Sachidanandan C., Bloch K.D.,
B505907K
Peterson R.T. // Bioorg. Med. Chem. Lett. 2008. Vol. 18.
22.
Chen H.J., Chew C.Y., Chang E.H., Tu Y.W., Wei L.Y.,
N 15. P. 4388. doi 10.1016/j.bmcl.2008.06.052
Wu B.H., Chen C.H., Yang Y.T., Huang S.C., Chen J.K.,
12.
Cherukupalli S., Karpoormath R., Chandrasekaran B.,
Chen I.C., Tan K.T. // J. Am. Chem. Soc. 2018. Vol. 140.
Hampannavar G.A., Thapliyal N., Palakollu V.N. // Eur.
N 15. P. 5224. doi 10.1021/jacs.8b01159
J. Med. Chem. 2017. Vol. 126. P. 298. doi 10.1016/j.
23.
Tribulovich V.G., Garabadzhiu A.V., Kalvin’sh I. //
ejmech.2016.11.019
Pharm. Chem. J. 2011. Vol. 45. N 4. P. 241. doi 10.1007/
13.
Arias-Gómez A., Godoy A., Portilla J. // Molecules.
s11094-011-0605-z
2021. Vol. 26. N 9. P. 2708. doi 10.3390/
24.
Barder T.E., Walker S.D., Martinelli J.R., Buchwald S.L. //
molecules26092708
14.
Moszczyński-Pętkowski R., Majer J., Borkowska M.,
J. Am. Chem. Soc. 2005. Vol. 127. N 13. P. 4685. doi
Bojarski Ł., Janowska S., Matłoka M., Stefaniak F.,
10.1021/ja042491j
Development of a Reproducible and Scalable Method
for the Synthesis of Biologically Active
Pyrazolo[1,5-a]pyrimidine Derivatives
D. S. Novikovaa,*, F. Darwisha, T. A. Grigorevaa, and V. G. Tribulovicha
St. Petersburg State Institute of Technology (Technical University), St. Petersburg, 190013 Russia
*e-mail: dc.novikova@gmail.com
Received April 24, 2023; revised April 24, 2023; accepted April 25, 2023
A reproducible and scalable method for the synthesis was developed, and a series of 3,6-substituted pyra-
zolo[1,5-a]pyrimidines, which are the basis for the rational design of selective inhibitors of AMP-activated
protein kinase, was obtained and characterized. In the course of the formation of new types of carbon skeleton,
the possibility of applying Suzuki-Miyaura cross-coupling with Buchwald ligands to form C-C bond in the
sterically hindered position 6 of 5,7-dimethyl-substituted pyrazolo[1,5-a]pyrimidine was shown.
Keywords: pyrazolo[1,5-a]pyrimidine, pyridine-1H-pyrazol-5-amine, AMPK inhibitor, Compound C,
cross-coupling
ЖУРНАЛ ОБЩЕЙ ХИМИИ том 93 № 5 2023