ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2022, том 58, № 11, с. 1207-1213
УДК 547-326
СИНТЕЗ ПРОПАРГИЛОВОГО ЭФИРА
(±)-(5-МЕТИЛИДЕН-4-ОКСОПЕНТ-2-ЕН-1-ИЛ)-
УКСУСНОЙ КИСЛОТЫ
© 2022 г. З. Р. Макаев*, Н. С. Востриков, М. С. Мифтахов
Уфимский Институт химии - обособленное структурное подразделение
ФГБНУ «Уфимского федерального исследовательского центра РАН»,
Россия, 450054 Уфа, просп. Октября, 71
*e-mail: z.makaev.orgsynthesis@gmail.com
Поступила в редакцию 23.02.2022 г.
После доработки 14.03.2022 г.
Принята к публикации 22.03.2022 г.
Описан синтез пропаргилового эфира (±)-(5-метилиден-4-оксоциклопент-2-ен-1-ил)уксусной кислоты
из метоксикарбонильного предшественника. Синтез включает стадии образования аддукта Михаэля с
тиофенолом по терминальной двойной связи, восстановления кетогруппы по Люшу (Luche), щелочного
гидролиза сложноэфирной функции, алкилирования кислоты пропаргилбромидом с получением карбок-
сизащищенного циклопентенола.Последний окислением реагентом Десса-Мартина и затем действием
30%-ной перекиси водорода превращен в кетосульфон. ЭлиминированиеPhSO2H из кетосульфона при-
водит к целевой молекуле.
Ключевые слова: циклопентеноновые простагландины, пропаргиловый эфир
DOI: 10.31857/S051474922211009X, EDN: LSRBMH
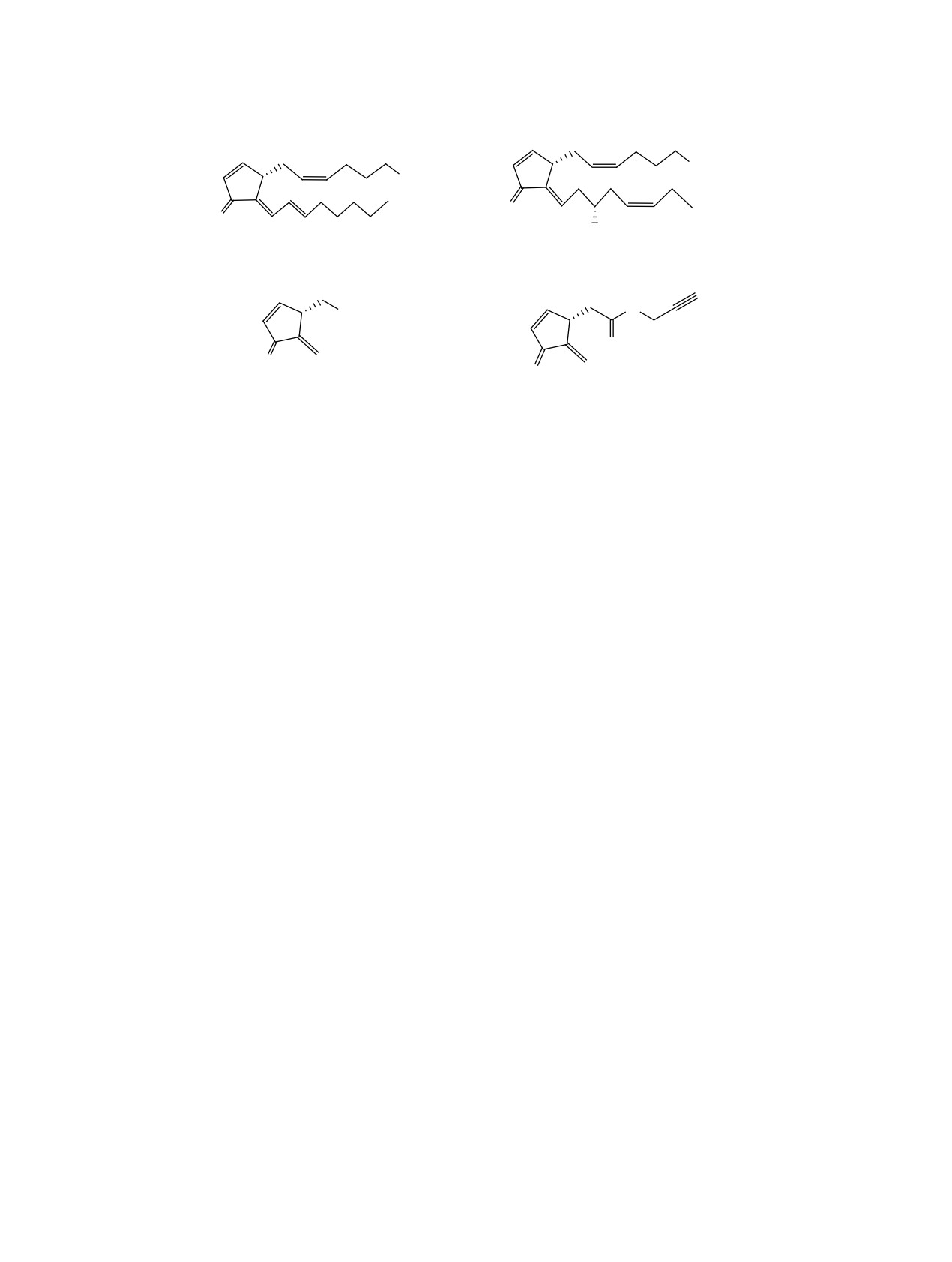
ВВЕДЕНИЕ
лиден циклопентенона [5]. Ранее мы предложили
более простого строения биоизостерный соеди-
Циклопентеноновые простагландины (CyPG),
нениям 1 и 2 экзометилиден циклопентеноны 3,
содержащие в своей структуре перекрестно сопря-
проявившие цитотоксические свойства
[6,
7].
женную с кетогруппой систему двойных связей,
Так, в некоторых тестах соединение (+)-3 имело
привлекают внимание как перспективный в ряду
сравнимую с соединением 2b активность, напри-
высокоактивных цитостатиков подкласс семей-
мер, для HEK293 в случае соединения (+)-3 IC50
ства простагландинов [1, 2]. Среди них примечате-
0.83 мM и для соединения 2b IC50 0.583 мM. На
лен 15-дезокси-Δ12,14-PGJ2 1, селективный лиганд
других испытанных линиях раковых клеток ци-
для PPARγ рецепторов ядра, ответственных за за-
тотоксичность соединения (+)-3 была в 2-5 раз
пуск апоптоза, воспалительных и других жизнен-
выше, чем рацемического соединения (±)-3. В дан-
но важных процессов (см. рисунок) [3]. Из этой
ной работе с целью поиска более активных струк-
серии отметим также Δ12-PGJ3 2а, известный как
тур было запланировано получение пропаргилово-
селективный ингибитор рака стволовых клеток,
го эфира 5 из соединения (±)-3. Выбор эфира 5 в
проявляющий активность в наномолярных кон-
качестве целевой структуры обоснован тем, что в
центрациях [4].
пропаргиловых эфирах кислот зачастую наблюда-
Как уже отмечалось, ответственным за био-
ется усиление цитотоксичности или, в случае от-
логическую активность фрагментом структуры в
сутствия этих свойств в родоначальных кислотах,
соединениях 1, 2 и родственных им соединениях
появление противораковых свойств в их пропарги-
является система кросс-сопряженного экзомети-
ловых эфирах [8, 9].
1207
1208
МАКАЕВ и др.
R
CO2
CO2H
O
O
OH
1
2a, b
R = H (a), CH3 (b).
O
CO2Me
O
O
O
ɢ )-3
)-5
Структуры CyPG 1, 2a, b и экзометилиденциклопентенонов 3 и 5
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
элиминированием из соединения 11 PhSO2H дей-
ствием DBU получили целевое соединение 5.
На пути к соединению 5 попытки прямой
транс-этерификации соединения 3, а также бо-
На пути к соединению 5 попытки прямой
лее мягкие варианты гидролиза сложноэфирной
транс-этерификации соединения 3, а также бо-
группы не увенчались успехом. Поэтому решено
лее мягкие варианты гидролиза сложноэфирной
было превратить диеноновую систему 3 в удобную
группы не увенчались успехом. Поэтому решено
для работы циклопентенольную. Для этого полу-
было превратить диеноновую систему 3 в удобную
ченное из соединения 3 фенилтиопроизводное 4
для работы циклопентенольную. Для этого полу-
восстановлением, обработкой NaBH4/CeCl3·7H2O
ченное из соединения 3 фенилтиопроизводное 4
по Люшу (Luche) [10], превратили в смесь эпимер-
восстановлением, обработкой NaBH4/CeCl3·7H2O
ных спиртов 6 и 7 и далее щелочным гидролизом
по Люшу (Luche) [10], превратили в смесь эпимер-
получили гидроксикислоту 8a, b (схема 1) в том же
ных спиртов 6 и 7 и далее щелочным гидролизом
эпимерном соотношении. На стадии восстановле-
получили гидроксикислоту 8a, b (схема 1) в том же
ния кетона 4 образуются 2 основных эпимерных
эпимерном соотношении. На стадии восстановле-
спирта 6 и 7 в соотношении 2:1 (подтверждено ме-
ния кетона 4 образуются 2 основных эпимерных
тодом спектроскопии ЯМР 1Н по интенсивности
спирта 6 и 7 в соотношении 2:1 (подтверждено ме-
сигналов 4-Н). Наблюдаемая стереоселективность
тодом спектроскопии ЯМР 1Н по интенсивности
восстановления в пользу α-эпимера 6 связана с
сигналов 4-Н). Наблюдаемая стереоселективность
тем, что атака BH4--аниона контролируется заме-
восстановления в пользу α-эпимера 6 связана с
стителем CH2SPh и осуществляется с противопо-
тем, что атака BH4--аниона контролируется заме-
ложной стороны. Заданный на этой стадии сте-
стителем CH2SPh и осуществляется с противопо-
реохимический состав (α-эпимер-β-эпимер, 2:1)
ложной стороны. Заданный на этой стадии сте-
практически сохраняется и в диастереомерах 8 и 9.
реохимический состав (α-эпимер-β-эпимер, 2:1)
В спектре ЯМР 13С изомера 6 сигналы С4 и С5
практически сохраняется и в диастереомерах 8 и
более сильнопольны из-за стерических факторов.
9. В спектре ЯМР 13С изомера 6 сигналы С4 и С5
Селективным алкилированием соединения 8 про-
более сильнопольны из-за стерических факторов.
паргилбромидом в кислотной части с выходом,
Селективным алкилированием соединения 8 про-
близким к количественному, был получен пропар-
паргилбромидом в кислотной части с выходом,
гиловый сложный эфир 9. Спирт 9 мягким окисле-
близким к количественному, был получен пропар-
нием реагентом Десса-Мартина трансформирова-
гиловый сложный эфир 9. Спирт 9 мягким окисле-
ли в енон 10. Селективное окисление последнего
нием реагентом Десса-Мартина трансформирова-
30%-ной H2O2 при катализе солями молибдена
ли в енон 10. Селективное окисление последнего
привело к кетосульфону 11 с выходом 90%. Затем
30%-ной H2O2 при катализе солями молибдена
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 58 № 11 2022
СИНТЕЗ ПРОПАРГИЛОВОГО ЭФИР
А
1209
Схема 1
CO2Me
CO2Me
CO2Me
i
ii
(±)-3
+
O
HO
HO
SPh
SPh
SPh
4
6
7
O
O
CO2H
iii
iv
v
6+7
O
O
HO
HO
O
SPh
SPh
SPh
8a, b
9
10
O
O
vi
vii
O
O
O
O
SO2Ph
11
5
Ɋɟɚɝɟɧɬɵ ɢ ɭɫɥɨɜɢɹ i 3K6+
ɷɤɜ (W31 ɋ+2Cl2
& ɱ
ii 1D%+4,
ɋH&O3Â +22 0H2+
& ɦɢɧ
iii /L2+ ɷɤɜ
7ȽɎ±+22
ɨɛɴɟɦɧɨɟ ɫɨɨɬɧɨɲɟɧɢɟ
& ɱ
iv %U&+2&Ł&+ ɞɢɢɡɨɩɪɨɩɢɥɷɬɢɥɚɦɢɧ ',3($
0H&1
& ɱ
v Ⱦɟɫɫɚ±Ɇɚɪɬɢɧɚ ɩɟɪɢɨɞɢɧɚɧ &+2Cl2
&
vi +2O2
ɜɨɞɧɵɣ
1+4) Mo O24,
0H2+
& ɱ
vii '%8 3K+
& ɱ
привело к кетосульфону 11 с выходом 90%. Затем
Метиловый эфир (1S*,5R*)-(4-оксо-5-фенил-
элиминированием из соединения 11 PhSO2H дей-
тиометилциклопент-2-ен-1-ил)уксусной кис-
ствием DBU получили целевое соединение 5.
лоты (4) получали согласно [7]. Выход 1350 мг
(75%), маслообразная жидкость, Rf 0.56 (петро-
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
лейный эфир-этилацетат, 7:3). К раствору 1033 мг
Растворители были очищены и высушены
(3.75 ммоль) соединения 3 и 0.62 мл (3.80 ммоль)
стандартными процедурами перед использо-
PhSH в 150 мл CH2Cl2 по каплям в течение 1 ч при-
ванием. Реагенты производства
«Alfa-Aestar»
бавляли 2.54 мл (0.18 ммоль) Et3N. По окончании
(США), «Sigma-Aldrich» (США), «Lancaster» (Ве-
реакции (контроль методом ТСХ), раствор упа-
ликобритания) были высшего качества и исполь-
ривали, остаток растворяли в EtOAc, промывали
зовались без дальнейшей очистки, если не указано
водой, объединенные органические слои сушили
иное. ИК спектры записывали на спектрофотоме-
над Na2SO4, отфильтровывали, упаривали, далее
тре IR Prestige-21 (Shimadzu, Япония) в тонком
очищали хроматографией на SiO2 (петролейный
слое или вазелиновом масле. Спектры ЯМР 1H
эфир-этилацетат, 20:1).
и 13С записаны на спектрометрах Bruker (США)
AM-300 [300 МГц (1Н)] и AM-500 [125 МГц (13С)]
Метиловый эфир
(1S*,4R*S*,5R*)-(4-гид-
для растворов веществ в CDCl3, (D3C)2CO, вну-
рокси-5-фенилтиометилциклопент-2-ен-1-ил)-
тренний стандарт - ТМС. Масс-спектры сняты в
уксусной кислоты (6+7, смесь эпимеров α:β = 2:1).
этаноле на спектрометре Shimadzu LCMS-2010 EV
К раствору кетосульфида 4 (116 мг, 0.42 ммоль) в
(Япония). Для ТСХ анализа применяли хромато-
15 мл MeOH при 0°C при перемешивании прибав-
графические пластины Sorbfil (Россия). Для ко-
ляли 157 мг (0.42 ммоль) CeCl3·7H2O, затем посте-
лоночной хроматографии применяли силикагель
пенно прибавляли 20 мг (0.42 ммоль) NaBH4, кон-
марки «Lancaster» (Великобритания).
тролируя ход реакции методом ТСХ. По окончании
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 58 № 11 2022
1210
МАКАЕВ и др.
реакции после стандартной обработки и очистки
1706, 1465, 1458, 1372, 1026, 736. Спектр ЯМР
на SiO2 получали 150 мг (95%) неразделимой на
1Н (CDCl3), δ, м.д.: 1.951 (1H, H5), 2.181 д.д (1Н,
SiO2 смеси диастереомерных спиртов 6 и 7 в соот-
CH2CO, J 15.7, 9.3 Гц), 2.38 д.д (1H, CH2CO, J
ношении 2:1. ИК спектр, ν, см-1: 3420, 3057, 2950,
16.0, 8.3 Гц), 2.60 д.д (1H, CH2CO2, J 6.5, 16.5 Гц),
2914, 1729, 1583, 1481, 1438, 1261. Масс-спектр,
2.651 д.д (1H, CH2CO, J 5.3, 11.7 Гц), 2.80-2.84 м
m/z (Iотн, %): 161 (100) [M + H - PhSH - H2O]+,
(1H, H1), 2.95-2.981 м (1H, H1), 3.15 д.д (1H, CH2S,
261 (30) [M + H - H2O]+. Найдено, %: С 64.65; H
J 8.3, 12.9 Гц), 3.19-3.231 д.д (1H, CH2S, J 8.2,
6.59; O 17.15; S 11.61. C15H18O3S. Вычислено, %: С
12.9 Гц), 3.25 д.д (1H, CH2S, J 6.7, 12.8 Гц), 3.301
64.72; H 6.52; О 17.24; S 11.52. M 278.36.
д.д (1H, CH2S, J 12.8, 9.0 Гц), 4.60 уш.с (1H, H4),
4.731 д (1H, H4, J 8.3 Гц), 5.73-5.77 м (1H), 5.81 д
Метиловый эфир {(1S*,4S*,5R*)-4-гидрокси-
(1H, H4, J 5.6 Гц), 5.861 д.д (1H, J 2.1, 5.5 Гц), 5.91
5-[(фенилтио)метил]циклопент-2-ен-1-ил}ук-
д.д (1H, CH=CH, J 1.4, 5.6 Гц), 7.15-7.16 м (1H),
сусной кислоты (6). Спектр ЯМР 1Н (CDCl3), δ,
7.16 т (1Н, Рh, J 7.4 Гц), 7.37-7.39 м (2Н, Рh), 7.30
м.д.: 1.92-2.30 м (2Н), 2.25 д.д (1Н, СH2CO, J 8.6,
т (2Н, Рh, J 7.8 Гц). Спектр ЯМР 13С (CDCl3), δ,
15.4 Гц), 2.54 д.д (1Н, CH2S, J 15.4, 6.0 Гц), 2.96-
м.д.: 31.601 (СН2S), 36.20 (CH2S), 37.971 (CH2CO),
3.02 м (1H, H5), 3.12-3.22 м (2H, CH2S), 3.60 S (3H,
39.54 (-CH2CO), 45.721 (C1), 46.31 (C1), 47.871
OMe), 4.72 д (1H, Н4, J 6.2 Гц), 5.90-5.92 м (1Н,
(C5), 52.66 (C5), 75.071 (C4), 81.04 (C4), 125.361
CH=), 5.95 д.д (1H, CH=, J 1.0, 6.6 Гц), 7.20-7.25
(C6H5, CH=CH), 125.43 (C6H5, CH=CH), 128.25
м (1H, Ph), 7.30-7.33 м (2H, Ph), 7.36 д (2H, Ph, J
(C6H5, CH=CH), 128.291 (C6H5, CH=CH), 128.831
7.7 Гц). Спектр ЯМР 13С (CDCl3), δ, м.д.: 32.52
(C6H5, CH=CH), 133.271 (C3), 133.67 (C3), 134.95
(CH2), 38.41 (CH2), 45.95 (CH), 47.33 (CH), 51.61
(C2), 137.37 (Cq, C6H5, CH=CH), 172.86 (CO2).
(OMe), 76.07 (C4), 129.00, 129.03, 132.50 (CH=),
Масс-спектр, m/z (Iотн, %): 247 (100) [M - H2O +
136.04 (Сq), 138.77 (C6H5, CH=CH), 172.70 (-CO2).
H]+, 263 (100) [M - H]-. Найдено, %: С 63.70; H
Метиловыйэфир {(1S*,4R*,5R*)-4-гидрокси-
6.01; O 18.16; S 12.13. C14H14O3S. Вычислено, %:
5-[(фенилтио)метил]циклопент-2-ен-1-ил}ук-
С 63.61; H 6.10; О 18.16; S 12.13. M 262.06.
суснойкислоты (7). Спектр ЯМР 1Н (CDCl3), δ,
Пропаргиловыйэфир (1S*,5R*)-{4-гидрокси-
м.д.: 1.92-2.03 м (2Н), 2.45 д.д (1H, CH2CO, J 8.8,
5-[(фенилсульфанил)метил]циклопент-2-ен-1-
16.0 Гц), 2.55 перекрывающиеся д.д (1Н, CH2S),
ил}уксусной кислоты (9). К раствору 50 мг
3.62 c (3H, OMe), 3.96-4.00 м (2H, CH2S), 4.60 д
(0.19 ммоль) кислоты 7 в 10 мл MeCN при пе-
(1Н, H4, J 3.6 Гц),5.70 т (2H, CH=CH, J 7.0 Гц),
ремешивании прибавляли 0.08 мл (0.76 ммоль)
7.20-7.23 м (2H), 7.20-7.25 м (1H, Ph), 7.30-7.33
пропаргила бромистого и затем DIPEA 0.07 мл
м (2H, Ph), 7.36 д (2H, Ph, J 7.7 Гц). Спектр ЯМР
(0.4 ммоль). По истечении 24 ч реакционную массу
13С (CDCl3), δ, м.д.: 37.50 (CH2), 39.38 (CH2), 47.66
упаривали, очищали остаток на SiO2 (петролейный
(CH), 52.74 (CH), 82.52 (C4), 126.21, 129.00, (CH,
эфир-этилацетат, 1:1). Выход 49 мг (85%), мас-
Ph), 129.24, 129.29, 133.08, 135.74 (CH=), 172.87 (-
лообразное соединение 9, Rf 0.6 (CHCl3-MeOH,
CO2).
20:1). ИК спектр, ν, см-1: 3419, 3289, 3057, 2916,
(1S*,4R*S*,5R*)-4-(Гидрокси-5-фенилтиоме-
2128, 1738, 1729, 1721, 1583, 1480, 1438, 1248,
тилциклопент-2-ен-1-ил)уксусная кислота 8а,
1147, 1025, 741, 691. Спектр ЯМР 1Н (ацетон-d),
b. К раствору 260 мг (0.09 ммоль) смеси соедине-
δ, м.д.: 1.95-2.2 м (1H, H5), 2.28 д.д (1H, CH2CO, J
ний 6 и 7 в водном ТГФ (1:1) прибавляли 119 мг
9.2, 15.6 Гц), 2.431 д.д (1Н, СН2СО, J 8.2, 15.4 Гц),
(0.27 ммоль) LiOH·H2O. Через 12 ч реакционную
2.651 д.д (1H, СН2СО, J 6.6, 15.8 Гц), 2.71 д.д (1Н,
массу нейтрализовали 1M раствором HCl, упари-
CH2CO, J 5.5, 15.6 Гц), 2.96-3.00 м (1H, H1), 3.0
вали и экстрагировали CH2Cl2 (3×10 мл), объеди-
к (1Н, С≡СН, J 2.5 Гц), 3.14 д.д (1Н, СН2S, J 6.3,
ненные органические слои сушили над Na2SO4,
12.9 Гц), 3.171 д.д (1H, CH2S, J 7.1, 12.8 Гц), 3.211
отфильтровывали, упаривали, остаток очища-
д.д (1H, СН2S, J 6.9, 12.8 Гц), 3.30 д.д (1Н, СН2S, J
ли на SiO2 (CHCl3-MeOH, 25:1). Выход 228 мг
8.9, 13.0 Гц), 3.65 д (1H, OH, J 6.7 Гц), 3.981 д (1Н,
(96%) диастереомерной смеси кислот 8a, b, т.пл.
121-122°C. ИК спектр, ν, см-1: 3312, 3047, 2954,
1 Значения сдвигов в спектрах ЯМР мажорного эпимера.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 58 № 11 2022
СИНТЕЗ ПРОПАРГИЛОВОГО ЭФИР
А
1211
OH, J 6.3 Гц),4.61 м (1Н, Н4), 4.70 д (2Н, OCH2, J
[М]+, 232 (100) [M + H + MeCN - PhSH]+, 191 (75)
2.4 Гц), 5.761 д.т (1Н, СН=СН, H2, J 5.5, 1.8 Гц),
[M + H - PhSH]+. Найдено, %: С 67.90; H 5.45; O
5.801 д (1Н, СН=СН, Н3, J 5.7 Гц), 5.87-5.88 м
16.00; S 10.66. C17H16O3S. Вычислено, %: С 67.98;
(1H, CH=CH, Н2), 5.89 д.д (1Н, СН=СН, Н3, J 5.9,
H 5.37; О 15.98; S 10.68. M 300.63.
1.0 Гц), 7.17 т (1Н, Hпара, Ph, J 7.3 Гц), 7.30 т (2Н,
Пропаргиловый эфир
(1S*,5R*)-{4-оксо-5-
Нмета, Ph, J 7.6 Гц), 7.38 д (2Н, Норто, Ph, J 7.9 Гц).
[(фенилсульфонил)метил]циклопент-2-ен-1-
Спектр ЯМР 13С (ацетон-d), δ, м.д.: 31.74 (CH2S),
ил}уксусной кислоты (11). К перемешиваемому
34.511 (CH2S), 38.08 (CH2CO), 39.731 (CH2CO),
раствору 22.4 мг (0.018 ммоль) (NH4)6Mo7O24·
45.83 (C1), 46.321 (C1), 47.83 (С5), 51.37 (CH2C≡),
4H2O в MeOH при 0°C по каплям прибавляли
52.721 (C5), 75.14 (≡CH), 75.421 (≡CH), 78.10 (≡C-),
0.42 мл 30%-ной H2O2. Полученную смесь пе-
81.041 (C4), 125.59 (C6H5, CH=CH), 125.771 (C6H5,
ремешивали еще 30 мин, после чего прибавляли
CH=CH), 128.44 (C6H5, CH=CH), 128.671 (C6H5,
56 мг (0.18 ммоль) сульфида 9. Через 1 ч темпера-
CH=CH), 128.92 (C6H5, CH=CH), 133.67 (=CH2,
туру поднимали до комнатной и реакционную
C2), 134.05 (=CH, C3), 134.63 (=CH, C3), 137.051
массу нейтрализовали добавлением 70 мг Na2SO3.
(Cq, Ph), 137.381 (Cq, Ph), 171.071 (CO2), 171.15
После обработки полученную сырую массу рас-
(CO2). Масс-спектр, m/z (Iотн, %): 285 (100) [M +
творяли в EtOAc, промывали водой (3×5 мл), ор-
H - H2O]+, 175 (65) [M + H - H2O - PhSH]+. Найдено,
ганический слой сушили над Na2SO4, упаривали.
%: С 67.46; H 6.06; O 15.95; S 10.52. C17H18O3S.
Выход 59 мг (90%). Rf 0.25 (петролейный эфир-
Вычислено, %: С 67.52; H 6.00; О 15.87; S 10.60.
этилацетат, 7:3). ИК спектр, ν, см-1: 3298, 3062,
M 302.23.
2926, 2129, 1707, 1701, 1590, 1447, 1306, 1151,
Пропаргиловый эфир
(1S*,5R*)-{4-оксо-5-
1085, 737, 527. Спектр ЯМР 1Н (ацетон-d), δ,
[(фенилсульфанил)метил]циклопент-2-ен-1-
м.д.: 2.60 д.т (1Н, Н5, J 10.6, 2.6 Гц), 2.73 д.д (1Н,
ил}уксусной кислоты (10). К раствору 17 мг
СН2СО, J 9.4, 16.5 Гц), 3.08 т (1Н, ≡СН, J 2.4 Гц),
(0.056 ммоль) спирта 8 в 10 мл CH2Cl2 при переме-
3.18 д.д (1Н, СН2СО, J 4.5, 16.6 Гц), 3.44 д.д (1Н,
шивании прибавляли 76 мг (0.179 ммоль) реаген-
СН2SO2, J 10.7, 14.3 Гц), 3.48 м (1Н, Н1), 3.60 д.д
та Десса-Мартина. Через 6 ч реакционную массу
(1Н, CH2SO2, J 2.4, 14.3 Гц), 4.65 д (2Н, OCH2C≡,
нейтрализовали добавлением 4.8 мл насыщенного
J 2.4 Гц), 6.20 д.д (1Н, Н3, J 5.8, 1.9 Гц), 7.70 т
раствора Na2S2O3. Органический слой отделяли,
(2Н, Hмета, Ph, J 7.9 Гц), 7.78 т (1Н, Hпара, Ph, J
сушили над Na2SO4, упаривали, остаток очищали
7.4 Гц), 7.84 д.д (1Н, Н2, J 5.8, 2.2 Гц), 8.0 д (2H,
на SiO2. Выход 33 мг (70%), Rf 0.6 (петролейный
Норто, Ph, J 8.7 Гц). Спектр ЯМР 13С (ацетон-d), δ,
эфир-этилацетат, 7:3). ИК спектр, ν, см-1: 3287,
м.д.: 37.09 (CH2CO), 44.87 (C5), 45.51 (C1), 52.50
3074, 3058, 2923, 2128, 1755, 1694, 1528, 1480,
(СН2О), 57.47 (СН2SO2), 76.51 (≡CH), 78.83 (-C≡),
1436, 1265, 1145, 1025, 741, 690. Спектр ЯМР 1Н
128.80 (C6H5, CH=CH), 130.43 (C6H5, CH=CH),
(CDCl3), δ, м.д.: 2.33 д.т (1Н, Н5, J 9.4, 3.1 Гц), 2.49
133.05 (C6H5, CH=CH), 134.86 (C3), 140.72 (Сq,
т (1Н, ≡СН, J 2.4 Гц), 2.50 д.д (1Н, СН2СО, J 8.9,
Ph), 166.99 (C2), 171.35 (CO2), 206.13 (CO). Масс-
16.5 Гц), 2.80 д.д (1Н, СН2СО, J 5.9, 16.1 Гц), 2.95
спектр, m/z (Iотн, %): 333 (100) [М + H]+, 232 (100)
д.д (1Н, CH2S, J 9.5, 13.3 Гц), 3.29-3.33 м (1H, H1),
[M + H + MeCN - PhSO2]+. Найдено, %: С 61.40; H
3.55 д.д (1Н, CH2S, J 3.8, 13.3 Гц), 4.70 д (2Н, СН2О,
4.90; O 24.10; S 9.60. C17H16O5S. Вычислено, %: С
J 2.4 Гц), 6.20 д.д (1Н, СН=СН, Н3, J 5.7, 1.9 Гц),
61.43; H 4.85; О 24.07; S 9.65. M 332.06.
7.20 т (1Н, Hпара, Ph, J 7.3 Гц), 7.28 т (2H, Hмета,
Ph, J 7.9 Гц), 7.38 д (2Н, Hорто, Ph, J 7.3 Гц), 7.68
Пропаргиловый эфир
(±)-(5-метилиден-4-
д.д (1Н, СН=СН, Н2, J 5.7, 2.3 Гц). Спектр ЯМР
оксоциклопент-2-ен-1-ил)уксусной кислоты (5).
13С (CDCl3), δ, м.д.: 34.86 (CH2S), 37.87 (CH2CO),
К перемешиваемому раствору 50 мг (0.15 ммоль)
43.77 (C5), 50.14 (C1), 52.22 (C1, CH2O), 75.24 (C2),
сульфона 10 в 10 мл PhH прибавляли 0.02 мл ДБУ
77.06 (C3', ≡CH), 126.57 (C6H5, CH=CH), 129.13
(0.15 ммоль) и через 1 ч массу упаривали, остаток
(C6H5, CH=CH), 129.59 (C6H5, CH=CH), 133.67
очищали на SiO2 (петролейный эфир-этилацетат,
(C3), 135.29 (Cq, Ph), 165.33 (C2), 170.38 (CO2),
10:1). Выход 12 мг (40%), Rf 0.4 (петролейный
207.87 (CO). Масс-спектр, m/z (Iотн, %): 300 (20)
эфир-этилацетат, 5:1). ИК спектр, ν, см-1: 3302,
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 58 № 11 2022
1212
МАКАЕВ и др.
3056, 2927, 2130, 1753, 1703, 1656, 1645, 1256,
Мифтахов Мансур Сагарьярович, ORCID:
1154, 737. Спектр ЯМР 1Н (ацетон-d), δ, м.д.:
2.68 д.д (1Н, CH2CO2, J 7.7, 16.5 Гц), 2.78 д.д (1Н,
КОНФЛИКТ ИНТЕРЕСОВ
СН2СО, J 6.8, 16.5 Гц), 3.08 т (1Н, ≡СН, J 2.5 Гц),
3.86-3.90 м (1Н, Н1), 4.75 д (2Н, OCH2C≡, J
Авторы заявляют об отсутствии конфликта ин-
2.3 Гц), 5.58 с (1Нa, =СH2), 5.97 c (1Hb, =CH2), 6.35
тересов.
д.д (1H, Н3, J 6.0, 1.8 Гц), 7.75 д.д (1Н, Н2, J 6.0,
СПИСОК ЛИТЕРАТУРЫ
1.6 Гц). Спектр ЯМР 13С (ацетон-d), δ, м.д.: 34.98
(CH2S), 37.01 (CH2CO), 40.49 (C1), 51.67 (CH2O),
1. Straus D.S., Glass C.K. Med. Res. Rev. 2001, 21,
75.62 (≡CH), 77.86 (≡C-), 116.01 (=CH2), 134.80
185-210. doi 10.1002/med.1006
(C3), 145.21 (C5), 161.25 (C2), 170.30 (CO2), 196.62
2. Loza V.V., Gimazetdinov А.М., Miftakhov M.S. Russ.
(CO). Масс-спектр, m/z (Iотн, %): 232 (100) [M + H +
J. Org. Chem. 2018, 54, 1575-1620. doi 10.1134/
MeCN]+, 191 (100) [М + H]+. Найдено, %: С 69.50;
S0514749218110018
H 5.26; O 25.24. C11H10O3. Вычислено, %: С 69.46;
3. Abbasi S., Kajimoto K., Harashima H. Int. Ed.
H 5.30; О 25.24. M 190.06.
Nanomed. 2016, 11, 2685. doi 10.2147/IJN.S106297
ЗАКЛЮЧЕНИЕ
4. Hegde S., Kaushal N., Ravindr K.C., Chiaro C.,
Синтезирован пропаргиловый эфир (±)-(5-ме-
Hafer K.T., Gandhi U.H., Thompson J.T., Van den
тилиден-4-оксоциклопент-2-ен-1-ил)ацетата
из
Heuvel J.P., Kennett M.J., Hankey P., Paulson R.F.,
соответствующего метилового эфира. Для стаби-
Prabhu K.S. Blood. 2011, 118, 6909. doi 10.1182/
лизации лабильной системы кросс-сопряжения
blood-2010-11-317750
вначале присоединением PhSH и восстановлением
5. Suzuki M., Mori M., Niwa N., Hirata T, Furuta K.,
кетогруппы трансофрмировали в пригодный для
Ishikawa T, Noyori R. J. Am. Chem. Soc. 1997, 119,
введения на стадию щелочного гидролизагидрок-
2376-2385. doi 10.1021/ja9628359
сиэфир. На финальном этапе полученную гидрок-
6. Vostrikov N.S., Spirikhin L.V., Lobov A.N., Gima-
сикислоту алкилировали пропаргилбромидом,
zetdinov A.M., Zileeva Z.R., Vakhitova Yu.V.,
последующим окислением и элиминированием
Macaev Z.R., Pivnitskyc K.K., Miftakhov M.S. Men-
сульфона получили целевой пропаргиловый эфир.
deleev Commun. 2019, 29, 372-374. doi 10.1016/
БЛАГОДАРНОСТИ
j.mencom.2019.07.003
В работе использовали оборудование ЦКП
7. Vostrikov N.S., Makaev Z.R., Zagitov V.V., Lakh-
«Химия» УфИХ РАН.
vich F.A. Pashkovsky F.S., Miftakhov M.S. Russ.
Chem. Bull. 2020, 69, 547-551. doi 10.1007/s11172-
ФОНДОВАЯ ПОДДЕРЖКА
020-2796-5
Работа выполнена по государственному за-
8. Siddiq A., Dembitsky V. Anti-Cancer Agents
данию № АААА-А17-117011910032-4 и АААА-
Med. Chem.
2008,
8,
132-170. doi
10.2174/
А17-117011910027-0.
187152008783497073
ИНФОРМАЦИЯ ОБ АВТОРАХ
9. Ott I., Kicher B., Dembinsky R. Just R. Expert
Макаев Зайнутдин Рамилевич, ORCID: https://
Opin. Ther. Pat.
2008,
18,
327-337. doi
orcid.org/0000-0002-0958-3164
10.1517/13543776.18.3.327
Востриков Николай Сергеевич, ORCID: https://
10. Luche J.L. J. Am. Chem. Soc. 1978, 100, 2226-2227.
orcid.org/0000-0003-1782-8675
doi 10.1021/ja00475a040
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 58 № 11 2022
СИНТЕЗ ПРОПАРГИЛОВОГО ЭФИР
А
1213
Synthesis of Propargyl Ether
(±)-(5-Methyliden-4-oxopent-2-en-1-yl) Acetic Acid
Z. R. Makaev*, N. S. Vostrikov, and M. S. Miftakhov
Ufa Institute of Chemistry is a separate structural subdivision of the
Ufa Federal Research Center of the Russian Academy of Sciences,
prosp. Oktyabrya, 71, Ufa, 450054 Russia
*e-mail: z.makaev.orgsynthesis@gmail.com
Received February 23, 2022; revised March 14, 2022; accepted March 22, 2022
The synthesis of (±)-(5-methylidene-4-oxocyclopent-2-en-1-yl) acetate propargyl ether from a methoxycarbonyl
precursor is described. The synthesis includes the stages of formation of the Michael adduct with thiophenol
at the terminal double bond, reduction of the keto group according to Luche, alkaline hydrolysis of the ester
function, alkylation of the acid with propargyl bromide to obtain carboxy-protected cyclopentenol. The latter is
converted into ketosulfone by oxidation with the Dess-Martin reagent and then by the action of 30% hydrogen
peroxide. Elimination of PhSO2H from ketosulfone results in the target molecule.
Keywords: cyclopentenone prostaglandins, propargyl ether
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 58 № 11 2022