ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2023, том 59, № 10, с. 1301-1318
ОБЗОРНАЯ СТАТЬЯ
УДК 547.7, 547.8, 544.43, 544.18
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ
МЕХАНИЗМОВ СБОРОК КАРБО- И ГЕТЕРОЦИКЛОВ
НА ОСНОВЕ РЕАКЦИЙ АЦЕТИЛЕНОВ
В СУПЕРОСНОВНЫХ СРЕДАХ1
© 2023 г. Н. М. Витковская*, В. Б. Орел, В. Б. Кобычев, А. С. Бобков
ФГБОУ ВО «Иркутский государственный университет», Россия,664003 Иркутск, ул. Карла Маркса, 1
*e-mail: vita@cc.isu.ru
Поступила в редакцию 27.07.2023 г.
После доработки 10.08.2023 г.
Принята к публикации 11.08.2023 г.
Значительное развитие химия ацетиленов получила в контексте применения суперосновных сред в
органическом синтезе. Изучение механизмов реакций требует применения комплекса химических, фи-
зико-химических и теоретических методов. В обзоре представлены результаты недавних квантовохими-
ческих исследований механизмов разрабатываемых в Иркутском институте химии им. А.Е. Фаворского
СО РАН сборок на базе реакций ацетилена и его производных, протекающих в суперосновных средах
и завершающихся образованием сложных глубоко функционализированных молекулярных структур.
Ключевые слова: ацетилены, диметилсульфоксид, суперосновные среды, каскадные сборки, механизмы
реакций, квантовохимические расчеты
DOI: 10.31857/S0514749223100038, EDN: OLXDHN
СОДЕРЖАНИЕ
ВВЕДЕНИЕ
1. МЕТОДЫ И ПОДХОДЫ
1.1. МОДЕЛИ ОПИСАНИЯ СУПЕРОСНОВНЫХ СРЕД
1.2. МЕТОДИКА РАСЧЕТА
2. РЕАКЦИИ ОБРАЗОВАНИЯ ПИРРОЛОВ И АННЕЛИРОВАННЫХ К НИМ ГЕТЕРОЦИКЛОВ
2.1. РЕАКЦИЯ ТРОФИМОВА
2.2. САМОСБОРКА N-ФЕНИЛ-2,5-ДИМЕТИЛПИРРОЛА ИЗ АНИЛИНА И АЦЕТИЛЕНА
2.3. ОБРАЗОВАНИЕ ПИРРОЛО[2,1-c][1,4]ОКСАЗИНОВ
3. РЕАКЦИИ КЕТОНОВ С АЦЕТИЛЕНАМИ
3.1. СБОРКИ 7-МЕТИЛЕН-6,8-ДИОКСАБИЦИКЛО[3.2.1]ОКТАНОВ И ЦИКЛОПЕНТЕНОЛОВ
3.2. СБОРКА ЗАМЕЩЕННЫХ ФУРАНОВ
3.3. СБОРКА Δ2-ИЗОКСАЗОЛИНОВ
4. РЕАКЦИИ ИМИНОВ С АЦЕТИЛЕНАМИ
1 Статья посвящается академику РАН Б.А. Трофимову в
связи с его 85-летием.
1301
1302
ВИТКОВСКАЯ и др.
4.1. ОБРАЗОВАНИЕ ИМИДАЗОПИРИДИНОВЫХ СИСТЕМ
4.2. ОБРАЗОВАНИЕ ПИРРОЛИНОВ
ЗАКЛЮЧЕНИЕ
СПИСОК ЛИТЕРАТУРЫ
ВВЕДЕНИЕ
Недавно под действием супероснований уда-
лось вовлечь в реакцию этинилирования азотистые
Уникальные свойства ацетилена и его произ-
аналоги кетонов - кетимины - с образованием тер-
водных обеспечивают многообразие реакций с их
минальных и интернальных пропаргиламинов
участием, предоставляя базу для разработки новых
(аза-реакция Фаворского) [7, 8]. Синтетический
реакций и новых путей синтеза сложных соедине-
потенциал реакций иминов с ацетиленами только
ний [1-3]. Одно из важных и активно развиваю-
начинает раскрываться, однако уже сегодня с их
щихся направлений в этой области - химия ацети-
помощью удаётся осуществлять в зависимости от
лена в суперосновных средах на основе суспензий
природы связи C=N однореакторные синтезы цен-
и растворов гидроксидов или алкоксидов щелоч-
ных 1- и 2-азадиенов [9, 10], 1-пирролинов, 1H- и
ных металлов в диметилсульфоксиде (DMSO) [4].
2H-пирролов [11, 12], имидазопиридинов [13] и др.
В этих системах наиболее отчетливо проявляет-
Экспериментальное изучение механизмов этих
ся двойственная природа ацетилена, способного
реакций затруднено вследствие их многостадий-
выступать как в качестве нуклеофила в реакциях
ности и высокой реакционной способности уча-
этинилирования, так и в качестве электрофильно-
ствующих в них соединений, что требует для по-
го субстрата в реакциях винилирования. Помимо
нимания их закономерностей проведения кванто-
этого, под действием сильных оснований алкины
вохимических расчетов высокого уровня с после-
способны подвергаться ацетилен-алленовой пере-
дующим детальным совместным анализом резуль-
группировке с образованием высокореакционных
татов расчета и экспериментальных данных. Здесь
алленовых структур, а также быстрой многопози-
будут представлены полученные в последние годы
ционной миграции тройной связи по углеводород-
результаты совместных исследований ИрИХ СО
ной цепи.
РАН и Лаборатории квантовой химии ИГУ.
Ярким примером проявления такой двойствен-
1. МЕТОДЫ И ПОДХОДЫ
ной природы ацетилена являются его реакции с
кетонами. Классическая реакция этинилирования
1.1. МОДЕЛИ ОПИСАНИЯ
кетонов с образованием пропаргиловых спир-
СУПЕРОСНОВНЫХ СРЕД
тов осуществляется легко, однако она обратима,
Одна из используемых в наших работах моде-
и при повышении температуры происходит дис-
лей сверхосновных сред исходит из представления
социация продуктов на исходные ацетилен и ке-
о том, что супероснование - это комплекс сильно-
тон. Оказалось, что при дальнейшем повышении
го ионизированного основания с лигандом, спец-
температуры в суперосновных средах становится
ифически взаимодействующим с катионом этого
возможным депротонирование кетона и его присо-
основания в среде, слабо сольватирующей анионы
единение к тройной связи с образованием β,γ-не-
(как правило, в среде полярного негидроксильного
насыщенных кетонов, которые далее могут пре-
растворителя) [14]. Это позволяет в первом при-
вращаться в α,β-ненасыщенные изомеры [5]. Эта
ближении рассматривать реакции с участием ани-
уникальная реакция даёт начало огромному мно-
онов без учета взаимодействия с сильно сольва-
гообразию сборок сложных молекулярных струк-
тированным катионом. Преимуществом такой
тур из нескольких молекул ацетилена и кетона
анионной модели является её компактность, что
(иногда с участием других нуклеофилов: гидрази-
позволяет использовать её для рассмотрения до-
на, гидроксиламина, гуанидина и т.п.), в которых
статочно больших систем. Она также может быть
ацетилен попеременно выступает в роли электро-
использована для оценки надежности выбранных
фила и нуклеофила [6].
приближений на примерах простейших реакций
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1303
исследуемых классов соединений путём сравне-
том числе переходных состояний, часто удовлет-
ния с результатами прецизионных подходов.
ворительно описываются уже на уровне газовой
фазы.
Слабая сольватация анионов связана с умень-
шением степени диссоциации солей и оснований в
В дальнейшем изложении мы будем использо-
DMSO. Так, согласно кондуктометрическим изме-
вать обозначения PENTA, MONO и ANION для
рениям, константа диссоциации t-BuOK в DMSO
этих трех моделей, добавляя к ним индексы, ука-
составляет лишь 3.7×10-3, и снижается до 10-6 при
зывающие на учет сольватационных эффектов на
переходе к t-BuONa; метоксиды щелочных метал-
стадии оптимизации, например, PENTAGAS или
лов диссоциированы в ещё меньшей степени [15];
MONOPCM.
для КОН K = 7.9×10-4 [16]. В связи с этим нами
1.2. МЕТОДИКА РАСЧЕТА
была предложена модель суперосновного центра,
включающая недиссоциированную молекулу ги-
В наших исследованиях главным образом
дроксида или алкоксида щелочного металла [17].
используется метод функционала плотности
Формирование ближайшего сольватного окруже-
B3LYP/6-31+G* [20, 21] для оптимизации геоме-
ния (специфическая сольватация), включающего в
трии и расчета колебательных поправок с уточ-
случае KOH и t-BuOK пять молекул растворителя,
нением энергий в рамках дабл-гибридного функ-
а в случае NaOH и t-BuONa - четыре, сопровожда-
ционала B2PLYP [22] с дисперсионной поправ-
ется значительным увеличением длины связи M-O
кой [23, 24] в расширенном базисе 6-311+G**.
(M = Na, K). Влияние оставшейся части раствори-
Комбинированный подход B2PLYP-D/6-311+G**//
теля (неспецифическая сольватация) учитывается
B3LYP/6-31+G* обеспечивает хорошее согласие
на уровне континуальной модели IEF PCM [18].
с данными прецизионных подходов CCSD(T)/6-
Рассмотрение в рамках такой пентасольватной (в
311+G**//CCSD/6-31+G* и CBS-Q//B3 для базо-
случае KOH и t-BuOK) модели типовых реакций
вых реакций ацетилена, этинилирования и вини-
этинилирования и винилирования показало, что
лирования, а также успешно справляется с такими
они осуществляются, как правило, на периферии
традиционно сложными для популярных подходов
реакционного комплекса, и это объясняет приме-
на основе DFT и MP2 задачами, как описание про-
нимость в ряде случаев простой анионной моде-
пин-алленовой перегруппировки [25-27] и аль-
ли. Стоит отметить, что реагирующая система при
дольной реакции [28].
этом сохраняет контакт с плотно окруженным мо-
Для учета сольватационных эффектов исполь-
лекулами растворителя катионом. Это позволяет,
зована модель IEFPCM [18] в сочетании с методом
в случае необходимости, учесть влияние катионов
B3LYP/6-31+G*, предоставляющая достаточно
различной природы.
сбалансированное описание нейтральных и ани-
По понятным причинам пентасольватная мо-
онных участников реакции. Кроме того, для кор-
дель оказывается достаточно ресурсоемкой, и для
ректного описания свободных энергий в растворе
случаев, когда учет катиона представляется необ-
были дополнительно учтены два фактора: измене-
ходимым, предложена моносольватная модель.
ние энтропии при переходе от идеального газа к
Она включает в явном виде одну молекулу раство-
1М раствору, которое составляет ΔS = R ln(1/22.4) =
рителя, и в сочетании с континуальной моделью
-25.86 Дж·моль-1·K-1, и изменение трансля-
PCM обеспечивает описание как специфических,
ционного и ротационного вкладов в энтропию.
так и неспецифических эффектов сольватации [17,
Последнее оценивалось на основании представле-
19], позволяя при этом экономить вычислительные
ний о том, что при переходе из газовой фазы в один
ресурсы. Особенностью моносольватной модели
и тот же растворитель различные вещества теряют
является необходимость учета сольватационных
одну и ту же долю энтропии [29, 30], это утверж-
эффектов уже на стадии оптимизации геометрии,
дение справедливо и для сольватации ионов [29].
тогда как для пентасольватной и анионной моде-
Следуя протоколу [31], мы в конечном итоге по-
лей геометрическое строение и частоты нормаль-
лучили для 1М раствора при температуре 300K
ных колебаний для всех стационарных структур, в
выражение Ssol = 0.74·Sharm - 3.2 кал·моль-1·K-1
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1304
ВИТКОВСКАЯ и др.
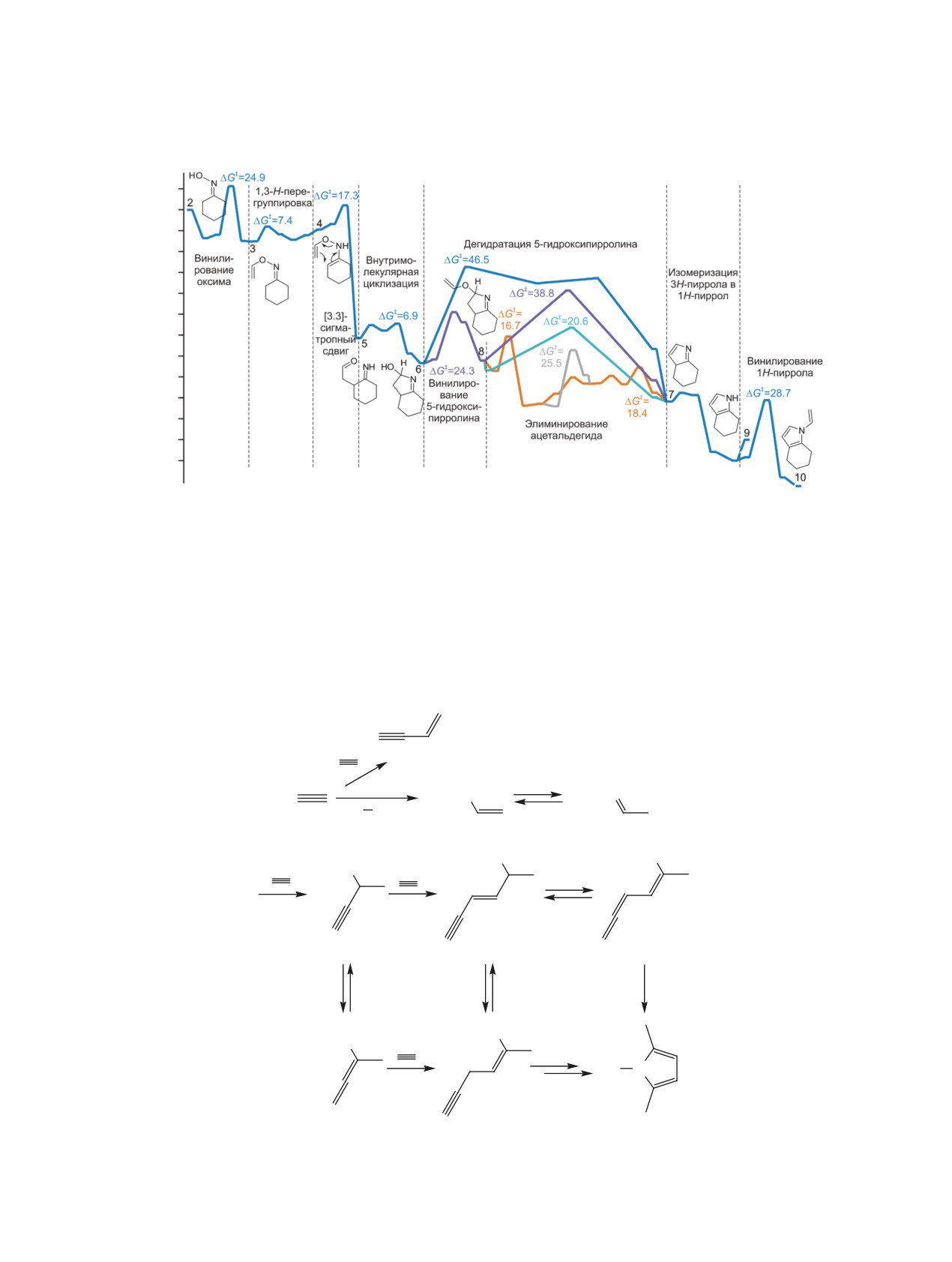
[32]. Отметим, что такой учет влияния раствори-
вание с формированием 5-винилоксипирролина 8,
теля в сочетании с расчетом свободных энергий
образование 3H-пиррола 7 путём элиминирова-
в газовой фазе на уровне B2PLYP-D/6-311+G**//
ния молекулы ацетальдегида при содействии ги-
B3LYP/6-31+G* обеспечивает оценку C-H, N-H,
дроксид-иона, перегруппировка в 1H-пиррол 9.
O-H и S-H кислотностей со средним отклонени-
Наиболее высокобарьерными оказываются ста-
ем, не превышающим двух единиц pKa (в данном
дии O-винилирования кетоксима (2→3, ΔG‡ =
случае для модели IEF PCM использовался мас-
24.9 ккал/моль) и 5-гидроксипирролина (6→8,
штабирующий множитель α = 1.35 [33]).
ΔG‡ = 24.3 ккал/моль). Показана неустойчивость
винилоксиамина 4 и иминоальдегида 5 относи-
Расчёты были проведены с использованием па-
тельно предшествующих и последующих интер-
кетов квантовохимических программ Gaussian 09
медиатов, что объясняет их отсутствие среди экс-
[34], Gaussian 16 [35] и GAMESS [36].
периментально выделенных.
2. РЕАКЦИИ ОБРАЗОВАНИЯ ПИРРОЛОВ И
Предложено и изучено несколько вариантов пе-
АННЕЛИРОВАННЫХ К НИМ ГЕТЕРОЦИКЛОВ
рехода 5-гидроксипирролина в 3H-пиррол. Уста-
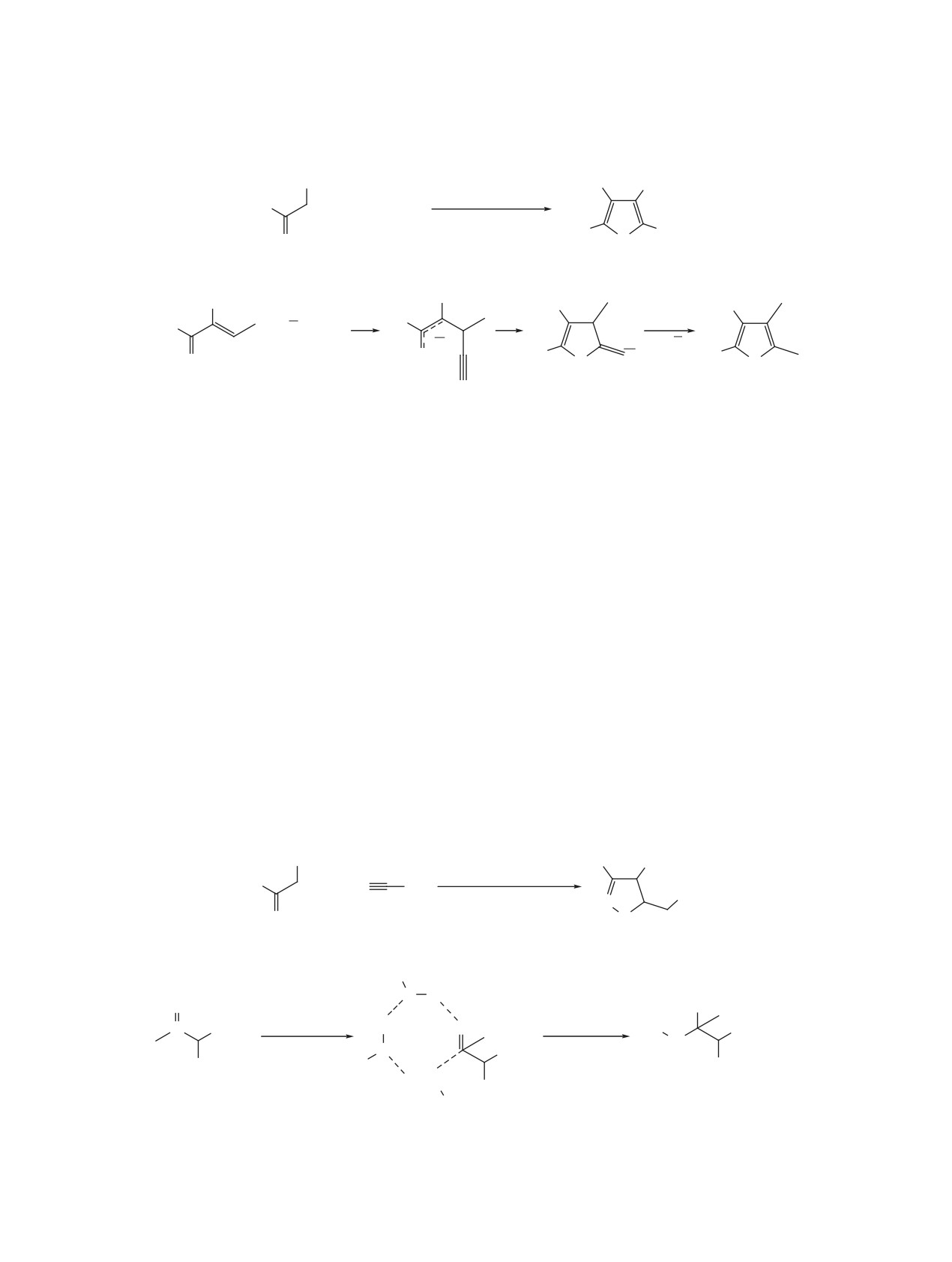
2.1. РЕАКЦИЯ ТРОФИМОВА
новлено, что дегидратация 5-гидроксипирролина
с образованием 3H-пиррола (6→7) невозможна в
Один из классических примеров сборки ге-
условиях эксперимента вследствие высокого ак-
тероциклических соединений на базе ацетилена
тивационного барьера (ΔG‡ = 46.5 ккал/моль). В
в суперосновных средах - реакция Трофимова.
этом ключе, одним из основных интермедиатов
Взаимодействие легкодоступных из кетонов и ги-
реакции становится 5-винилоксипирролин 8, кото-
дроксиламина кетоксимов с ацетиленом обеспечи-
рый позволяет миновать высокобарьерную стадию
вает простой и эффективный путь к 2,3-замещен-
дегидратации 5-гидроксипирролина и провести
ным 1H-пирролам [37]. Механизм, предложенный
реакцию энергетически более выгодными марш-
экспериментаторами, был доказан выделением
рутами через его винилирование и последующее
интермедиатов, однако некоторые из постулиро-
элиминирование молекулы ацетальдегида (ΔG‡
ванных промежуточных соединений, такие как
~ 18.5-20.5 ккал/моль). Последующее винили-
винилоксиамин и иминоальдегид до сих пор экс-
рование 4,5,6,7-тетрагидро-1H-индола (в случае
периментально зафиксированы не были. Хотя дан-
избытка ацетилена) осуществляется с большим
ная реакция открыта более 40 лет назад, она при-
активационным барьером, чем лимитирующая
влекает внимание исследователей и по сей день.
стадия его образования, что качественно согла-
Изучению механизма реакции Трофимова посвя-
суется с кинетическими исследованиями
[44].
щен ряд теоретических работ [38-42], однако все
Проведенное теоретическое исследование позво-
они описывали лишь отдельные стадии, которые
лило значительно дополнить данные о механизме
не позволяли сформировать целостную картину
реакции Трофимова.
взаимодействий. Мы впервые на едином теоре-
тическом уровне (B2PLYP/6-311+G**//B3LYP/6-
2.2. САМОСБОРКА N-ФЕНИЛ-
31+G*) с использованием модели ANIONPCM из-
2,5-ДИМЕТИЛПИРРОЛА ИЗ АНИЛИНА
учили все стадии реакции Трофимова на примере
И АЦЕТИЛЕНА
сборки 4,5,6,7-тетрагидро-1H-индола из ацетиле-
Недавно открытая однореакторная самосбор-
на 1 и оксима циклогексанона 2, а также его после-
ка N-фенил-2,5-диметилпиррола из ацетилена и
дующее винилирование ацетиленом [43] (рис. 1).
анилина в присутствии супероснования KOH/
Продемонстрировано, что наиболее пред-
DMSO [45] изучена методами квантовой хи-
почтительный путь реализуется через следую-
мии (B2PLYP(D3)/6-311+G**//B3LYP/6-31+G* +
щий каскад превращений: O-винилирование ке-
IEFPCM) с использованием моделей PENTAGAS
токсима
2,
1,3-прототропная перегруппировка
и MONOPCM [46]. Было предложено два вариан-
O-винилоксима 3 в винилоксиамин 4, [3,3]-сигма-
та механизма данной реакции: в одном случае ре-
тропный сдвиг с образованием иминоальдегида 5,
акция запускается присоединением ацетилена 1 к
циклизация в 5-гидроксипирролин 6, O-винилиро-
ацетилену 1 с формированием винилацетилена 10,
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1305
ΔG, ккал/моль
10
0
-10
-20
-30
-40
-50
-60
-70
-80
-90
-100
-110
-120
Рис. 1. Энергетический профиль реакции Трофимова и N-винилирования пиррола с основными стадиями и интермедиатами
в другом - присоединением анилина 11 к ацетиле-
Близкая величина получена и с использованием
ну 1 с образованием N-виниланилина 12 (схема 1).
модели MONOPCM (ΔΔG‡ = 3.6 ккал/моль. С боль-
шей вероятностью реализуется рассмотренный
Моделирование присоединения аниона ани-
далее механизм через N-виниланилин. Все даль-
лина и аниона ацетилена к ацетилену с исполь-
нейшие стадии рассмотрены в модели MONOPCM.
зованием модели PENTAGAS (рис. 2) показало
значительную кинетическую предпочтительность
За стадией винилирования анилина ацетиленом
N-винилирования (ΔΔG‡ = 5.1 ккал/моль).
следует енамин-иминная изомеризация (12→13)
Схема 1. Основные стадии и интермедиаты самосборки N-фенил-2,5-диметилпиррола
10
Ph NH
Ph N
Ph
NH2
1
12
13
11
Ph NH
Ph NH
Ph NH
14
16
18
Ph NH
Ph NH
Ph
N
15
17
19
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1306
ВИТКОВСКАЯ и др.
превращаться как в N-гекс-2-ен-5-ин-2-ил-анилин
17, так и в N-гекса-2,4,5-триен-2-ил-анилин 18.
На заключительной стадии циклизации, наобо-
рот, кинетически более благоприятным (ΔΔG‡ =
9.4 ккал/моль) по сравнению с пропаргильным
2.135 Å
(17→19)оказывается алленильныйпуть(18→19)об-
разования N-фенил-2,5-диметилпиррола 19 с реге-
нерацией суперосновного комплекса KOH·DMSO.
Весь путь каскадной самосборки N-фенил-
2,5-диметилпиррола в случае KOH/DMSO имеет
два близких лимитирующих барьера: винилиро-
вание анилина (1+11→12, ΔG‡ = 22.6 ккал/моль)
и этинилирование промежуточного пропаргил-
1+11→12
TS
амина (14+1→16, ΔG‡ = 22.8 ккал/моль). Рас-
ΔG‡ = 21.2 ккал//моль
K+∙PhNH-∙∙∙HCCH∙H2O∙5DMSO
считанные маршруты образования N-фенил-2,5-
диметилпиррола показывают способность ацети-
лена реагировать в рамках одной сборки как элек-
трофил и как нуклеофил, а также объясняют от-
сутствие промежуточных продуктов в каскадной
сборке.
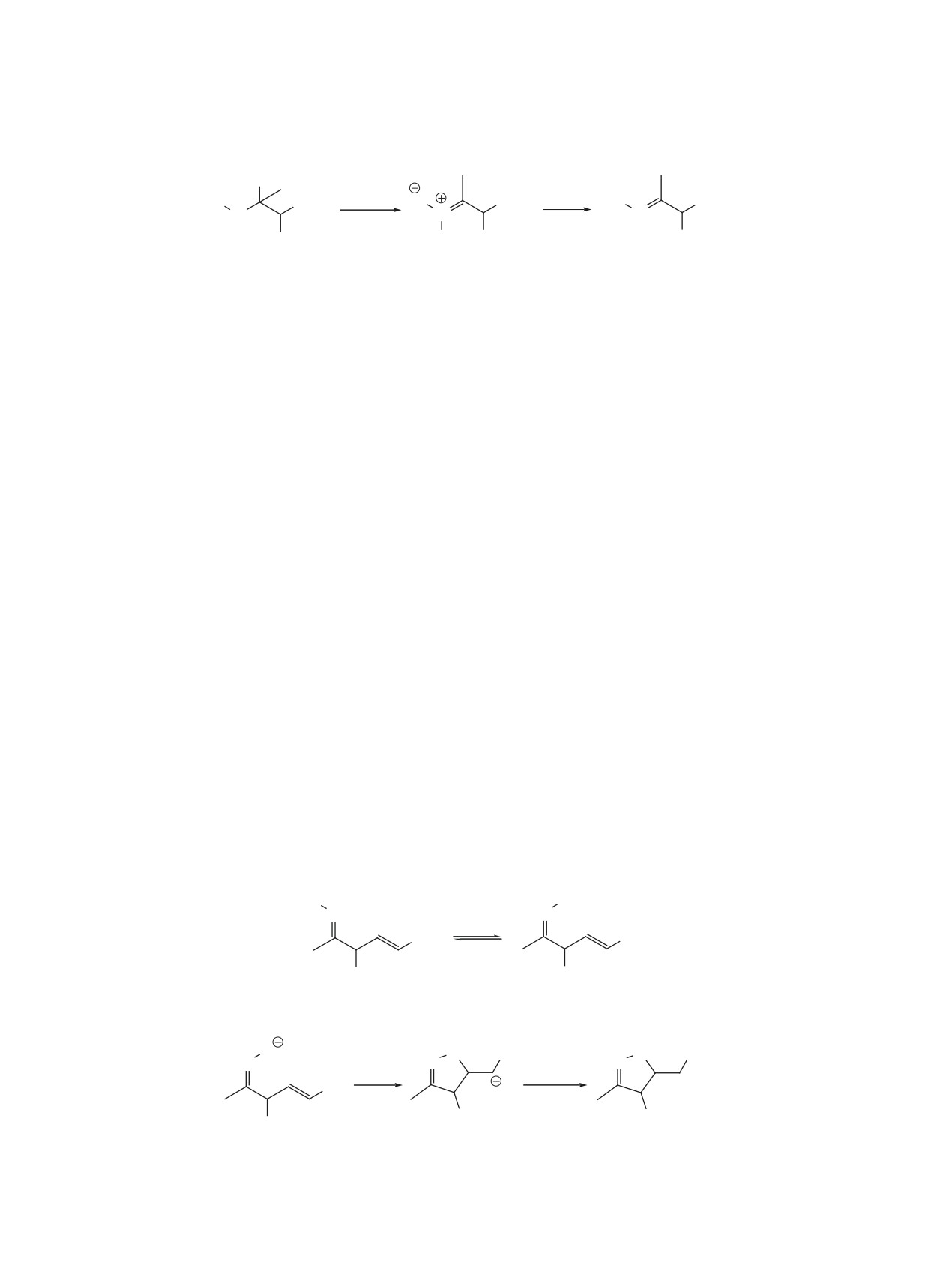
2.3. ОБРАЗОВАНИЕ ПИРРОЛО[2,1-c][1,4]-
ОКСАЗИНОВ
Перспективным направлением органического
синтеза в области создания аннелированных к пир-
ролу гетероциклов является введение в структуру
2.089 Å
1H-пирролов алленильной группы в положение 1
и альдегидной в положение 2. Восстановлением
1H-пиррол-2-илкарбальдегида был получен 1H-
TS1+1→10
пиррол-2-илметанол 20, обладающий сразу дву-
ΔG‡ = 26.3 ккал//моль
мя потенциальными нуклеофильными центрами:
O∙5DMSO
K+∙HCC-∙∙∙HCCH∙H2
атом кислорода спиртовой группы и атом азота
пиррольного цикла. Его взаимодействие с про-
Рис. 2. Структуры переходных состояний присоедине-
ния анионов анилина и ацетилена к ацетилену в при-
паргилхлоридом 21 может приводить к широкому
сутствии суперосновного комплекса KOH·5DMSO
многообразию реакций и продуктов за счет введе-
и этинилирование альдимина 13 по связи C=N
ния пропаргильной группы в структуру пиррола
(аза-реакция Фаворского). Полученный N-бут-3-
(схема 2, 20+21→22). Пропаргильная группа в ус-
ин-2-ил-анилин 14 может напрямую присоединять
ловиях суперосновных систем может легко изоме-
еще одну молекулу ацетилена по тройной связи
ризоваться в алленильную (22→23). Затем каждая
(пропаргильный путь) или может подвергаться
из этих форм может участвовать в циклизации за
пропин-алленовой перегруппировке в N-бут-2,3-
счет присоединения O-нуклеофила к терминаль-
диен-2-иланилин 15, который затем этинилирует-
ному (22→25, 23→27) атому углерода с формиро-
ся (алленильный путь). На стадии этинилирова-
ванием семичленного оксазепанового цикла или к
ния с участием 14 или 15 кинетически выгодным
интернальному (22→24, 23→26) с образованием
оказался маршрут через пропаргильную структу-
шестичленного оксазинового цикла. Кроме того,
ру (14→16). Полученный N-гекс-3-ен-5-ин-2-ил-
изучена возможность образования дизамещённых
анилин 16 может через 1,3-прототропный сдвиг
продуктов (22→28, 23→29).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1307
Схема 2. Изученные взаимодействия 1H-пиррол-2-илметанола и пропаргилхлорида с различными заместителями
в пиррольном цикле и при С≡С связи: a) R1 = H, R2 = H; b) R1 = Ph, R2 = H; c) R1 = H, R2 = Me
Cl
R1
+
N
R2
H
OH
20
21
R1
N
Cl
O
R2
R1
R1
R2
N
R2
N
24
OH
O
R2
R2
R1
N
22
28
O
R2
25
R1
Cl
N
O
R2
1
R1
R
N
N
R2
R2
OH
O
26
R2
R2
R1
N
23
29
O
R2
27
Совместное
теоретическое
(ANIONGAS,
наиболее вероятна изомеризация пропаргильной
CBS-Q//B3) и экспериментальное исследование
группы в алленильную (ΔG‡ = 11.0 ккал/моль) с
позволило ответить на вопрос о наиболее веро-
последующей развилкой. N-алленил-1H-пиррол-
ятных путях химических превращений 20a и 21a
2-илметанол 23a может как циклизоваться с об-
[47]. Показано, что в суперосновной среде на пер-
разованием 3-Me-1H-пирроло[2,1-с][1,4]оксазина
вой стадии образуется исключительно N-анион, а
26a (ΔG‡ = 13.2 ккал/моль), так и атаковать вторую
депротонирование спиртовой группы не осущест-
молекулу пропарилхлорида с образованием диза-
вляется. Это объясняется большей кислотностью
мещенного продукта 29a (ΔG‡ = 12.0 ккал/моль).
1H-пиррола в DMSO по сравнению с метанолом.
Близкие активационные барьеры обеспечивают
Нуклеофильное замещение хлора пирролид-ио-
вероятность образования в реакционной смеси
ном с формированием N-пропаргил-1H-пиррол-
сразу нескольких продуктов. Экспериментально
2-илметанола 22a оказывается скорость-опреде-
коллегами из ИрИХ СО РАН показано, что реакция
ляющей стадией реакции (ΔG‡ = 21.5 ккал/моль).
управляется, и можно с высокой селективностью
Анализ дальнейших превращений показал, что
получить каждый из трёх продуктов: 23a, 26a, 29a.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1308
ВИТКОВСКАЯ и др.
В рамках дальнейшего комплексного исследо-
дели ANIONGAS на примере систем ацетофенон/
вания данной реакции изучено влияние заместите-
ацетилен (a) [17, 51] и циклогексанон/ацетилен (b)
лей на энергетические характеристики отдельных
[52]. В первой системе диоксабицикло[3.2.1]окта-
стадий [48]. Введение фенильного заместителя
ны образуются диастереоселективно (в структуре
в пятое положение пиррольного цикла (20b) на
продукта может присутствовать до пяти асимме-
1.3 ккал/моль снижает активационный барьер
трических центров), а во второй диастереоселек-
циклизации (ΔG‡
=
11.7 ккал/моль) в целе-
тивность нарушается - образуется смесь (обыч-
вой пирролооксазин 26b, повышая при этом на
но трех) изомеров диоксабицикло[3.2.1]октанов
1.0 ккал/моль барьер конкурирующего процесса
(тетрациклических фронталинов) с преобладани-
образования дизамещенного продукта 29b (ΔG‡ =
ем одного. Сборка диоксабицикло[3.2.1]октанов
13.0 ккал/моль). Эксперимент подтвердил, что ис-
имеет каскадный характер и запускается реакцией
пользование арилзамещенных пирролов приводит
С-винилирования кетонов ацетиленом 1 (присо-
к селективному образованию 26.
единением карбанионов кетонов 30 к ацетилену)
Метильная группа при тройной связи пропар-
с образованием β,γ-ненасыщенных кетонов 31,
гилхлорида (21с) приводит не только к повыше-
которые далее под действием гидроксид-иона без
нию активационных барьеров ключевых стадий
активационного барьера претерпевают 1,3-прото-
реакции на 2-5 ккал/моль, но и к практически пол-
тропную перегруппировку в α,β-ненасыщенные
ной потере селективности за счёт выравнивания
кетоны 33, через диенолят-ионы 32 (схема 4). Эта
активационных барьеров образования из аллено-
стадия является самой высокобарьерной и при-
вой формы 23c оксазинового 26c и оксазепанового
водит к существенному понижению свободной
27c циклов. Стоит отметить, что в литературе нами
энергии (для системы a ΔG‡ = 22.5 ккал/моль,
не было найдено примеров образования пирроло-
ΔG = -24.6 ккал/моль, для системы b ΔG‡ =
оксазинов из структур с алкильным заместителем
18.7 ккал/моль, ΔG = -29.1 ккал/моль).
при кратной связи [49, 50]. Мы показали, что полу-
На последующих стадиях (рис. 3): присоеди-
чение таких структур указанным выше способом
нение второго карбаниона кетона 30 по β-атому
малоперспективно из-за падения селективности
углерода 33 с образованием 1,5-дикетона 34 (ре-
реакции.
акция Михаэля), присоединение этинид-иона 35
3. РЕАКЦИИ КЕТОНОВ С АЦЕТИЛЕНАМИ
по карбонильной группе дикетона 34 с образова-
нием аниона полукеталя 36 (реакция этинилиро-
3.1. СБОРКИ 7-МЕТИЛЕН-6,8-ДИОКСАБИЦИК-
вания), внутримолекулярное O-винилирование в
ЛО[3.2.1]ОКТАНОВ И ЦИКЛОПЕНТЕНОЛОВ
анионе 36 с образованием целевого диоксабици-
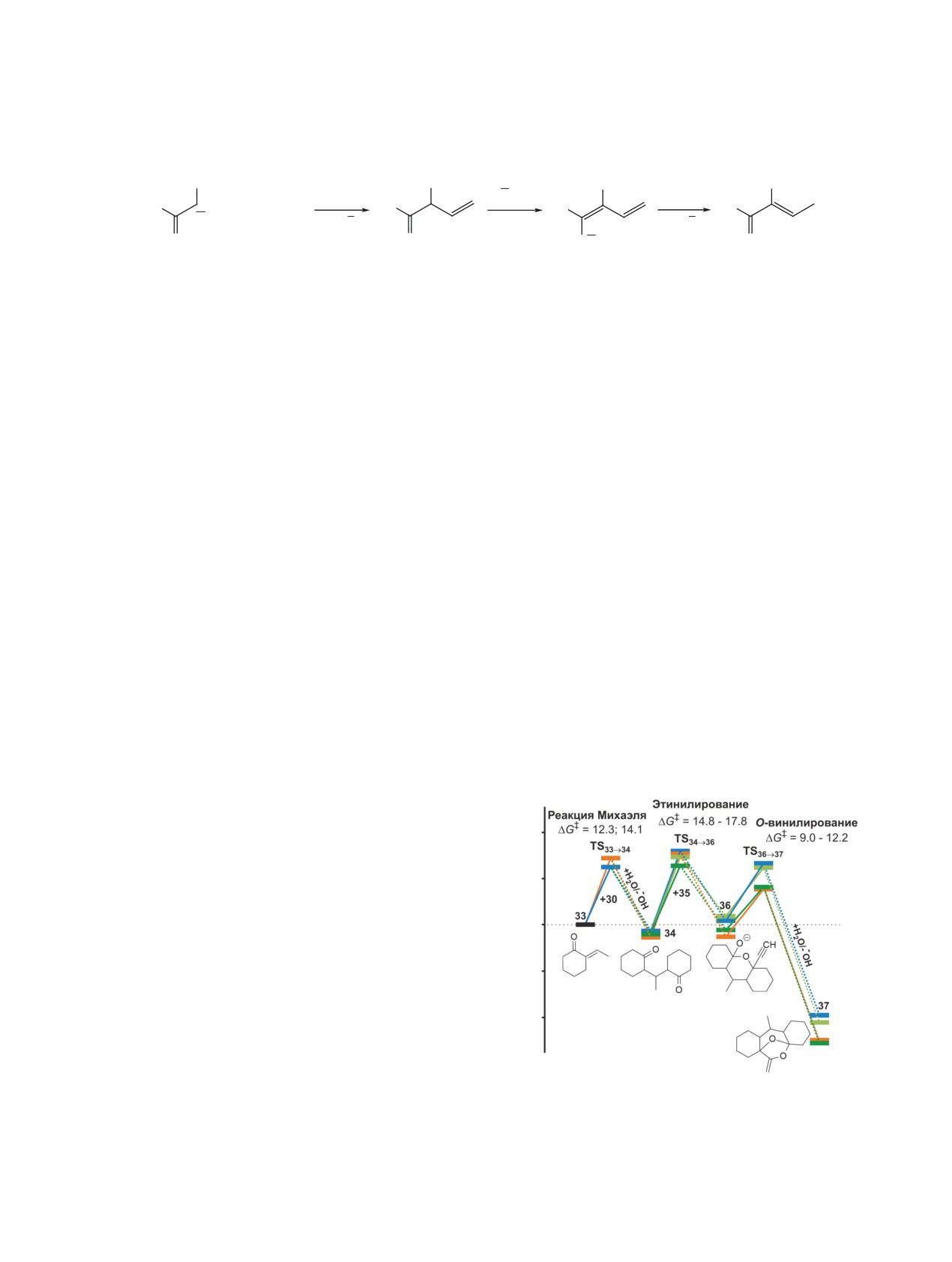
Сборка 7-метилен-6,8-диоксабицикло[3.2.1]ок-
кло[3.2.1]октана 37 - возникают оптические ак-
танов осуществляется в результате катализиру-
тивные центры. При этом диастереомерный со-
емого основанием KOH/DMSO взаимодействия
став диоксабицикло[3.2.1]октанов, образованных
двух молекул ацетилена и двух молекул кетона
из алкиларилкетонов и ацетилена определяется
(схема 3) [6].
на завершающей стадии внутримолекулярного
Нами было проведено квантовохимическое мо-
O-винилирования (TS36a→37a), а эксперименталь-
делирование механизма этой реакции в рамках мо-
но наблюдаемая диастереоселективность обеспе-
Схема 3. Образование 7-метилен-6,8-диоксабицикло[3.2.1]октанов из ацетилена и кетонов
R1
R2
R2
KOH/DMSO
R1
+ HC CH
O Me
70-80°C, 0.5-1 ч
O
O
R2
R1
R1 = Alk, (Het)Ar; R2 = H, Alk.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1309
Схема 4. Реакция С-винилирования кетонов ацетиленом с образованием α,β-ненасыщенных кетонов
R2
R2
R2
R2
H2O
OH
H2O
R1
R1
R1
R1
+ HC CH
-OH
-H2O
−OH
O
O
O
O
30
1
31
32
33
чивается за счет существенных различий в вели-
чески выгоднее оказывается сборка диоксабици-
чинах активационных барьеров этой стадии для
клооктанов, и это согласуется с его 86% выходом.
различных диастереомеров.
Для R = Thiophenyl [система тиенилметилкетон/
ацетилен (с)] соотношение барьеров становится
В случае сборки тетрациклических фронтали-
противоположным: 19.1 и 17.8 ккал/моль, и, со-
нов диастереомерный состав определяется уже на
ответственно, экспериментальные выходы диок-
стадии этинилирования промежуточного 1,5-дике-
сабициклооктана и циклопентенола меняются и
тона 34b (TS34b→36b). Причем наблюдаемое нару-
составляют 34 и 55%.
шение диастереоселективности сборки происхо-
дит из-за сопоставимых активационных барьеров
Показано, что диастереоселективность сборки
и скоростей этой реакции для разных диастерео-
циклопентенолов обеспечивается на завершающей
меров (рис. 3).
стадии внутримолекулярного С-винилирования за
счет различий (ΔΔG‡ = 1.6 ккал/моль) в величинах
Наряду с диоксабицикло[3.2.1]октанами в ре-
активационных барьеров образования различных
акции метиларилкетонов с ацетиленами могут ди-
диастереомеров.
астереоселективно образовываться циклопентено-
лы (схема 5) [6].
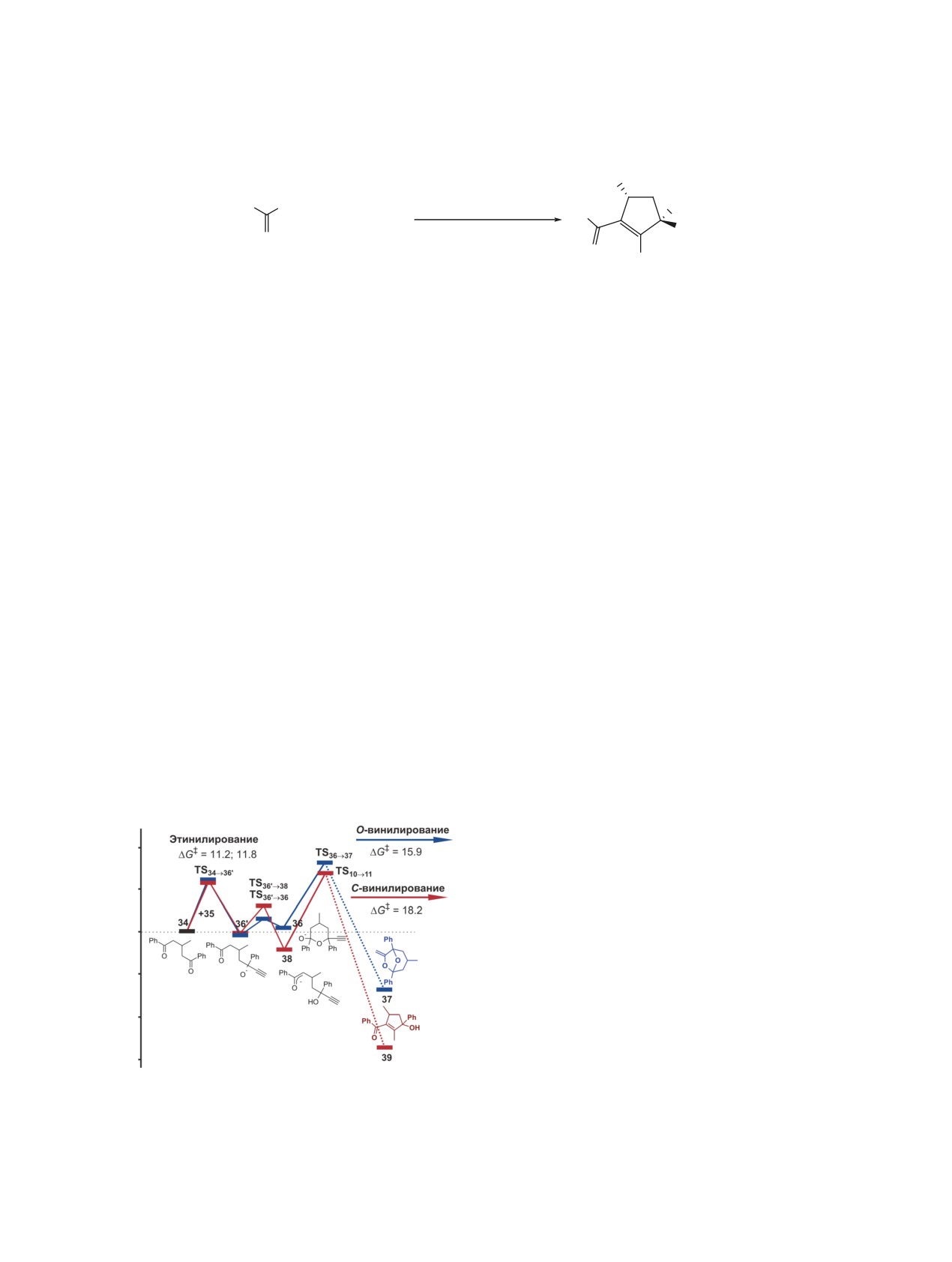
3.2. СБОРКА ЗАМЕЩЕННЫХ ФУРАНОВ
Ключевыми интермедиатами этой каскадной
Кетоны с объемными заместителями при кар-
сборки являются 1,5-дикетоны 34, присоедине-
бонильной группе R1 или при α-атоме углерода
ние этинид-иона 35 по карбонильной группе ко-
R2 в реакции с ацетиленом превращаются по дру-
торых приводит к структурному изомеру аниона
гому пути, с образованием замещенных фуранов
полукеталя 36 - δ-ацетиленовому енолят-иону 38
(схема 6) [6].
(рис. 4). В енолят-ионе 38 осуществляется вну-
Точка ветвления здесь возникает в результате
тримолекулярное C-винилирование с образова-
конкуренции этинид-аниона 35 и карбаниона кето-
нием метилиденциклопентанола, который после
1,3-прототропной перегруппировки превращается
ΔG, ккал/моль
в циклопентенол 39.
20
В рамках модели ANIONGAS мы показа-
ли [17, 51], что δ-ацетиленовый енолят-ион 38a
10
термодинамически выгоднее аниона полуке-
таля 36a и относительно легко образуется из
O-центрированного аниона третичного ацетилено-
0
вого спирта (предшественника 36a). Соотношение
диоксабицикло[3.2.1]октанов и циклопентенолов
-10
в смеси продуктов каскадной сборки алкиларилке-
тонов с ацетиленом определяется различиями в ак-
-20
тивационных барьерах последних стадий их сбор-
ки - реакций внутримолекулярного O- (TS36a→37a)
и С-винилирования (TS38a→39a), соответствен-
Рис. 3. Реакционный профиль образования четырех
но (рис. 4). При R = Ph (система a) барьеры ак-
(из восьми возможных) диастереомеров тетрацикли-
тивации этих стадий сборок составляют ΔG‡ =
ческих фронталинов для системы циклогексанон/аце-
15.9 ккал/моль и ΔG‡ = 18.2 ккал/моль, т.е. кинети-
тилен (b)
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1310
ВИТКОВСКАЯ и др.
Схема 5. Образование циклопентенолов из ацетилена и кетонов
Me
R Me
6.5 mol % KOH/DMSO
R
R
+ HC CH
70°C, 8 ч
OH
O
O
Me
R = (Het)Ar.
на 30 в реакции присоединения к активированной
соединения карбаниона кетона на 1.7 ккал/моль
C=C связи α,β-ненасыщенного кетона 33 (реакция
ниже, чем при присоединении этинид-иона, и
Михаэля). Присоединение этинид-аниона 35 при-
реакция Михаэля дает промежуточный 1,5-ди-
водит к образованию β-ацетиленового енолят-и-
кетон 34. Напротив, когда R1 = Mes, R2 = H (d),
она 40, в котором далее осуществляется внутри-
присоединение этинид-иона связано с меньшим
молекулярное O-винилирование с образованием
на 3.1 ккал/моль барьером, чем присоединение
метилиденового карбаниона дигидрофурана 41 и,
карбаниона кетона [17, 53]. Было показано, что
в результате прототропных перегруппировок, це-
предпочтительность присоединения карбанионов
левого замещенного фурана 42 (схема 7).
кетонов или этинид-ионов к двойной С=С связи
ненасыщенного кетона может быть предсказана
Результаты сравнительного изучения термо-
на основании комбинированных индексов реакци-
динамичесских характеристик сборок диоксаби-
онной способности. Для реакций с карбанионами
цикло[3.2.1]октанов, циклопентенолов и фуранов
кетонов это произведение индекса локальной элек-
показали, что термодинамически наиболее выгод-
трофильности ω+ субстрата и мультифильности
ным является сборка замещенных фуранов (ΔG =
Δωk карбаниона кетона, а для реакций с этинида-
-29.7÷-34.6 ккал/моль). При этом для кетонов с
ми - произведение заряда β атома на глобальную
объемными заместителями R1 = Mes, R2 = H или
электрофильность субстрата [q(β)×ω] и на индекс
R1 = Ph, R2 = Bn образование диоксабицикло-
мультифильности Δωk этинид-иона [53].
[3.2.1]октанов вообще не выгодно термодинами-
чески (ΔG = 10.0 ккал/моль и ΔG = 2.0 ккал/моль)
На примере мезитилметилкетона в рамках
модели ANIONGAS был изучен весь механизм
[53]. Помимо термодинамических факторов, клю-
чевую роль играет соотношение активационных
сборки фуранов [17, 53]. Наиболее высокоба-
рьерной стадией является внутримолекулярное
барьеров в реакции Михаэля. Для случая, когда
O-винилирование в 40d с образованием дигидро-
R1 = Ph, R2 = H (a), активационный барьер при-
фурана 41d, при этом величина барьера хорошо
ΔG, ккал/моль
согласуется с более жесткими условиями сборки
20
фуранов (90°С) по сравнению со сборками диокса-
бицикло[3.2.1]октанов и циклопентенолов.
10
3.3. СБОРКА Δ2-ИЗОКСАЗОЛИНОВ
Синтетические возможности, связанные с на-
0
чальным образованием ненасыщенных кетонов,
могут быть расширены при введении в реакцию
-10
еще одного нуклеофила. Механизм реакции ке-
тонов с арилацетиленами и гидроксиламином в
-20
присутствии t-BuOK/DMSO с последующей об-
работкой реакционной смеси H2O и KOH с полу-
-30
чением Δ2-изоксазолинов (схема 8) исследован на
Рис.
4. Реакционный профиль образования R,R,S-
примере сборки (4R,5S)-5-бензил-4-этил-3-метил-
диастереомера диоксабицикло[3.2.1]октана и S,S-
4,5-дигидроизоксазола из пентан-2-она, фенилаце-
диастереомера циклопентенола для системы ацетофе-
нон/ацетилен (a)
тилена и NH2OH [54].
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1311
Схема 6. Образование фуранов из ацетилена и кетонов
R2
R2
Me
KOH/DMSO
R1
+ HC CH
90°C, 15 min
R1
O
O Me
Схема 7. Схема образования фуранов из α,β-ненасыщенных кетонов и ацетилена
R2
R2
R2
R2
1
R
H2O
R1
+ C CH
-OH
O
R1
R1
O
O
O
33
35
40
41
42
Оценены энергии активации последователь-
диметилсульфоксида, наоборот, депротонирование
ных стадий образования карбаниона кетона и его
метильной группы с образованием С1-карбаниона
нуклеофильного присоединения к тройной связи
более выгодно на 0.7 ккал/моль. В то же время
(винилирование); присоединения гидроксиламина
присоединение карбаниона С3 к фенилацетилену
к карбонильной группе и последующей дегидра-
связано с более низким активационным барьером
тации образованного карбиноламина (оксимиро-
(ΔG‡ = 14.3 ккал/моль), что определяет наблюда-
вание); E/Z-изомеризации оксимной группы под
емое [55] осуществление реакции винилирования
действием основания; предполагаемой прототроп-
исключительно по атому углерода С3 пентанона-2
ной перегруппировки β,γ-ненасыщенного оксима
с образованием (4Е)-3-этил-5-фенилпент-4-ен-2-
в α,β-ненасыщенную структуру и, наконец, вну-
она.
тримолекулярного нуклеофильного присоедине-
Стадия оксимирования (схема 9) - присоедине-
ния к двойной связи с образованием пятичленного
ние гидроксиламина к (4E)-3-этил-5-фенилпент-
гетероцикла.
4-ен-2-ону осуществляется через восьмичленное
На стадии винилирования пентан-2-она фени-
переходное состояние с участием молекул воды и
лацетиленом могут образовываться как С1-, так
трет-бутанола (ΔG‡ = 18.8 ккал/моль).
и С3-карбанионы. В газовой фазе отрыв протона
от С3-положения пентанона-2 термодинамически
Наиболее предпочтительным является участие
предпочтительнее на 0.9 ккал/моль, а в растворе
воды в качестве донора протонов и трет-бутано-
Схема 8. Образование Δ2-изоксазолинов из арилацетиленов, кетонов и гидроксиламина
1. t-BuOK/DMSO
R2
2. NH2OH·HCl
R1
R2
3. KOH
R1
+ HC
R3
R3
N
O
O
Схема 9. Оксимирование через восьмичленное переходное состояние
H
O
H
O
OH
H2N OH
C
Et
H
O
HO
Et
N
H2O (ROH)
−H2O (ROH)
O
Et
H
CHCHPh
R
CHCHPh
H NH
CHCHPh
OH
R = t-Bu.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1312
ВИТКОВСКАЯ и др.
Схема 10. Дегидратация карбиноламина
OH
HO
Et
O
Et
HO
Et
N
N
N
-H2O
H
CHCHPh
H
CHCHPh
CHCHPh
ла в качестве акцептора. Участие молекул воды и
Последующее протонирование промежуточ-
трет-бутанола также понижает активационный
ного карбаниона завершает сборку 5-бензил-4-
барьер последующей дегидратации образующе-
этил-3-метил-4,5-дигидро-1,2-оксазола.
гося карбиноламина до ΔG‡ = 18.4 ккал/моль.
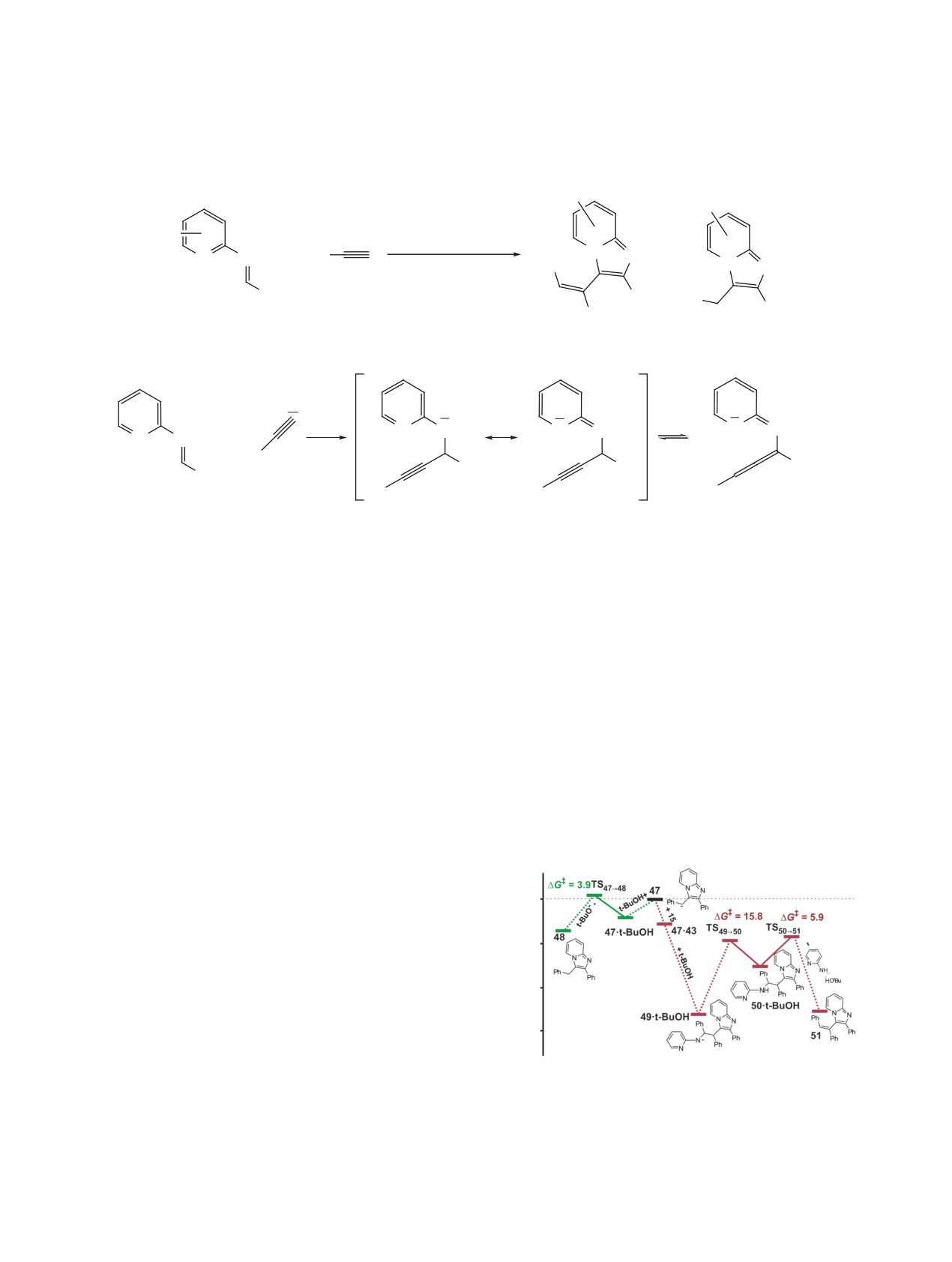
4. РЕАКЦИИ ИМИНОВ С АЦЕТИЛЕНАМИ
Примечательно, что эта дегидратация осуществля-
4.1. ОБРАЗОВАНИЕ ИМИДАЗОПИРИДИНО-
ется через образование промежуточного нитрона,
ВЫХ СИСТЕМ
который далее изомеризуется в (2Е,4Е)-3-этил-N-
Новые возможности для построения азотсодер-
гидрокси-5-фенилпент-4-ен-2-имин с активацион-
жащих гетероциклических систем предоставляет
ным барьером ΔG‡ = 6.0 ккал/моль (схема 10).
недавно открытая аза-реакция Фаворского. Так,
Различие энергий активации образования
при вовлечении 2-пиридил(гет)арилиминов в ре-
(2Е,4Е)- и (2Z,4Е)-изомеров β,γ-ненасыщенного
акцию этинилирования с (гет)арилацетиленами
оксима обусловливает наблюдаемое в экспери-
было неожиданно обнаружено образование ими-
менте [55] образование исключительно (2Е,4Е)-
дазопиридиновых структур (схема 13) [13].
изомеров из диалкилкетонов.
В рамках NBO (B3LYP/6-31+G*) анализа мы
показали, что в анионе пропаргиламина 45, об-
Легкость отрыва протона от C3-положения
разующемся при этинилировании 2-пиридилфе-
(2E,4E)-3-этил-N-гидрокси-5-фенилпент-4-ен-2-
нилимина 43 фенилэтинидом 44, отрицательный
имина позволяет превратить его в (2Z,4E)-изомер
заряд распределен между атомами азота аминово-
(схема 11).
го и пиридинового фрагментов (схема 14). Таким
Предполагавшаяся изначально [55] прототроп-
образом, формируется диазатриенильная систе-
ная перегруппировка β,γ-ненасыщенного оксима в
ма, в которой происходит внутримолекулярное
α,β-ненасыщенный
(2Z,3Z)-3-этил-N-гидрокси-5-
N-винилирование с образованием имидазопири-
фенилпент-3-ен-2-имин термодинамически невы-
динов. В рамках B2PLYPD/6-311+G**//B3LYP/6-
годна, а циклизация соответствующего О-аниона
31+G* подхода в модели ANIONGAS мы оценили
связана с высоким активационным барьером
относительную термодинамическую устойчи-
ΔG‡ = 27.9 ккал/моль. Напротив, замыкание β,γ-не-
вость аниона пропаргиламина 45 и его аллениль-
насыщенного оксимат-иона (схема 12) происходит
ного изомера 46, а также изучили весь механизм
легко (ΔG‡ = 16.3 ккал/моль).
сборки имидазопиридинов [13].
Схема 11. Изомеризация β,γ-ненасыщенного оксима
HO
OH
N
N
Ph
Ph
Et
Et
2E,4E
2Z,4E
Схема 12. Циклизация оксимат-иона
O
O
Ph
O
Ph
N
N
N
H2O
Ph
-
-OH
C2H5
C2H5
C2H5
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1313
Схема 13. Образование (Z)-стильбен/имидазопиридиновых ансамблей и бензилимидазопиридинов
из (гет)арилацетиленов и 2-пиридил(гет)арилиминов
R1
R1
R1
t-BuONa/DMSO
N N
+
R3
N N
+
R2
N
N
rt, 1 ч
R2
R2
R3
R2
R3
Схема 14. Аза-реакция Фаворского между 2-пиридилфенилимином 43 и фенилэтинидом 44
с образованием диазатриенильных анионов пропаргил- (45) и аллениламина (46)
N
N
N
N
N
N
N
N
+
Ph
Ph
Ph
Ph
Ph
Ph
Ph
Ph
43
44
45
46
Алленильная форма 46 оказалась на ΔΔG =
наблюдаемый в эксперименте сдвиг соотношения
8.8 ккал/моль выгоднее, чем пропаргильная форма
продуктов в сторону стильбена 51 при добавлении
45, при этом барьер внутримолекулярного присое-
MeOH или t-BuOH.
динения по пиридиновому атому азота в 46 с обра-
4.2. ОБРАЗОВАНИЕ ПИРРОЛИНОВ
зованием аниона бензилимидазопиридина 47 со-
N-бензилимины в реакциях с ацетиленами не
ставляет лишь ΔG‡ = 4.6 ккал/моль. На следующей
стадии возникает конкуренция между протониро-
проявляют ожидаемых электрофильных свойств и
не вступают в реакцию этинилирования, посколь-
ванием аниона 47 трет-бутанолом с образовани-
ку кислотность этих соединений в диметилсуль-
ем бензилимидазопиридина 48 и присоединением
фоксиде на несколько порядков выше кислотно-
47 к ещё одной молекуле имина 43 с образованием
сти ацетилена и фенилацетилена [56, 57]. В при-
через промежуточные анионные аддукты 49 и 50
сутствии супероснований они депротонируются
(Z)-стильбен/имидазопиридинового ансамбля 51.
с образованием высоко реакционноспособных
На этом этапе определяется конечный состав про-
азааллильных анионов. При взаимодействии их с
дуктов и их соотношение. Результаты нашего ис-
ацетиленом образуются 2-азабутадиены [10, 57].
следования показали, что присоединение имина 43
кинетически и термодинамически более предпоч-
ΔG, ккал/моль
тительно (рис. 5). Это согласуется с эксперимен-
тальными данными о преобладании в продуктах
0
реакции стильбена 51 над бензилимидазопириди-
ном 48 при эквимольном соотношении исходных
-10
имина и ацетилена, причем при недостатке имина
43 удается увеличить выходы бензилимидазопири-
-20
динов.
Показано, что на пути образования стильбе-
-30
на 51 возникает высокобарьерная стадия ΔG‡ =
Рис. 5. Реакционный профиль конкурирующих реак-
15.8 ккал/моль, которая связана с изомеризацией
ций протонирования аниона бензилимидазопиридина
49 в 50 с участием t-BuOH. При увеличении числа
и его присоединения к молекуле имина с образовани-
переносчиков протонов этот высокобарьерный пе-
ем бензилимидазопиридина и (Z)-стильбен/имидазо-
ренос протона происходит быстрее, что объясняет
пиридинового ансамбля
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1314
ВИТКОВСКАЯ и др.
Схема 15. Образование пирролинов из арилацетиленов и N-бензилиминов
R4
R2
t-BuOK/DMSO
R3
+
R4
R1
60°С, 15 мин
N
R1
N
R3
R2
R1, R3, R4 = Ar, Hetar; R2 = Alk, Ar.
R3
R3
t-BuOK/DMSO
R1
N
R2
+
R3
R2
+
R2
60°С, 15 мин
N
N
R1
R1
R1, R2, R3 = Ar, Hetar.
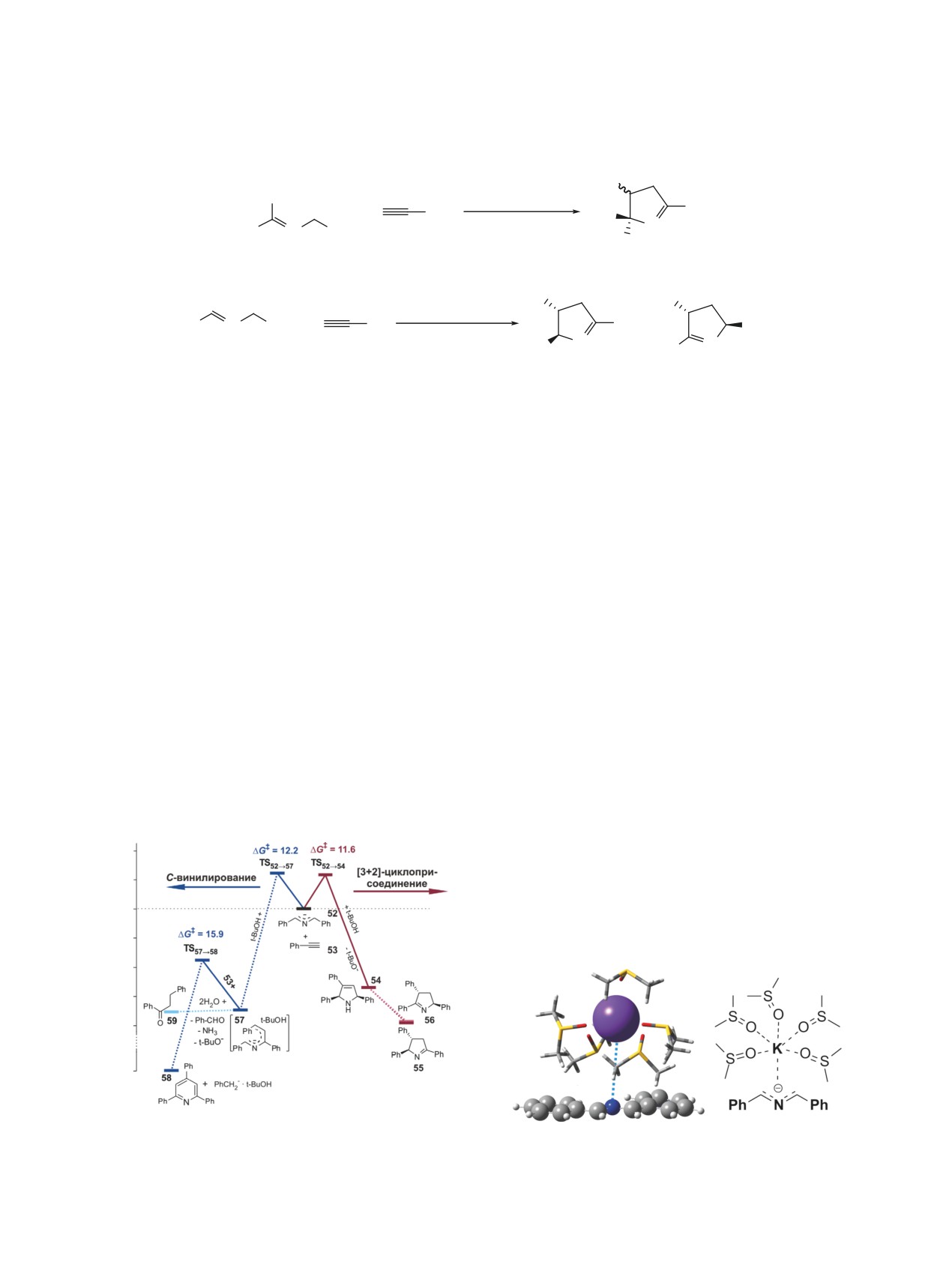
Добавление же к таким азааллильным анионам
приводит к целевым пирролинам 55 и 56 (рис. 6) с
арилацетиленов приводит к образованию смеси
преобладанием 55 за счет большей термодинами-
пирролинов (схема 15) [11, 12].
ческой устойчивости.
Мы провели квантовохимическое моделиро-
Мягкие условия сборки 1-пирролинов (60°C,
вание механизма сборки пирролинов в рамках
15 мин) обеспечивает невысокий активационный
моделей ANIONGAS и PENTAGAS из N-бензил-1-
барьер циклоприсоединения ΔG‡ = 11.6 ккал/моль,
фенилметанимина 52 и фенилацетилена 53 [58].
достигаемый за счет формирования сольватных
Было показано, что кинетически более предпочти-
комплексов, в которых азааллильный анион бо-
тельным является не стадийный, а согласованный
лее открыт для атаки молекулой фенилацетилена
механизм циклоприсоединения. При этом для осу-
(рис. 7).
ществления согласованного циклоприсоединения
Вместе с тем было получено, что близкой
необходима стабилизация азааллильного аниона
энергией активации характеризуется реакция
в переходном состоянии водородной связью моле-
С-винилирования альдимина 52, которая приводит
кулы t-BuOH или растворителя DMSO При спуске
к аниону 2-аза-1,4-пентадиена 57. Теоретически
из переходного состояния происходит не только
и экспериментально было показано, что анион
[3+2]-циклоприсоединение, но и протонирование
57 является интермедиатом побочных процессов
трет-бутанолом формирующегося N-аниона, что
образования триарилпиридинов 58 через согласо-
в конечном итоге приводит к 3-пирролину 54. Ряд
ванное циклоприсоединение следующей молеку-
последующих прототропных перегруппировок
лы фенилацетилена, а также 1,3-диарилпропан-1-
онов 59 в результате гидролиза.
ΔG, ккал/моль
20
ЗАКЛЮЧЕНИЕ
10
Подводя итоги, можно с уверенностью утвер-
0
ждать, что химия ацетилена в суперосновных
средах предоставляет большую свободу хими-
-10
ку-синтетику и дает обширный материал для ис-
-20
-30
-40
-90
5.698 Å
Рис. 6. Реакционный профиль образования 1-пирроли-
нов и побочных продуктов
Рис. 7. Устройство комплекса азааллильного аниона
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1315
следования химику-теоретику. Резкий рост вычис-
СПИСОК ЛИТЕРАТУРЫ
лительных мощностей в последние два десятиле-
1.
Alabugin I.V., Gold B. J. Org. Chem. 2013, 78, 7777-
тия позволил в полной мере использовать допол-
7784. doi 10.1021/jo401091w
нительные инструменты изучения механизмов
2.
Trotuş I.T., Zimmermann T., Schüth F. Chem. Rev.
химических реакций, предоставляемые квантовой
2014, 114, 1761-1782. doi 10.1021/cr400357r
химией.
3.
Voronin V.V., Ledovskaya M.S., Bogachenkov A.S.,
Rodygin K.S., Ananikov V.P. Molecules. 2018, 23,
В данном обзоре обобщены последние дости-
2442. doi 10.3390/molecules23102442
жения в исследованиях механизмов сборок кар-
4.
Trofimov B.A. Curr. Org. Chem. 2002, 6, 1121-1162.
бо- и гетероциклов на основе реакций ацетиленов
doi 10.2174/1385272023373581
в суперосновных средах. Нам удалось создать мо-
5.
Trofimov B.A., Schmidt E.Y., Zorina N.V., Ivano-
дели суперосновных центров для описания харак-
va E.V., Ushakov I.A. J. Org. Chem. 2012, 77, 6880-
терных для таких сред реакций: винилирования,
6886. doi 10.1021/jo301005p
этинилирования, 1,3-прототропных перегруппи-
6.
Trofimov B.A., Schmidt E.Y. Acc. Chem. Res. 2018,
ровок, реакции Михаэля, и успешно применить их
51, 1117-1130. doi 10.1021/acs.accounts.7b00618
для изучения механизмов различного рода реак-
7.
Bidusenko I.A., Schmidt E.Y., Ushakov I.A., Trofi-
ций, включающих ряд быстро протекающих эле-
mov B.A. Eur. J. Org. Chem. 2018, 2018, 4845-4849.
ментарных стадий. Мы надеемся, что изложенные
doi 10.1002/ejoc.201800850
результаты будут способствовать лучшему пони-
8.
Schmidt E.Y., Bidusenko I.A., Protsuk N.I., Demya-
манию особенностей этих интересных сложных
nov Y.V., Ushakov I.A., Trofimov B.A. Eur. J.
превращений.
Org. Chem. 2019, 2019, 5875-5881. doi 10.1002/
ejoc.201900932
БЛАГОДАРНОСТИ
9.
Schmidt E.Y., Bidusenko I.A., Protsuk N.I., Demya-
Авторы глубоко признательны академику
nov Y.V., Ushakov I.A., Vashchenko A.V., Trofi-
Борису Александровичу Трофимову и его учени-
mov B.A. J. Org. Chem. 2020, 85, 3417-3425. doi
кам за интересные химические реакции, полезные
10.1021/acs.joc.9b03192
дискуссии и плодотворное сотрудничество.
10.
Bidusenko I.A., Schmidt E.Y., Protsuk N.I., Usha-
kov I.A., Vashchenko A.V., Afonin A.V., Trofi-
ФОНДОВАЯ ПОДДЕРЖКА
mov B.A. Org. Lett. 2020, 22, 2611-2614. doi 10.1021/
Обзор выполнен при финансовой поддержке
acs.orglett.0c00564
Министерства науки и высшего образования РФ в
11.
Bidusenko I.A., Schmidt E.Y., Ushakov I.A.,
рамках государственного задания № FZZE-2020-
Vashchenko A.V., Trofimov B.A. Org. Lett. 2021, 23,
4121-4126. doi 10.1021/acs.orglett.1c01009
0025.
12.
Bidusenko I.A., Yu. Schmidt E., Protsuk N.I., Usha-
ИНФОРМАЦИЯ ОБ АВТОРАХ
kov I.A., Trofimov B.A. Mendeleev Commun. 2023,
33, 24-26. doi 10.1016/j.mencom.2023.01.007
Витковская Надежда Моисеевна, ORCID:
13.
Bidusenko I.A., Schmidt E.Y., Ushakov I.A.,
Vashchenko A.V., Protsuk N.I., Orel V.B., Vitkovs-
Орел Владимир Борисович, ORCID: https://
kaya N.M., Trofimov B.A. J. Org. Chem. 2022, 87,
orcid.org/0000-0003-2834-6700
12225-12239. doi 10.1021/acs.joc.2c01372
14.
Трофимов Б.А., Гусарова Н.К. Усп. хим. 2007,
Кобычев Владимир Борисович, ORCID: https://
76,
550-570.
[Trofimov B.A., Gusarova N.K.
orcid.org/0000-0003-2936-1865
Russ. Chem. Rev. 2007, 76, 507-527.] doi 10.1070/
Бобков Александр Сергеевич, ORCID: https://
RC2007v076n06ABEH003712
orcid.org/0000-0001-8901-3577
15.
Exner J.H., Steiner E.C. J. Am. Chem. Soc. 1974, 96,
1782-1787. doi 10.1021/ja00813a022
КОНФЛИКТ ИНТЕРЕСОВ
16.
Васильцов А.М., Трофимов Б.А., Амосова С.В. Изв.
Авторы заявляют об отсутствии конфликта ин-
АН СССР. Сер. хим. 1987, 36, 1785-1791. [Vasil’-
тересов.
tsov A.M., Trofimov B.A., Amosova S.V. Bull. Acad.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1316
ВИТКОВСКАЯ и др.
Sci. USSR Div. Chem. Sci. 1987, 36, 1653-1658.] doi
to M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J.,
10.1007/BF00960125
Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyo-
ta K., Fukuda R., Hasegawa J., Ishida M., Nakajima T.,
17.
Vitkovskaya N.M., Orel V.B., Kobychev V.B., Bob-
Honda Y., Kitao O., Nakai H., Vreven T., Montgome-
kov A.S., Absalyamov D.Z., Trofimov B.A. Int. J.
ry Jr. J.A., Peralta J.E., Ogliaro F., Bearpark M.J.,
Quantum Chem. 2020, 120, 26158 (1-12). doi 10.1002/
Heyd J.J., Brothers E.N., Kudin K.N., Staroverov V.N.,
qua.26158
Keith T.A., Kobayashi R., Normand J., Raghavacha-
18.
Tomasi J., Mennucci B., Cancès E. J. Mol. Struct.
ri K., Rendell A.P., Burant J.C., Iyengar S.S., Toma-
THEOCHEM. 1999,
464,
211-226. doi
10.1016/
si J., Cossi M., Rega N., Millam J.M., Klene M.,
S0166-1280(98)00553-3
Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramil-
19.
Vitkovskaya N.M., Larionova E.Y., Kobychev V.B.,
lo J., Gomperts R., Stratmann R.E., Yazyev O., Aus-
Kaempf N.V, Trofimov B.A. Int. J. Quantum Chem.
tin A.J., Cammi R., Pomelli C., Ochterski J.W., Mar-
2011, 111, 2519-2524. doi 10.1002/qua.22643
tin R.L., Morokuma K., Zakrzewski V.G., Voth G.A.,
20.
Becke A.D. Phys. Rev. A. 1988, 38, 3098-3100. doi
Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D.,
10.1103/PhysRevA.38.3098
Farkas O., Foresman J.B., Ortiz J.V., Cioslowski J., Fox
21.
Lee C., Yang W., Parr R.G. Phys. Rev. B. 1988, 37,
D.J. Gaussian 09, revision C.01 software. Gaussian
785-789. doi 10.1103/PhysRevB.37.785
Inc., 2010.
22.
Grimme S. J. Chem. Phys. 2006, 124, 034108. doi
35.
Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E.,
10.1063/1.2148954
Robb M.A., Cheeseman J.R., Scalmani G., Barone V.,
Petersson G.A., Nakatsuji H., Li X., Caricato M.,
23.
Schwabe T., Grimme S. Phys. Chem. Chem. Phys.
Marenich A.V., Bloino J., Janesko B.G., Gomperts R.,
2007, 9, 3397-3406. doi 10.1039/b704725h
Mennucci B., Hratchian H.P., Ortiz J.V., Izmay-
24.
Grimme S., Ehrlich S., Goerigk L. J. Comput. Chem.
lov A.F., Sonnenberg J.L., Williams-Young D., Ding F.,
2011, 32, 1456-1465. doi 10.1002/jcc.21759
Lipparini F., Egidi F., Goings J., Peng B., Petrone A.,
25.
Кобычев В.Б., Витковская Н.М., Клыба Н.С., Тро-
Henderson T., Ranasinghe D., Zakrzewski V.G., Gao J.,
фимов Б.А. Изв. АН, Сер. хим. 2002, 51, 713-720.
Rega N., Zheng G., Liang W., Hada M., Ehara M.,
[Kobychev V.B., Vitkovskaya N.M., Klyba N.S.,
Toyota K., Fukuda R., Hasegawa J., Ishida M.,
Trofimov B.A. Russ. Chem. Bull. 2002, 51, 774-782.]
Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T.,
doi 10.1023/A:1016068313892
Throssell K., Montgomery J.A.J., Peralta J.E., Oglia-
26.
Woodcock H.L., Schaefer H.F., Schreiner P.R. J.
ro F., Bearpark M.J., Heyd J.J., Brothers E.N., Ku-
Phys. Chem. A. 2002, 106, 11923-11931. doi 10.1021/
din K.N., Staroverov V.N., Keith T.A., Kobayashi
jp0212895
R., Normand J., Raghavachari K., Rendell A.P., Bu-
rant J.C., Iyengar S.S., Tomasi J., Cossi M., Mil-
27.
Navarro-Vázquez A. Beilstein J. Org. Chem. 2015, 11,
lam J.M., Klene M., Adamo C., Cammi R., Ochters-
1441-1446. doi 10.3762/bjoc.11.156
ki J.W., Martin R.L., Morokuma K., Farkas O., Fores-
28.
Wheeler S.E., Moran A., Pieniazek S.N., Houk K.N.
man J.B., Fox D.J. Gaussian 16, revision C.01 software.
J. Phys. Chem. A. 2009, 113, 10376-10384. doi
Gaussian Inc., 2019.
10.1021/jp9058565
36.
Schmidt M.W., Baldridge K.K., Boatz J.A., Elbert S.T.,
29.
Wertz D.H. J. Am. Chem. Soc. 1980, 102, 5316-5322.
Gordon M.S., Jensen J.H., Koseki S., Matsunaga N.,
doi 10.1021/ja00536a033
Nguyen K.A., Su S., Windus T.L., Dupuis M.,
30.
Abraham M.H. J. Am. Chem. Soc. 1981, 103, 6742-
Montgomery J.A. J. Comput. Chem. 1993, 14, 1347-
6744. doi 10.1021/ja00412a036
1363. doi 10.1002/jcc.540141112
31.
Cooper J., Ziegler T. Inorg. Chem. 2002, 41, 6614-
37.
Трофимов Б.А., Михалева А.И. ХГС.
1980,
6622. doi 10.1021/ic020294k
1299-1312. [Trofimov B.A., Mikhaleva A.I. Chem.
32.
Vitkovskaya N.M., Kobychev V.B., Bobkov A.S.,
Heterocycl. Compd. 1980, 16, 979-991.] doi 10.1007/
Orel V.B., Schmidt E.Y., Trofimov B.A. J. Org. Chem.
BF00496592
2017, 82, 12467-12476. doi 10.1021/acs.joc.7b02263
38.
Sączewski J., Fedorowicz J., Gdaniec M., Wiśniews-
33.
Kobychev V.B. J. Phys. Conf. Ser. 2021, 1847, 012054.
ka P., Sieniawska E., Drażba Z., Rzewnicka J., Balews-
doi 10.1088/1742-6596/1847/1/012054
ki Ł. J. Org. Chem. 2017, 82, 9737-9743. doi 10.1021/
acs.joc.7b01851
34.
Frisch M.J., Trucks G.W., Schlegel H., Scuseria G.E.,
Robb M.A., Cheeseman J.R., Scalmani G., Barone V.,
39.
Ларионова Е.Ю., Витковская Н.М., Кобычев В.Б.,
Mennucci B., Petersson G.A., Nakatsuji H., Carica-
Скитневская А.Д., Шмидт Е.Ю., Трофимов Б.А.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
КВАНТОВОХИМИЧЕСКИЕ ИССЛЕДОВАНИЯ МЕХАНИЗМОВ СБОРОК
1317
Докл. АН. 2011, 438, 765-767. [Larionova E.Y.,
48.
Ivanov A.V., Bobkov A.S., Martynovskaya S.V.,
Vitkovskaya N.M., Kobychev V.B., Skitnevskaya A.D.,
Budaev A.B., Vitkovskaya N.M. Asian J. Org. Chem.
Shmidt E.Y., Trofimov B.A. Dokl. Chem. 2011, 438,
2023. doi 10.1002/ajoc.202300153
167-169.] doi 10.1134/S001250081106005X
49.
Vandavasi J.K., Hu W.-P., Senadi G.C., Boomina-
40.
Шагун В.А., Васильцов А.М., Иванов А.В., Миха-
than S.S.K., Chen H.-Y., Wang J.-J. Eur. J. Org. Chem.
лева А.И., Трофимов Б.А. ЖСХ. 2013, 54, 25-33.
2014, 2014, 6219-6226. doi 10.1002/ejoc.201402818
[Shagun V.A., Vasil’tsov A.M., Ivanov A. V., Mikhale-
va A.I., Trofimov B.A. J. Struct. Chem. 2013, 54, 17-
50.
Abbiati G., Canevari V., Caimi S., Rossi E.
25.] doi 10.1134/S0022476613010034
Tetrahedron Lett. 2005, 46, 7117-7120. doi 10.1016/
41.
Shabalin D.A., Dvorko M.Y., Schmidt E.Y., Usha-
j.tetlet.2005.08.102
kov I.A., Protsuk N.I., Kobychev V.B., Soshnikov D.Y.,
51.
Vitkovskaya N.M., Orel V.B., Kobychev V.B.,
Trofimov A.B., Vitkovskaya N.M., Mikhaleva A.I.,
Schmidt E.Y., Trofimov B.A. Int. J. Quantum Chem.
Trofimov B.A. Tetrahedron. 2015, 71, 3273-3281. doi
2018, 118, e25689 (1-10). doi 10.1002/qua.25689
10.1016/j.tet.2015.03.111
52.
Orel V.B., Manzhueva A.A. Tetrahedron. 2021, 89,
42.
Kuzmin A.V., Shabalin D.A. J. Phys. Org. Chem. 2018,
31, e3829. doi 10.1002/poc.3829
132164. doi 10.1016/j.tet.2021.132164
43.
Bobkov A.S., Vitkovskaya N.M., Trofimov B.A.
53.
Orel V.B., Vitkovskaya N.M. J. Phys. Conf. Ser. 2021,
J. Org. Chem. 2020, 85, 6463-6470. doi 10.1021/
1847, 012056. doi 10.1088/1742-6596/1847/1/012056
acs.joc.0c00353
54.
Kobychev V.B., Pradedova A.G., Trofimov B.A.
44.
Васильцов А.М., Полубенцев Е.А., Михалева А.И.,
J. Mol. Struct. 2021, 1246, 131185. doi 10.1016/
Трофимов Б.А. Известия АН СССР. Сер. хим. 1990,
j.molstruc.2021.131185
39, 864-867. [Vasil’tsov A.M., Polubentsev E.A.,
Mikhaleva A.I., Trofimov B.A. Bull. Acad. Sci. USSR
55.
Schmidt E.Y., Tatarinova I.V, Ivanova E. V, Zori-
Div. Chem. Sci. 1990, 39, 773-776.] doi 10.1007/
na N.V., Ushakov I.A., Trofimov B.A. Org. Lett. 2013,
BF00960344
15, 104-107. doi 10.1021/ol303132u
45.
Schmidt E.Y., Semenova N.V., Ivanova E.V., Bidusen-
56.
Bordwell F.G. Acc. Chem. Res. 1988, 21, 456-463. doi
ko I.A., Trofimov B.A. Mendeleev Commun. 2020, 30,
10.1021/ar00156a004
109-111. doi 10.1016/j.mencom.2020.01.036
57.
Прадедова А.Г., Кобычев В.Б. ЖСХ. 2023, 64, 107402.
46.
Vitkovskaya N.M., Orel V.B., Absalyamov D.Z.,
[Pradedova A.G., Kobychev V.B. J. Struct. Chem.
Trofimov B.A. J. Org. Chem. 2020, 85, 10617-10627.
2023, 64, 386-397.] doi 10.1134/S0022476623030058
doi 10.1021/acs.joc.0c01185
58.
Orel V.B., Zubarev A.A., Bidusenko I.A., Usha-
47.
Vitkovskaya N.M., Bobkov A.S., Kuznetsova S.V.,
Shcherbakova V.S., Ivanov A.V. Chempluschem. 2020,
kov I.A., Vitkovskaya N.M. J. Org. Chem. 2023, 88,
85, 88-100. doi 10.1002/cplu.201900407
7058-7069. doi 10.1021/acs.joc.3c00333
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023
1318
ВИТКОВСКАЯ и др.
Quantum Chemical Investigations of the Mechanisms
of Carbo- and Heterocycles Assemblies Based
on Acetylenes Reactions in Superbasic Media
N. M. Vitkovskaya*, V. B. Orel, V. B. Kobychev, and A. S. Bobkov
Irkutsk State University, ul. Karla Marksa, 1, Irkutsk, 664003 Russia
*e-mail: vita@cc.isu.ru
Received July 27, 2023; revised August 10, 2023; accepted August 11, 2023
The chemistry of acetylenes has received significant development in the context of the use of superbasic media
in organic synthesis. The study of reaction mechanisms requires the use of a complex of chemical, physico-
chemical, and theoretical methods. This review presents the results of recent quantum-chemical studies on
mechanisms of assemblies based on the reactions of acetylene and its derivatives occurring in superbasic media
and yielding in the formation of complex deeply functionalized molecular structures, which are being developed
at the Irkutsk Institute of Chemistry named after A.E. Favorsky SB RAS.
Keywords: acetylenes, dimethyl sulfoxide, superbasic media, cascade assemblies, reaction mechanisms, quan-
tum chemical calculations
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 10 2023