ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ, 2023, том 59, № 2, с. 165-179
УДК 547.917 + 54.057
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ
ЛИГАНДОВ АСИАЛОГЛИКОПРОТЕИНОВОГО
РЕЦЕПТОРА
© 2023 г. Р. А. Петров*, С. А. Петров, Д. А. Гришин, И. Г. Колмаков, Д. С. Абрамчук,
В. Т. Ткаченко, Е. А. Власова, С. Ю. Маклакова, А. В. Лопухов,
Н. Л. Клячко, Е. К. Белоглазкина
ФГБОУ ВО «Московский государственный университет им. М.В. Ломоносова»,
химический факультет, Россия, 119991 Москва, Ленинские горы, 1, стр. 3
*e-mail: petrovrostaleks@gmail.com
Поступила в редакцию 22.03.2022 г.
После доработки 11.04.2022 г.
Принята к публикации 13.04.2022 г.
Синтезированы новые триантеннарные производные N-ацетилгалактозамина на основе трис(гидрок-
симетил)аминометана для связывания с асиалогликопротеиновым рецептором, экспрессируемым
гепатоцитами. Значения измеренных методом спектроскопии поверхностного плазмонного резонанса
равновесных констант диссоциации комплексов асиалогликопротеинового рецептора и полученных
соединений находятся в субнаномолярном диапазоне, что на 6 порядков ниже равновесных констант
диссоциации комплекса рецептора и природного лиганда N-ацетилгалактозамина. Полученные соеди-
нения характеризуются образованием гораздо более прочных комплексов с рецептором, чем природный
лиганд, что позволяет рассматривать их как перспективные средства для адресной доставки различных
лекарственных соединений в гепатоциты.
Ключевые слова: адресная доставка, лиганды асиалогликопротеинового рецептора, гепатоцеллюлярная
карцинома, производные N-ацетилгалактозамина
DOI: 10.31857/S0514749223020027, EDN: QIWWQA
ВВЕДЕНИЕ
В литературе широко представлены примеры
наноразмерных систем, использующих ASGPR
Асиалогликопротеиновый рецептор (ASGPR),
в качестве мишени (полимеры, наночастицы, ли-
располагающийся на поверхности гепатоци-
посомы и мицеллы [4-7]), несмотря на то, что
тов, представляет собой удобную мишень для
использование наноразмерных средств адресной
направленной доставки лекарственных средств
доставки имеет ряд недостатков: затрудненную
в печень, поскольку его структура подробно из-
проницаемость мембран клеток и стенок сосудов,
учена, известна его селективность в отноше-
нии производных галактозы и галактозамина,
возможность иммунного ответа, неустойчивость
а также возможность реализации с его участи-
в плазме крови, недостаточную растворимость
ем процесса рецептор-опосредованного эн-
[8-13]. В то же время низкомолекулярные кова-
доцитоза [1-3]. Успешный подбор лигандов к
лентные конъюгаты с лигандами ASGPR, не обла-
ASGPR может иметь значение для разработки
дающие подобными недостатками, исследованы
новых эффективных целевых систем для тера-
существенно меньше [14-18]. При этом фарма-
пии и диагностики различных заболеваний пече-
цевтические компании активно ведут разработки
ни.
низкомолекулярных гликоконъюгатов олигону-
165
166
ПЕТРОВ и др.
клеотидов и моносахаридов с лигандами ASGPR в
триантеннарных лигандов на связывание с ASGPR
области генной терапии заболеваний печени, кото-
[38-43]. В качестве фрагмента ветвления исполь-
рые находятся на разных стадиях доклинических
зовали различные молекулярные платформы: три-
и клинических исследований [7, 19-26]; в насто-
с(гидроксиметил)аминометан (TRIS) [38, 40-43],
ящее время управление по санитарному надзору
лизин и содержащий его дипептид [39], а также
за качеством пищевых продуктов и медикаментов
последовательность монономерных звеньев на
США (FDA, USA) одобрило уже 3 подобных ле-
основе 1-гидроксиметил-3-гидроксипирролидина,
карственных препарата [22, 27-29].
связанных фосфодиэфирной связью [42]. При ва-
рьировании длины спейсера было установлено,
ASGPR представляет собой трансмембранный
что наибольшим сродством к ASGPR обладают ли-
белок, отвечающий за распознавание и поглоще-
ганды с линейным расстоянием от моносахарида
ние клеточного фибронектина, протромбических
до точки ветвления ~ 20 Å [38]. Исследования вли-
компонентов, печеночных липопротеинов и имму-
яния количества углеводных фрагментов в струк-
ноглобулина А. Поэтому природные, а также син-
туре лиганда показывают нелинейное увеличение
тетические гликопротеины и углеводы, содержа-
сродства при переходе от моно- к тривалентным
щие десиалированные галактозу или галактозамин
лигандам, так называемый «кумулятивный эф-
в качестве концевого фрагмента, могут выступать
фект», в то же время дальнейшее увеличение ко-
лигандами асиалогликопротеинового рецептора.
личества углеводных фрагментов сказывается на
Внеклеточный домен ASGPR имеет 3 сайта связы-
аффинности незначительно [44, 45]. Важно, что
вания, каждый из которых способен одновременно
удалось показать влияние увеличения сродства к
связываться с углеводом [30], что делает перспек-
ASGPR у лигандов на поглощение конъюгатов in
тивной разработку так называемых триантеннар-
vitro и in vivo [43].
ных лигандов ASGPR с 3 углеводными (галактоза,
N-ацетилгалактозамин) фрагментами, находящи-
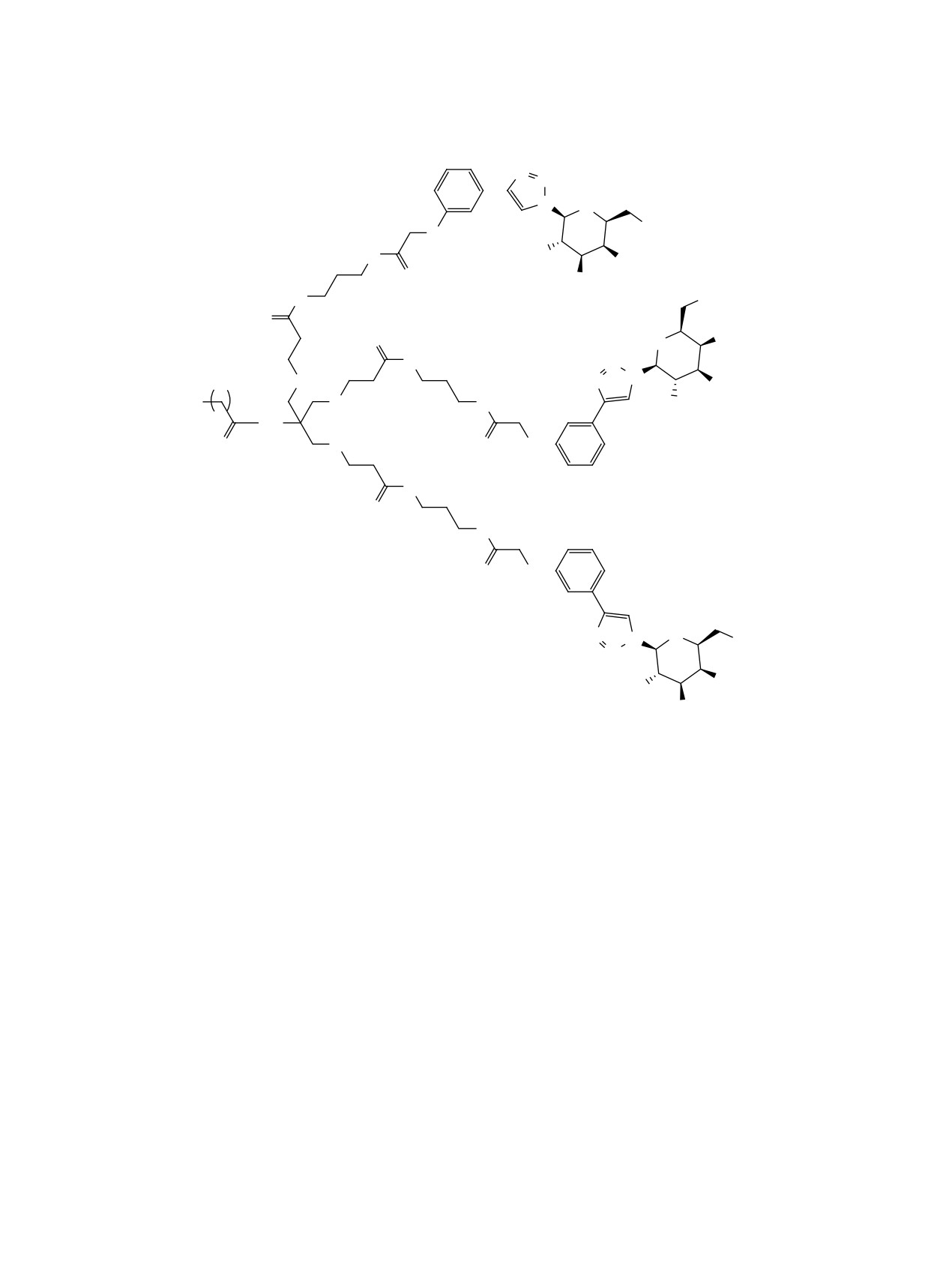
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
мися на определенном расстоянии от точки вет-
В данной работе нами предложены новые три-
вления. Исходя из исследований кристаллической
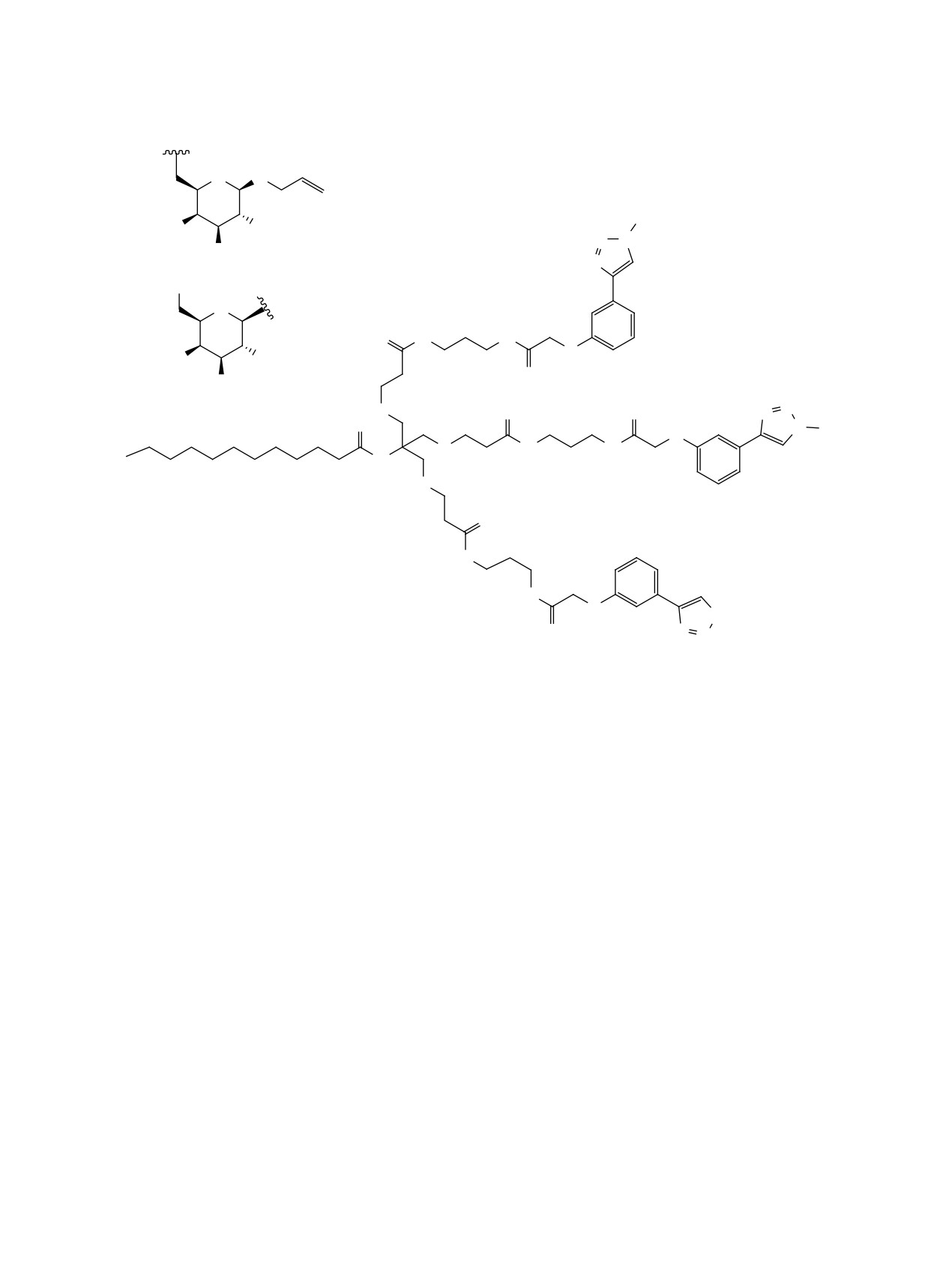
антеннарные лиганды ASGPR 1 и 2 общей струк-
структуры углевод-распознающего домена белка
туры, показанной на рисунке. Лиганд 1, содер-
[31] и результатов докинга [32-34], было высказа-
жащий галактозный фрагмент с аллилоксильным
но предположение о том, что ключевую роль в свя-
заместителем в первом положении шестичленного
зывании играют гидроксильные группы в положе-
углеводного цикла и ароматическим заместите-
ниях 3 и 4 моносахарида. Поиски оптимальной для
лем в шестом, синтезирован на основе TRIS. Мы
связывания структуры моносахарида указывают
выбрали соединение 1 в качестве целевой струк-
на важность ацетамидного фрагмента в положе-
туры на основании данных о высоком сродстве к
нии 2 (который увеличивает сродство к рецептору
ASGPR подобного моносахарида [37] и незначи-
примерно в 50 раз относительно галактозы) [35,
тельном различии в аффинности при выборе фраг-
36], а также возможность модификаций первого и
ментов ветвления. Лиганд 2 имеет сходную струк-
шестого положения моносахарида, которые могут
туру, однако синтез его углеводного фрагмента
как положительно, так и отрицательно сказываться
включает меньшее количество стадий (3 вместо 6;
на аффинности к рецептору [33, 37]. Значительно
см. рисунок), имеет больший суммарный выход и
увеличивает связывание аллилоксильный замести-
не включает стадий выделения при помощи коло-
тель в первом положении моносахарида; при одно-
ночной хроматографии.
временном присутствии триазольного фрагмента
в шестом положении моносахарида равновесная
Углеводные фрагменты целевых соединений 5,
константа диссоциации комплекса лиганд-рецеп-
10 были синтезированы по схеме 1 [15, 17, 18].
тор (KD) уменьшается еще в 5 раз [37].
Финальной стадией синтеза (схема 2) фрагмен-
Параллельно с варьированием заместителей в
тов молекулы, отвечающих за связывание с рецеп-
моносахаридном фрагменте проводилось иссле-
тором, являлась реакция медь-катализируемого
дование влияния пространственной структуры
[3+2]-азид-алкинового циклоприсоединения меж-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
167
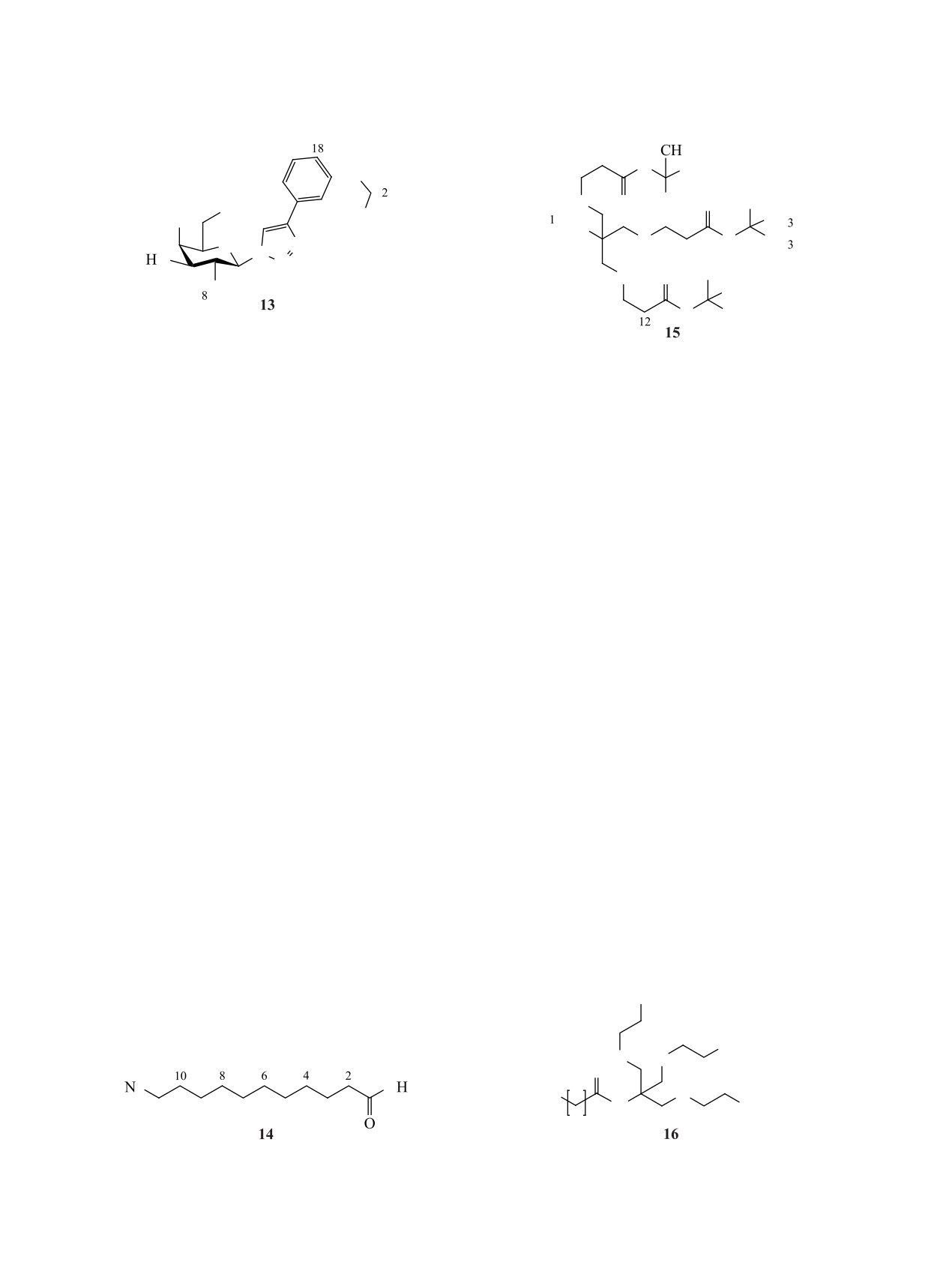
O
O
R=
1
R
HO
NHAc
N
N
OH
N
OH
O
R=
2
H
H
O
N
N
O
HO
NHAc
O
OH
N
O
O
O
N
O
N
R
O
O
N
N
N3
N
H
H
H
O
O
HN
HN
O
N R
O
N N
Структура целевых триантеннарных лигандов
ду полученными азидами 5, 10 и (мета-этинилфе-
цию проводили в щелочной среде. После разделе-
нокси)уксусной кислотой 11, которую синтезиро-
ния реакционной смеси методом колоночной хро-
вали из этил-2-бромацетата и мета-этинилфенола.
матографии среди продуктов не было обнаружено
Увеличение нуклеофильности фенольной группы
продуктов присоединения аминогруппы к α,β-не-
происходило за счет ее депротонирования в ос-
насыщенному эфиру, однако были выделены по-
новной среде, полученный таким образом фенолят
бочные продукты моно- и диалкилирования TRIS
вступал в реакцию SN2 замещения с этил-2-бром-
по гидроксильным группам.
ацетатом и в дальнейшем подвергался гидролизу
Введение фрагмента
11-азидоундекановой
по сложноэфирной связи. Реакцию [3+2]-азид-
кислоты 14 позволяет создать в векторной моле-
алкинового циклоприсоединения проводили для
куле необходимое расстояние между лигандом
азидов 5, 10 и алкина 11 в присутствии сульфа-
и доставляемым лекарственным веществом для
та меди и аскорбата натрия. Получение целевого
нивелирования возможных негативных простран-
продукта реакции подтверждается наличием син-
ственных эффектов; азидогруппа дает в даль-
глета триазольного цикла в спектрах ЯМР 1H по-
нейшем возможность соединять получаемые ли-
лученных соединений (8.53 м.д. - соединение 12,
ганды с лекарственными препаратами реакцией
8.60 м.д. - соединение 13).
[3+2]-азид-алкинового циклоприсоединения, не
затрагивая другие функциональные группы.
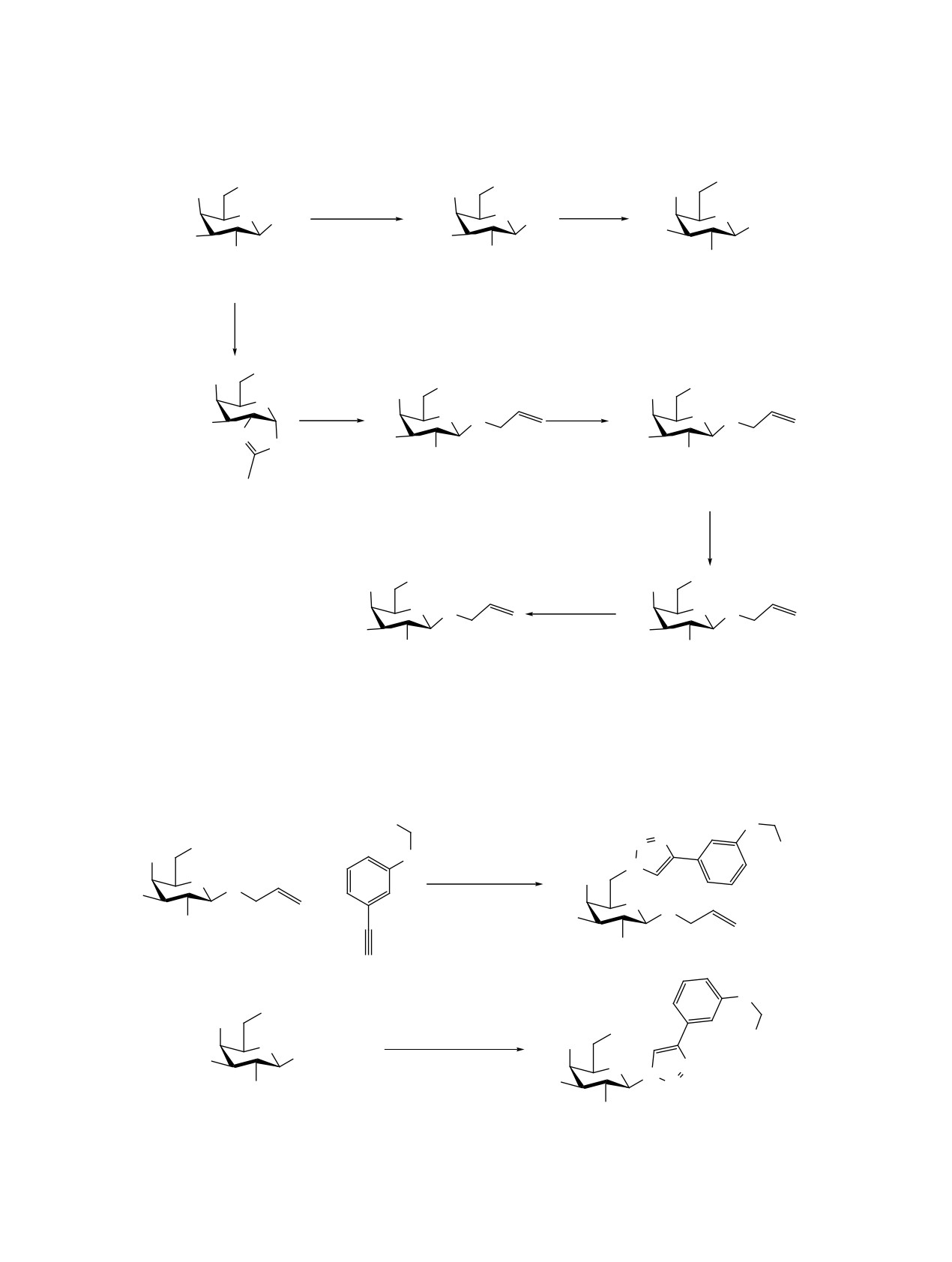
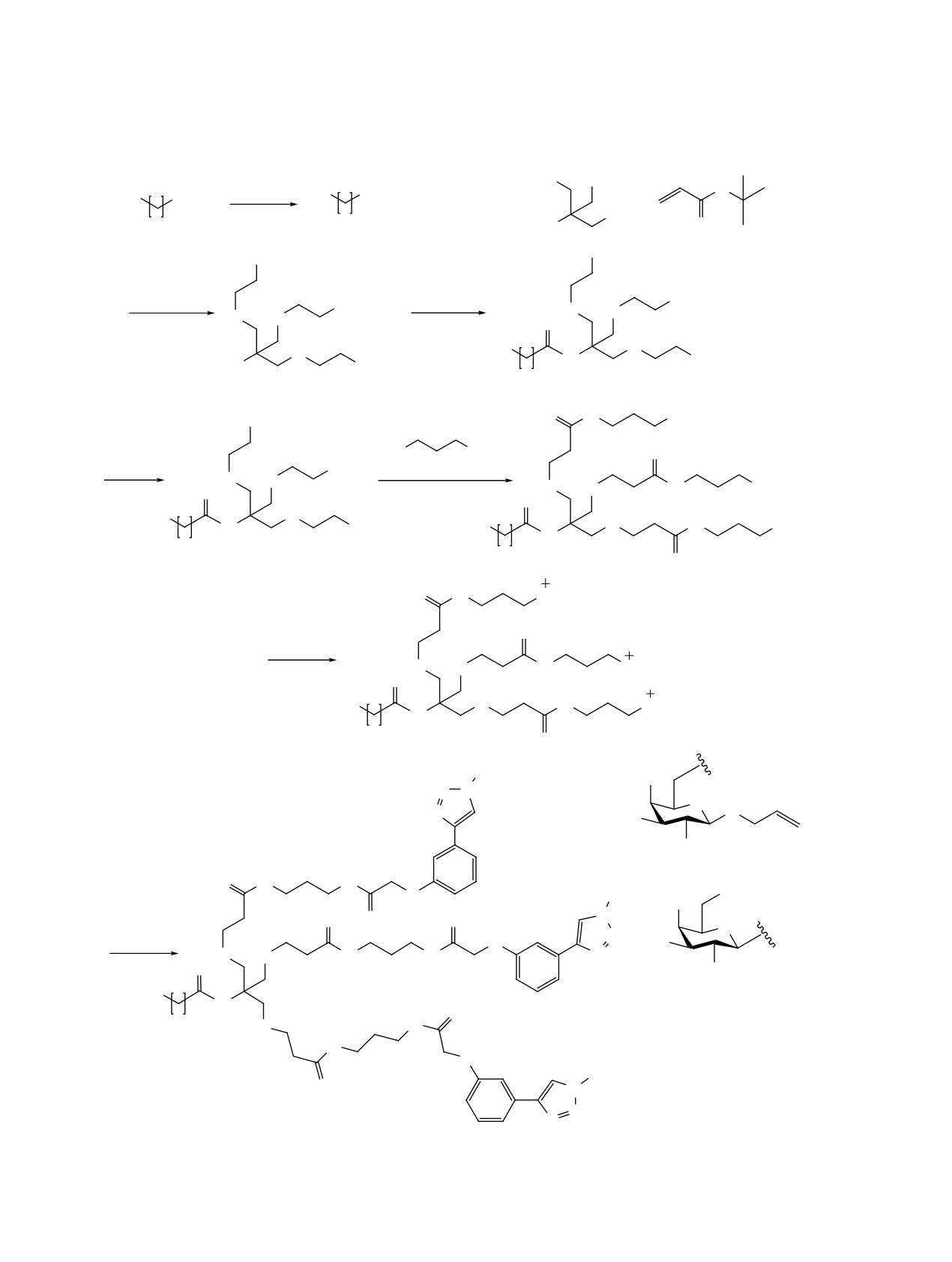
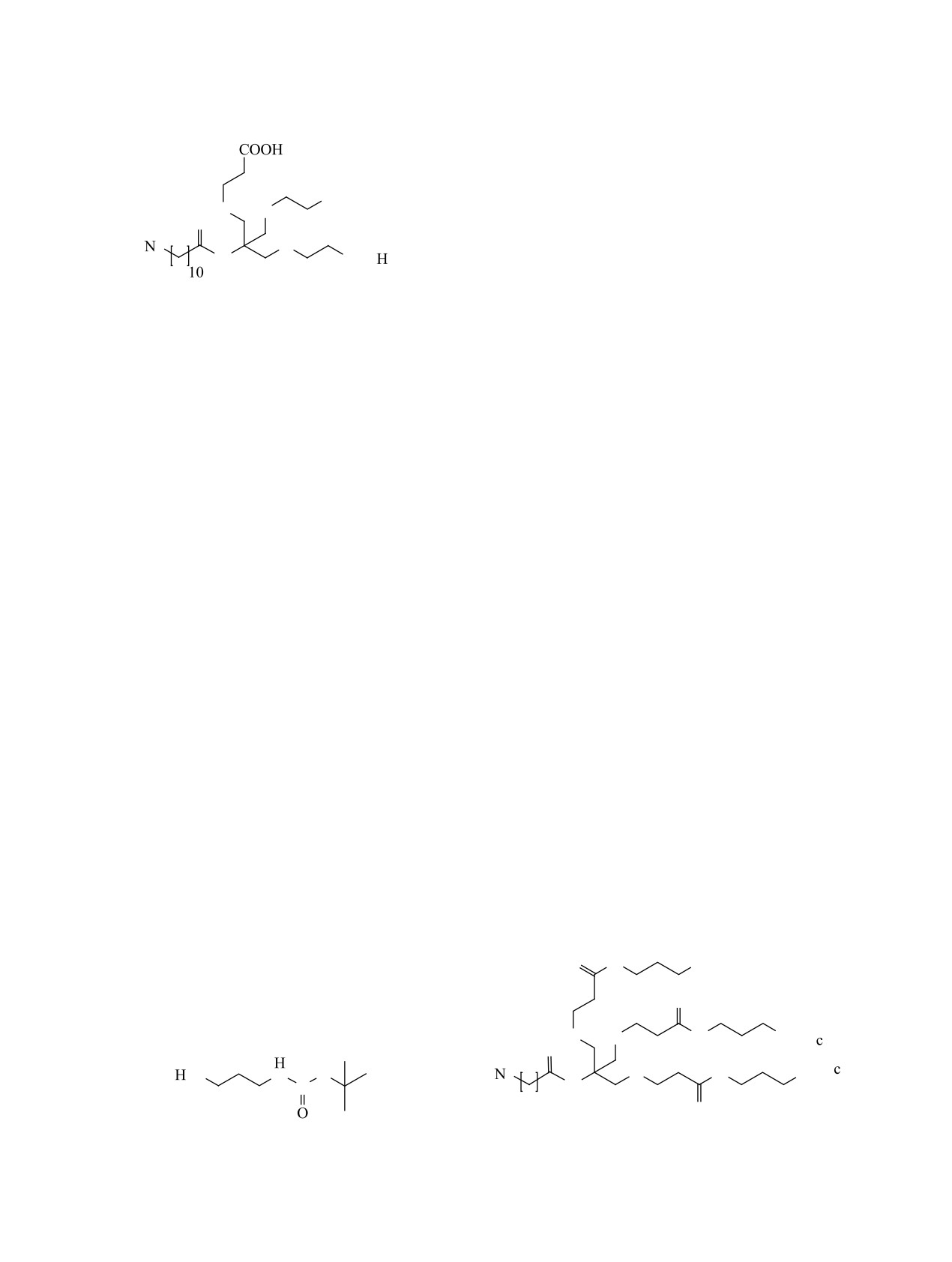
Синтез фрагмента ветвления начали с реакции
1,4-присоединения по Михаэлю трис(гидроксиме-
Соединение 16 синтезировали методом кар-
тил)аминометана (TRIS) к трет-бутилакрилату.
бодиимидного синтеза. В качестве систем для
Чтобы увеличить нуклеофильность гидроксиль-
активации карбоксильной группы в реакции об-
ных групп по сравнению с аминогруппой, реак-
разования амидной связи применяли 3-[бис(ди-
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
168
ПЕТРОВ и др.
Схема 1. Синтез углеводных фрагментов
OH
OAc
OAc
AcO
CH2Cl2, TMSN3,
AcO
MeONa
OH
FeCl3
MeOH
O OAc
O N3
O
N3
HO
AcO
AcO
NHAc
NHAc
NHAc
3, 89%
4, 73%
5, 98%
TMSOTf
(CH2Cl)2
OAc
AcO
AllOH
OAc
OH
TMSOTf
AcO
MeONa
HO
O
CH2Cl
2
MeOH
AcO
O O
O O
N
AcO
HO
O
NHAc
NHAc
6, 92%
7, 71%
8, 98%
TsCl, Py
40°C
N3
OTs
HO
HO
NaN3 DMF
80°C
O O
O O
HO
HO
NHAc
NHAc
10, 68%
9, 63%
метиламино)метил]-3H-бензотриазол-1-оксида
выше. О полноте протекания реакции свидетель-
гексафторфосфат (HBTU) и 3-(этилиминометиле-
ствует отсутствие в спектрах ЯМР 1H выделенного
намино)-N,N-диметилпропан-1-амин (EDC), при
продукта характерного сигнала исходного амина
использовании последнего выходы реакции были
в области 2.59 м.д. (уширенный синглет -NH2),
Схема 2. Финальная стадия синтеза углеводных фрагментов
HOOC
O
N
N3
N
COOH
OH
O CuSO4*5H2O,
N
sodium ascorbate
OH
O
+
O
HO
DMF
O
O
HO
NHAc
NHAc
10
11
12, 48%
O
OH
OH
CuSO4*5H2O,
OH
sodium ascorbate
HOOC
OH
O
+
N3
11
HO
DMF
O
N
N
HO
NHAc
N
NHAc
5
13, 65%
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
169
а также наличие характерного сигнала амидной
N-ацетилгалактозамина (448±43 мкМ), что явля-
группы при 6.47 м.д.
ется проявлением кооперативного эффекта, опи-
санного для мультивалентных лигандов [42]. Это
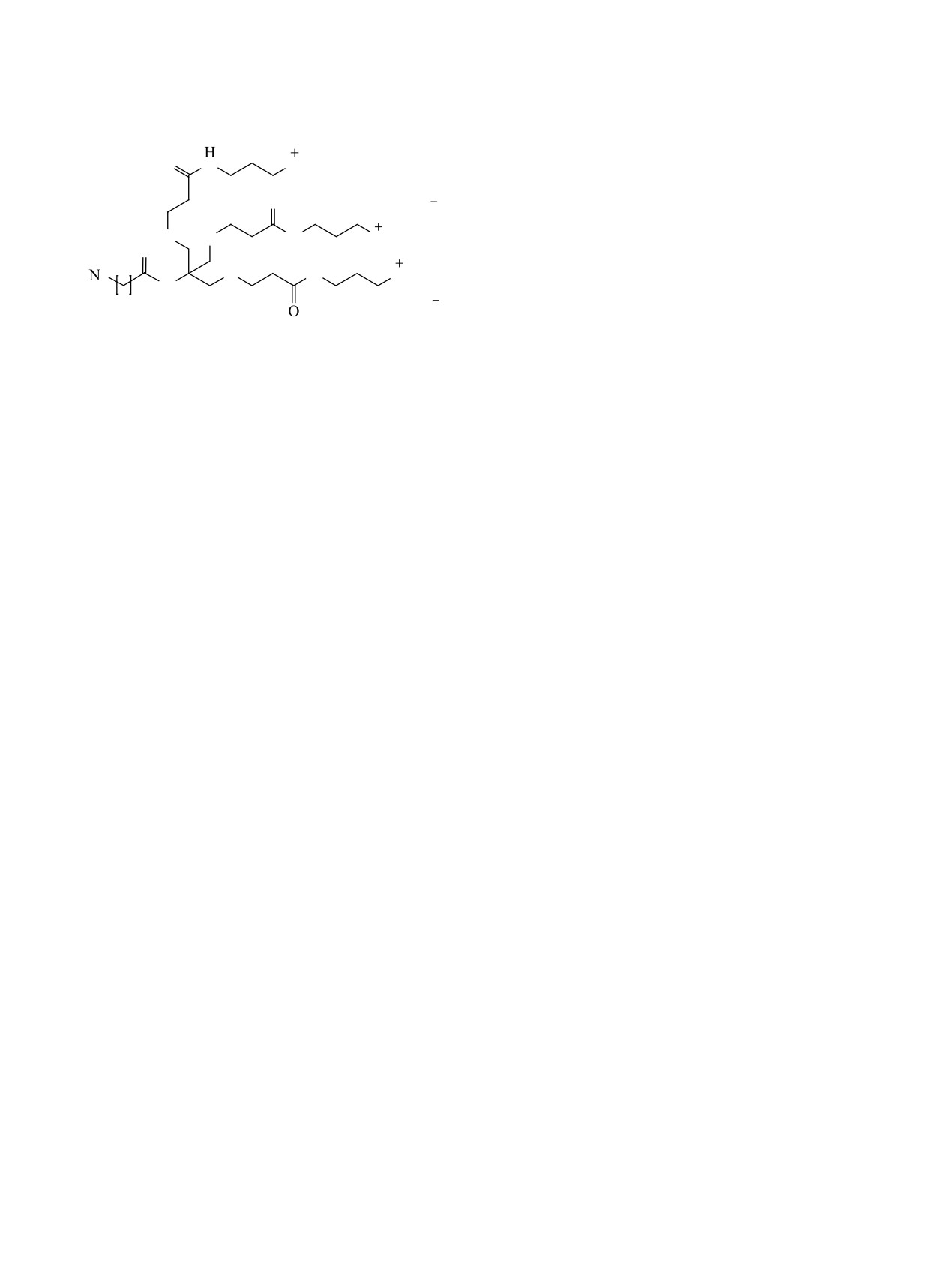
Отщепление трет-бутильных групп с получе-
свидетельствует о существенном улучшении пара-
нием трикарбоновой кислоты 17 проводили обра-
метров связывания полученных в данной работе
боткой продукта 16 муравьиной кислотой.
соединений по сравнению с природным лигандом.
Далее соединения 17 и 18 вводили в реакцию
Измеренные KD комплексов для соединений 1 и 2
карбодиимидного синтеза, используя систему
всего на порядок уступают значениям таковой для
EDC/NHS. Для последующего удаления защитных
триантеннарных лигандов на основе бицикличе-
Boc-групп использовали трифторуксусную кис-
ских углеводных фрагментов (KD = 0.03±0.01 нМ)
лоту. Сигналы трет-бутильных групп в спектре
[43]. Важно также отметить, что 2 полученных
ЯМР 1H (1.40-1.50 м.д.) продукта 20 отсутствуют,
триантеннарных лиганда ASGPR 1 и 2 показали
однако присутствует сигнал аммонийной группы
сходные значения равновесных констант диссоци-
(NH3+) (7.78 м.д.), что подтверждает протекание
ации лиганд-рецепторных комплексов, существен-
реакции.
но не зависящих от расстояния между углеводным
и триазольным циклами и места присоединения
Конечная стадия синтеза целевых триантеннар-
триазольного цикла к пиранозному, то есть дан-
ных лигандов (схема 3) заключалась в образова-
ные параметры не оказывают заметного влияния
нии амидных связей между полученными галакто-
на сродство к сайту связывания. Это предполагает
зными производными 12, 13 и фрагментом ветвле-
возможность использовать для получения галакто-
ния 20. Амид получали в присутствии EDC/NHS.
заминных лигандов более простые методы синте-
Дополнительно для активации протонированной
за, сократить количество стадий и увеличить ко-
аминогруппы соединения 20 прибавляли третич-
нечные выходы.
ный амин (DIPEA). Продукты реакции выделяли
с использованием колоночной хроматографии.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Образование целевого продукта подтверждали ме-
Исходные соединения представляют собой
тодами масс-спектрометрии высокого разрешения
коммерчески доступные реагенты.
и спектроскопии ЯМР 1Н.
Контроль за ходом реакций осуществляли ме-
Аффинность полученных лигандов 1 и 2 к
тодом ТСХ на закрепленном слое силикагеля
ASGPR была исследована методом спектроскопии
(Merck).
поверхностного плазмонного резонанса. Данный
Спектры ЯМР 1Н были зарегистрированы на
метод позволяет изучать межмолекулярное вза-
приборе Bruker Avance с рабочей частотой 400 МГц
имодействие без необходимости дополнитель-
(Германия). В качестве растворителей использова-
ной модификации исследуемых соединений и его
ли CDCl3, ДМСО-d6, CD3OD. Химические сдвиги
применимость также не зависит от спектральных
приведены в миллионных долях по шкале δ отно-
характеристик исследуемых соединений; чувстви-
сительно гексаметилдисилоксана как внутренне-
тельность метода позволяет работать с малыми
го стандарта. Спектры ЯМР 13С зарегистрирова-
количествами веществ. В процессе анализа опре-
ны на приборе Bruker Avance с рабочей частотой
деляли корреляцию между изменением аналити-
100 МГц (Германия).
ческого сигнала и изменением массы иммобилизо-
ванного на подложке рецептора в ходе обратимого
Масс-спектры высокого разрешения регистри-
образования лиганд-рецепторного комплекса в
ровали на масс-спектрометре высокого разреше-
контролируемом проточном режиме.
ния Orbitrap Elite (США). Растворы образцов в
ацетонитриле с 1%-ной муравьиной кислотой вво-
В проведенных тестах для полученных про-
дили в источник ионизации методом электрорас-
изводных 1 и 2 определена равновесная констан-
пыления.
та диссоциации комплекса с рецептором, на 6
порядков меньшая [KD(1) = 0.8±0.3 нМ, KD(2) =
Для очистки и анализа образцов методом ВЭЖХ
1.1±0.3 нМ], чем константа природного лиганда
использовали систему Shimadzu Prominence LC-20
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
170
ПЕТРОВ и др.
Схема 3. Получение целевых триантеннарных лигандов
HO OH
NaN3
N
COOH
O
Br
COOH
3
+
10
OH
10
H
O
14
2N
COOt-Bu
COOt-Bu
NaOH
14
COOt-Bu
COOt-Bu
O O
O O
DMSO H2O
EDC, NHS
O
THF
N3
O
O
N
COOt-Bu
H2N
COOt-Bu
10
H
15, 56%
16, 62%
H
O
N
NHBoc
COOH
H2N
NHBoc
O
HCOOH
COOH
18
O O
O
EDC, NHS,
O O
N
NHBoc
O
H
THF
N3
O
H
N3
O
N
NHBoc
N
COOH
N
10
H
10
H
O
17, 100%
19, 66%
H
O
N
NH
3
CF3COO
O
CF3COO
TFA
O O
N
NH3
CH2Cl2
O
H
H
N3
O
N
NH
3
N
10
H
CF3COO
O
20, 100%
R
OH
N
N
O
N
R =
O
HO
NHAc
H
H
1, 22%
O
N
N
R
OH
12 ɢɥɢ 13,
O
OH
EDC, NHS,
O
O
O
N
DIPEA
O
DMF
N
HO
O
O O
N
N
N
O
H
H
NHAc
N3
2, 7%
N
H
O
10
H
O
N
H
N
O
R
O
N
N
N
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
171
(Япония) с конвекционной колонкой и коллекто-
2-[3-(1-{[(2R,3R,4R,5R,6R)-5-Ацетамидо-6-
ром фракций, соединенным с одиночным квадру-
(аллилокси)-3,4-дигидрокситетрагидро-2H-
польным масс-спектрометром Shimadzu LCMS-
пиран-2-ил]метил}-1H-1,2,3-триазол-4-ил)фе-
2020 с двойным источником ионизации DUIS-ESI-
нокси]уксусная кислота (12). Соединение
10
APCI (Япония). Аналитическая и препаративная
(0.155 г, 0.00054 моль) и соединение 11 (0.0955 г,
колонка - Phenomenex Luna 3u C18 100A.
0.00054 моль) растворяли в 10 мл смеси тетра-
гидрофуран-вода. В инертной атмосфере при-
Препаративное хроматографическое разделе-
бавляли пентагидрат сульфата меди
(0.135 г,
ние веществ проводили при помощи хроматогра-
0.00054 моль) и аскорбат натрия
(0.107 г,
фа INTERCHIM puriFlash 430 на обращенно-фа-
0.00054 моль). Реакционную смесь перемешива-
зовой колонке PURIFLASH C18-HP 15UM F0012
ли в течение суток. После прохождения реакции
(Франция).
растворители удаляли при пониженном давлении,
(3-Этинилфенокси)уксусная кислота
(11).
смесь растворяли в метаноле и отфильтровывали
Смесь 3-этинилфенола (1.02 г, 0.01 моль), этил-
через тонкий слой цеолита. Из фильтрата удаляли
2-бромацетата (1.67 г, 0.01 моль) и K2CO3 (3.036 г,
избыток растворителя на роторном испарителе и
0.022 моль, 2.2 экв) в 12.5 мл ДМФА перемешива-
полученную смесь очищали методом колоночной
ли при температуре 60°C в течение 8 ч. Затем смесь
хроматографии в системе с градиентом концен-
разбавляли водой и перемешивали ещё 16 ч при
траций метанол-дихлорметан (1:20) → метанол-
60°C. Полноту протекания реакции контролиро-
дихлорметан (1:2). Выход 0.120 г (48%), тем-
вали с помощью метода ТСХ. После полного про-
но-коричневое твердое вещество. Спектр ЯМР 1Н
текания реакции смесь охлаждали до комнатной
(ДМСО-d6), δ, м.д.: 1.80 с (3H, NHAc), 3.54-3.72 м
температуры и в реакционную смесь осторожно
(4Н, H2,3,5,6), 3.77-3.81 м (1H, H4), 4.02-4.13 м (2Н,
прибавляли воду (50 мл), экстрагировали диэтило-
H1,6), 4.25 с (2Н, H25), 4.54 д (2Н, H13, J 6.5 Гц),
вым эфиром (2×50 мл), органическую фазу суши-
4.98 д.д (1Н, H15, J 1.8, 10.5 Гц), 5.06 д.д (1Н, H15,
ли над безводным MgSO4, после чего растворитель
J 1.8, 17.4 Гц), 5.55-5.68 м (1Н, H14), 6.77 д (1Н,
удаляли при пониженном давлении. Полученную
NH, 8.0 Гц), 7.26 т (1Н, H20, 8.0 Гц), 7.29-7.36 м
смесь очищали методом колоночной хромато-
(2Н, H21,22), 7.79 д (1Н, H19, J 8.2 Гц), 8.53 с (1Н,
графии в системе гексан-этилацетат (4:1). Далее
H16). Спектр ЯМР 13C (CD3OD), δ, м.д.: 22.1, 48.8,
выделенный промежуточный продукт добавляли
50.8, 52.1, 66.7, 67.8, 70.4, 70.9, 73.2, 100.0 (СН=),
в раствор (50 мл) 1 M NaOH в смеси ТГФ-H2O
111.4 (СН=), 114.7 (СН=), 118.5 (СН=),
123.3
(1:1). Реакционную смесь перемешивали в течение
(СН=), 130.5 (СН=), 130.6 (СН=), 147.0 (СН=),
12 ч при комнатной температуре, затем добавляли
158.2 (СН=),174.8 [C(O)NH], 197.0 (СООН). Масс-
ионообменную смолу Dowex 50W до получения
спектр (МСВР), m/z: 463.1832 [M + H]+, 485.16492
нейтрального значения pH, дополнительно смесь
[M + Na]+. C21H26N4O8. [М + H] 463.1829, [M +
перемешивали 10 мин, после чего ионообменную
Na]+ 485.1648.
смолу отфильтровывали. Растворитель удаляли на
роторном испарителе. Выход 1.34 г (76%), свет-
2-(3-{1-[(2R,3R,4R,5R,6R)-3-Ацетамидо-4,5-
ло-коричневое кристаллическое вещество
[46].
дигидрокси-6-(гидроксиметил)тетрагидро-2H-
Спектр ЯМР 1Н, CDCl3, δ, м.д.: 3.01 с (1Н, Н13),
пиран-2-ил]-1H-1,2,3-триазол-4-ил}фенокси)-
4.51 с (2Н, О-СН2), 6.86 д (1Н, H3, J 8.3 Гц), 6.94 с
уксусная кислота (13). Соединение 5 (0.420 г,
(1Н, H5), 7.03 д (1Н, H1, J 7.6 Гц), 7.15 т (1Н, H2, J
0.0017 моль) и соединение 11 (0.299 г, 0.0017 моль)
7.9 Гц). C10H8O3.
O
22
23
N
10
OH
N
8
18
COO
O
N
17
21
26
6
6
O
OH
5
11
16
19
20
5
4
O
14
4
1
O
7
O
2
13
3
3
15
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
172
ПЕТРОВ и др.
17
4
3
O
CH3
20 O
3
16
2
15
19
O
O
CH3
O
CH3
OH
6
14
HOOC
OH
'
2
CH
13
23
H2N
5
6
O
7
O
CH
4
O
N
8
1
N
O
N
10
2
CH3
3
O O
NHAc
CH
3
11
O CH
3
растворяли в 10 мл смеси тетрагидрофуран-вода
(в соотношении 1:1). В инертной атмосфере до-
ным раствором NaCl и сушили над Na2SO4, затем
бавляли пентагидрат сульфата меди
(0.425 г,
удаляли растворитель при пониженном давлении.
0.0017 моль) и аскорбат натрия
(0.333 г,
Выход 2.162 г (84%), прозрачная жидкость [49].
0.0017 моль). Реакционную смесь перемешива-
Спектр ЯМР 1Н, CDCl3, δ, м.д.: 1.23-1.42 м (12Н,
ли в течение суток. После прохождения реакции
H4-9), 1.54-1.69 м (4Н, H3,10), 2.35 т (2Н, H2, J
растворители удаляли при пониженном давлении,
7.5 Гц), 3.25 т (2Н, H11, J 7.0 Гц). C11H21N3O2.
смесь растворяли в метаноле и отфильтровыва-
Трис{[2-(трет-бутоксикарбонил)этокси}ме-
ли через тонкий слой цеолита. Фильтрат удаляли
тил}метиламин (15). Трисгидроксиметиламино-
при пониженном давлении и полученную смесь
метан (2.4 г, 0.0198 моль) растворяли в 10 мл
очищали методом колоночной хроматографии в
1 М раствора NaOH в ДМСО. Смесь охлаждали
системе с градиентом концентраций метанол-
до температуры ~ 17-18°C, после чего по каплям
дихлорметан (1:20) → метанол-дихлорметан (1:2).
в течение 1 ч прибавляли трет-бутилакрилат
Выход 0.468 г (65%), темно-коричневое твердое
(8 мл), после прикапывания охлаждение прекра-
вещество. Спектр ЯМР 1Н (ДМСО-d6), δ, м.д.: 1.60
щали. Реакционную смесь перемешивали в те-
с (3H, NHAc), 3.43 к (1H, H6, J 7.0 Гц), 3.48-3.60
чение суток. Смесь разбавляли водой (75 мл),
м (2H, H2,6), 3.66-3.73 м (1H, H5),3.75 д.д (1H, H3,
J 2.4, 10.4 Гц), 3.81 д (1H, H4, J 2.4 Гц), 4.52 с (2Н,
проводили экстракцию с этилацетатом (2×75 мл)
и промывали органическую фазу насыщенным
H22), 5.66 д (1Н, H1, J 9.9 Гц), 6.83 д (1Н, NH, J
8.1 Гц), 7.31 т (1H, H17, J 8.0 Гц), 7.36 с (1H, H19),
раствором NaCl (75 мл). Растворители и остатки
7.39 д (1H, H16, J 7.6 Гц), 7.85 д (1Н, H18, J 9.1 Гц),
трет-бутилакрилата удаляли при пониженном
8.61 с (1Н, H13). Спектр ЯМР 13C (CD3OD), δ, м.д.:
давлении. Дальнейшую очистку проводили мето-
21.6, 48.8, 52.1, 60.9, 67.7, 70.6, 78.3, 87.0, 111.5
дом препаративной колоночной хроматографии
(СН=), 114.7 (СН=), 118.7 (СН=), 120.9 (СН=),
в системе этилацетат-гексан (1:2 → 2:1). Выход
130.4 (СН=), 147.1 (СН=), 157.9 [NHC(О)], 174.2
5.516 г (56%), вязкая прозрачная маслянистая жид-
(СООН). Масс-спектр (МСВР), m/z:
423.1510
кость [47]. Спектр ЯМР 1Н (CDCl3), δ, м.д.: 1.40 с
[M + H]+, 445.1330 [M + Na]+. C18H23N4O8. [М + H]
(27Н, t-Bu), 1.99 уш.с (2Н, NH2), 2.41 т (6Н, H4,8,12,
423.1510, [M + Na]+ 445.1330.
J 6.4 Гц), 3.28 с (6Н, H2,6,10), 3.60 т (6Н, H3,7,11, J
6.4 Гц). C25H47NO9.
11-Азидоундекановая кислота (14). 11-Бром-
ундекановую кислоту (3 г, 0.0113 моль) и азид на-
N-Трис{[2-(трет-бутоксикарбонил)этокси]-
трия (3.68 г, 0.0566 моль) растворяли в ДМФА.
метил}метиламид
11-азидоундекановой кис-
Реакционную смесь перемешивали при темпера-
COOt-Bu
туре 50°C в течение 3 ч. Растворитель удаляли при
3
пониженном давлении. Оставшуюся реакционную
2
5
6
COOt-Bu
смесь растворяли в CHCl3, промывали насыщен-
O O
O
1
4
1
O
9
3
N3
O
11
9
7
5
3
N
COOt-Bu
7
8
10
H
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
173
(20 мл) и смесь охлаждали до 0°С. Ди-трет-
3
бутилдикарбонат (2.98 г, 0.0137 моль), растворен-
2
5
ный в 30 мл диоксана, прибавляли по каплям в
6
COOH
O O
течение 2 ч. Затем реакционную смесь оставляли
O
1
4
9
при перемешивании на ночь при комнатной тем-
3
O
N
COO
пературе. Растворитель удаляли при пониженном
7
8
H
давлении. Реакционную смесь разбавляли водой.
17
Ди-Boc-производное, выпавшее в осадок, отделя-
лоты (16). 11-Азидоундекановую кислоту
(14)
ли фильтрованием на стеклянном фильтре. Моно-
(0.4945 г, 0.00218 моль) растворяли в дихлор-
Boc-производное экстрагировали из водной фазы
метане (25 мл), к раствору прибавляли N-гид-
хлороформом (5×50 мл). Остатки воды из органи-
роксисукцинимид (0.276 г, 0.0024 моль, 1.1 экв)
ческой фазы удаляли Na2SO4, после чего раство-
и EDC·HCl (0.373 г, 0.0024 моль, 1.1 экв). Реак-
ритель удаляли при пониженном давлении. Выход
ционную смесь перемешивали 15 мин, после чего
1.43 г (60%), прозрачная маслянистая жидкость
прибавляли амин 15 (1.1 г, 0.00218 моль), пере-
[48]. Спектр ЯМР 1Н (CDCl3), δ, м.д.: 1.43 с (9Н,
мешивание продолжали в течение суток. Раство-
t-Bu), 1.60-1.70 м (2Н, H2), 2.59 уш.с (2Н, NH2),
ритель отделяли на роторном испарителе, реакци-
2.79 т (2Н, H1, J 6.5 Гц), 3.17-3.27 м (2Н, H3), 5.00
онную смесь разделяли методом препаративной
уш.с [1Н, С(О)NH]. C8H18N2O2.
колоночной хроматографии в системе хлористый
N-(11-Азидоундеканоил)амино-трис-(1-{3-
метилен-метанол (50:1). Выход 0.960 г (62%), про-
[N-(трет-бутоксикарбонил)аминопропил]-
зрачная маслянистая жидкость [50]. Спектр ЯМР
амино}-3-оксопропокси}метил)-метан (19). Со-
1Н (CDCl3), δ, м.д.: 1.21-1.38 м (12Н, H4'-9'), 1.44 с
единение 17 (0.350 г, 0.00064 моль) растворя-
(27Н, t-Bu), 1.53-1.65 м (4Н, H10',3', J 6.9 Гц), 2.20
ли в хлористом метилене, прибавляли N-гид-
т (2Н, H2', J 7.7 Гц), 2.44 т (6Н, H3,6,9, J 6.2 Гц),
роксисукцинимид (0.243 г, 0.002112 моль, 3.3 экв)
3.24 т (2Н, H11', J 7.0 Гц), 3.63 т (6Н, H2,5,8, J
и EDC·HCl (0.329 г, 0.002112 моль, 3.3 экв).
6.4 Гц), 3.69 с (6Н, H1,4,7), 6.22 уш.с [1Н, С(О)NH].
Реакционную смесь перемешивали в течение
C27H48N4O10.
15 мин, после чего прибавляли соединение 18
N-Трис[(2-карбоксиэтокси)метил]метил-
(0.368 г, 0.002112 моль, 3.3 экв). Перемешивали
амид
11-азидоундекановой кислоты
(17).
12 ч, после чего растворитель удаляли при пони-
Соединение 16 (0.960 г, 0.00134 моль) растворяли
женном давлении, а реакционную смесь разделя-
в 100%-ной муравьиной кислоте (20 мл) и пере-
ли методом препаративной колоночной хромато-
мешивали в течение 12 ч. Муравьиную кислоту
графии в системе метанол-хлористый метилен
удаляли на роторном испарителе. Выход 0.732 г
(1:50→1:20→1:10). Выход 0.429 г (66%), вяз-
(100%), прозрачная жидкость [50]. Спектр ЯМР 1Н
кая прозрачная жидкость [50]. Спектр ЯМР 1Н
(CDCl3), δ, м.д.: 1.18-1.42 с (12Н, H4'-9'), 1.49-1.67
(CDCl3), δ, м.д.: 1.20-1.37 м (12Н, H4'-9'), 1.42 с
(27Н, t-Bu), 1.51-1.69 м (10Н, H3',10',5,11,17), 2.18 т
м (4Н, H10',3'), 2.15 т (2Н, H2', J 7.3 Гц), 2.59 т (6Н,
H3,6,9, J 6.1 Гц), 3.26 т (2Н, H11', J 6.9 Гц), 3.58-
(2Н, H2', 7.6 Гц), 2.44 т (6Н, H3,9,15, J 5.4 Гц), 3.14 т
3.85 м (12Н, H1,2,4,5,7,8), 6.04 уш.с [1Н, С(О)NH].
H
5
C15H24N4O10.
O
N
NHBoc
4
6
N-(трет-Бутилоксикарбонил)-1,3-диамино-
3
O
2
пропан
(18).
1,3-Диаминопропан
(2.04
г,
8
10
12
9
0.0274 моль) растворяли в сухом
диоксане
O O
N
NHBo
11
O
H
1
7
15
H
5
2
NHBo
N
O
O
N
16
18
2N
3
4
C
N
17
1
3
10
13
14
H
O
18
19
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
174
ПЕТРОВ и др.
5
енте концентраций. Выход 0.025 г (22%). Спектр
O
N
NH3
CF3COO
ЯМР 1Н (CD3OD), δ, м.д.: 1.17-1.34 м (12Н, H4'-9'),
4
6
1.46-1.58 м (4Н, H3',10'), 1.66-1.75 м (6Н, H25,30,35),
3
O
COO
CF3
2
8
10
12
1.98 с (9Н, NHAc), 2.14 т (2Н, H2', J 6.5 Гц), 2.41
9
т (6Н, H12,16,20, J 6.1 Гц), 3.21 т (6Н, H26,31,36, J
O O
N
NH3
11
O
H
6.7 Гц), 3.60-3.75 м (18Н, H9,10,11,72,77,91,96,110,115,-
1
7
H
15
O
N
16
18
NH3
11',3,5,7), 3.84 д.д (3Н, H75,94,113, J 3.2, 11.1 Гц), 3.96 д
3
N
17
13
14
10
H
(3H, Н73,92,111, J 3.2 Гц), 4.26 д.д (3Н, H71, 90, 109, J 4.2,
CF3COO
8.7 Гц), 4.32 д.д (3Н, H71,90,109, J 3.7, 11.1 Гц), 4.56 с
20
(6Н, H39,42,57), 4.57-4.63 м (6Н, H79,98,117), 4.69 д.д
(6Н, H6,12,18, J 5.4 Гц), 3.21-3.33 м (8Н, H11',4,10,16),
(3Н, H75,96,114, J 6.0 Гц), 5.04 д.д (3Н, H81,100,119, J
3.61-3.74 м (12Н, H1,7,13,2,8,14), 5.29 уш.с [3Н,
1.5, 17.2 Гц), 5.59-5.72 м (3Н, H80,99,118), 6.98 д.д
С(О)NH-(Boc)], 6.47 уш.с [1Н, С(О)NH], 7.19 уш.с
(3Н, H50,56,65; J 1.9, 8.1 Гц), 7.19 с (1Н, С(О)NH),
[3Н, С(О)NH]. C39H72N10O13.
7.37 т (3Н, H47,54,63, J 8.1 Гц), 7.43-7.50 м (6Н,
2,2,2-Трифторацетат N-(11-азидоундекано-
H61,62,46,47,52,53), 8.35 с (3Н, H68,87,105). Масс-спектр
ил)амино-трис-(1-{3-[(3-аммониопропил)ами-
(МСВР), m/z: 2048.0059 [M + H]+, 2069.9876 [M +
но]-3-оксопропокси}метил)-метана (20). Соеди-
Na]+. C96H138N22O28. [М + H] 2048.0129, [M + Na]+
нение 19 (0.4286 г, 0.000423 моль) растворяли в
2069.9949.
60 мл смеси трифторуксусная кислота-хлористый
N-(11-азидоундеканоил)амино-трис-{3-[(3-
метилен (1:4) , реакционную смесь перемешивали
{2-[3-(1-{[3-ацетамидо-4,5-дигидрокси-6-(ги-
в течение 3 ч, после чего растворители удаляли
дроксиметил)тетрагидро-2H-пиран-2-ил]-1H-
при пониженном давлении. Вещество в дальней-
1,2,3-триазол-4-ил}фенокси)ацетамидо]про-
ших реакциях использовали без дополнительной
пил}амино)-3-оксопропокси]метил}-метан
(2).
отчистки. Выход 0.4469 г (100%), белое аморфное
Соединение 13 (0.0591 г, 0.00014 моль, 3.3 экв)
вещество [50]. Спектр ЯМР 1Н (CDCl3), δ, м.д.:
растворяли в
10 мл ДМФА, добавляли NHS
1.19-1.27 м (12Н, H4'-9'), 1.34-1.45 м (2Н, H3', J
(16 мг, 0.00014 моль, 3.3 экв) и EDC (22 мг,
5.3 Гц), 1.45-1.55 м (2Н, H10', J 6.9 Гц), 1.59-1.71 м
0.00014 моль, 3.3 экв). Реакционную смесь пере-
(6Н, H5,11,17), 2.03 т (2Н, H2', J 6.2 Гц), 2.29 т (6Н,
мешивали в течение 15 мин, после чего прибавля-
H3,9,15, J 6.2 Гц), 2.68-2.81 м (6Н, H4,10,16), 3.04-
ли раствор соединения 20 (0.050 г, 43 мкмоль) и
3.13 м (6Н, H6,12,18), 3.28 т (2Н, H11', J 6.9 Гц), 7.01
25 мкл DIPEA (3.3 экв) в 5 мл ДМФА. Оставляли
уш.с [1Н, С(О)NH], 7.76 уш.с (9Н, NH3+), 8.01-8.07
перемешиваться в течение ночи, после чего рас-
м [3Н, С(О)NH]. C33H66N10O7·3CF3COOH.
творитель удаляли при пониженном давлении, а
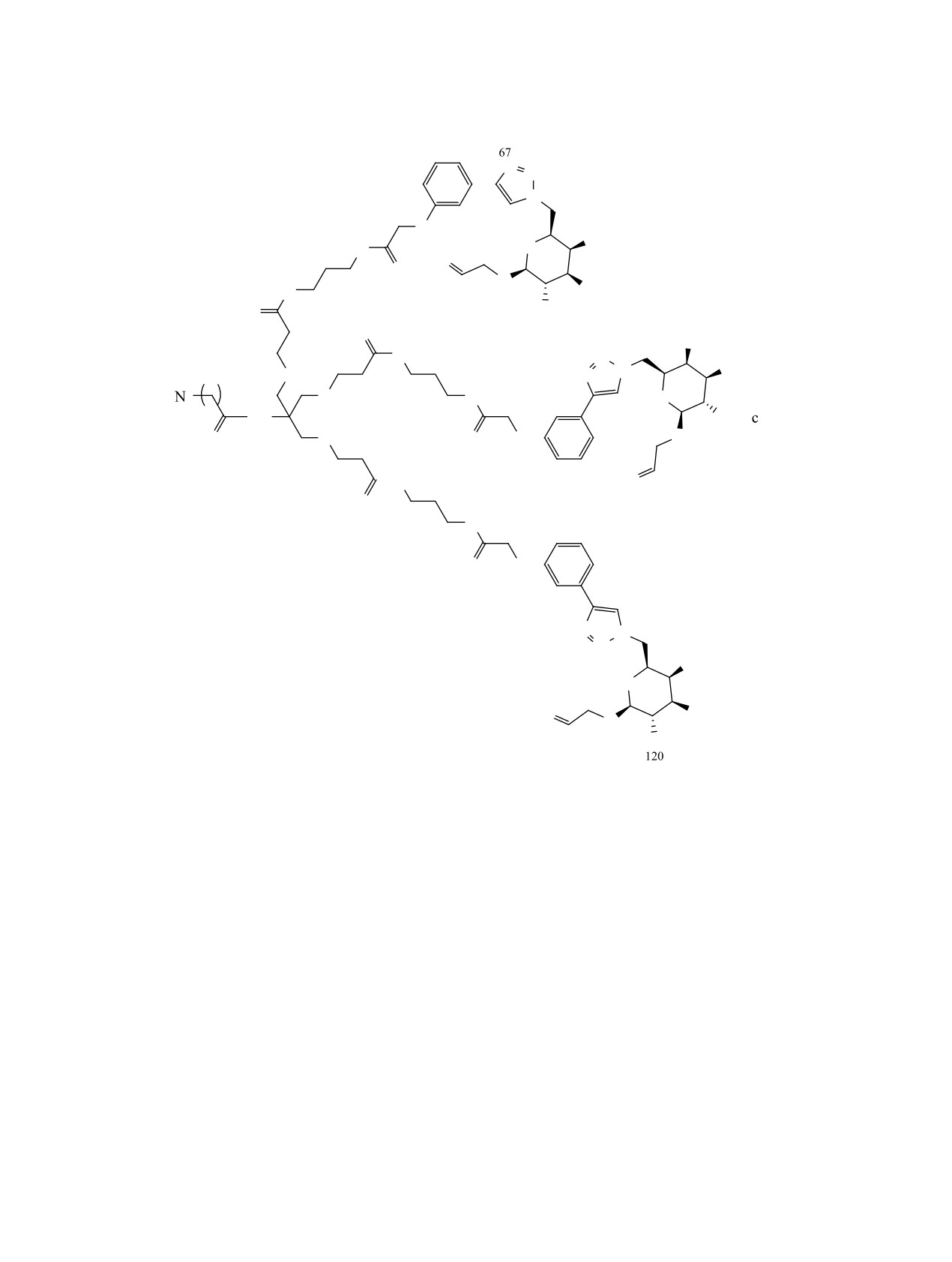
N-(11-азидоундеканоил)амино-трис-({3-[(3-
реакционную смесь разделяли методом обращен-
{2-[3-(1-{[5-ацетамидо-6-(аллилокси)-3,4-ди-
но-фазовой ВЭЖХ в системе вода-ацетонитрил
гидрокситетрагидро-2H-пиран-2-ил]метил}-
в градиенте концентраций. Выход 0.008 г (7%).
1H-1,2,3-триазол-4-ил)фенокси]ацетамидо}-
Спектр ЯМР 1Н (CD3OD), δ, м.д.: 1.15-1.33 м
пропил)амино]-3-оксопропокси}метил)-метан
(12Н, H4'-9'), 1.43-1.57 м (4Н, H3',10'), 1.62-1.72 м
(1). Соединение
12
(0.065 г,
0.00014 моль,
(6Н, H25,30,35), 1.77 c (9H, NHAc), 2.12 т (2Н, H2', J
3.3 экв) растворяли в 10 мл ДМФА, прибавляли
7.3 Гц), 2.31-2.43 м (6Н, H12,16,20), 3.11-3.23 м (6Н,
NHS (16 мг, 0.00014 моль, 3.3 экв) и EDC (22 мг,
H24,29,34), 3.41 т (2Н, H11', J 5.3 Гц), 3.55-3.70 м
0.00014 моль, 3.3 экв). Реакционную смесь пере-
(18Н, H9,10,11,3,5,7,5'',10'',133), 3.75-3.89 м (9Н, H4'',9'',-
мешивали в течение 15 мин, после чего прибавля-
132,2'',7'',130,1'',6'',129),
4.06 д (3H, H3'',8'',131), 4.40-4.61
ли раствор соединения 20 (0.050 г, 43 мкмоль) и
c (6Н, H39,42,57), 5.77 д (3Н, H71,90,109, J 9.5 Гц),
25 мкл DIPEA (3.3 экв) в 5 мл ДМФА. Оставляли
6.86 д (3Н, H50,56,65, J 8.0 Гц), 7.29 т (3Н, H49,54,63,
перемешиваться в течение 12 ч после чего раство-
J 8.0 Гц), 7.32-7.38 м (3Н, H46,53,62), 8.04 с (3Н,
ритель удаляли при пониженном давлении, а реак-
H68,87,105). Масс-спектр (МСВР), m/z:
1927.9127
ционную смесь разделяли методом обращенно-фа-
[M + H]+, 1949.8998 [M + Na]+. C87H126N22O28. [М +
зовой ВЭЖХ в системе вода-ацетонитрил в гради-
H] 1927.9190, [M + Na]+ 1949.9010.
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
175
54
56
N
69
66
N
52
55
N70
51
53
68
71
39
O
72
37
38
40
74
OH84
HN
81
O
79
73
35
H2C
76
O
75
18
36
41
80
O
HN
77
OH83
34
78
19
O
17
NHAc
15
82
16
O
14
88
103OH
9
NH
89
90
102
13
N
91
92
11
25
86
N
OH
O8
N
12
24
27
94
5
7
O
NH
85
3
6
26
87
93
O
95
10
28
42
46
96
HN1
2
48
NHA
45
101
O
O4
O
O
50
O97
3
98
126
20
44
43
10
47
49
99
21 NH22
H2C
100
30
O
29
32
23
NH
31
33 57
61
63
60
O
O
65
58
59
64
62
105
104
107
106
N
N
109
N
108
110
122OH
112
O
111
119
117
114
115
H2C
113
OH121
O
118
116
NHAc
1
Анализ аффинности к рецептору методом
TRIS, pH 7.4. Исследуемые соединения растворя-
спектроскопии поверхностного плазмонного
ли в ДМСО и полученные растворы разбавляли
резонанса. Определение KD in vitro методом спек-
рабочим буферным раствором так, чтобы концен-
троскопии поверхностного плазмонного резонан-
трация органического растворителя была < 1%
са проводили на приборе Biacore X100 (Biacore
(v/v). Концентрация исследуемых лигандов была
AB, Швеция) с использованием чипа-носителя
представлена в широком диапазоне от 10-2 М до
CM5, состоящего из золотой пластины, покры-
5×10-11 М. Раствор исследуемого соединения по-
той слоем карбоксиметилированного декстрана.
давали в течение 180 с (время связывания) при
Поверхность чипа включает в себя 2 проточных
скорости потока 20 мкл/мин и далее в течение 60
ячейки. На поверхности одной из ячеек был им-
с изучали диссоциацию комплекса. Термодина-
мобилизован фермент ASGPR из печени кролика
мическую константу диссоциации KD определяли,
[Generic Assays GmbH, Германия, чистота > 95%
используя модель изотермы адсорбции Ленгмюра
(SDS-PAGE)]. Для иммобилизации рецептора ис-
(1:1 Langmuir association). Для восстановления
пользовали буферную смесь 10 мМ HEPES pH 7.0.
чип-носитель обрабатывали 20 мкл 20 мМ раство-
Для экспериментов по определению аффинности
ра ЭДТА. Полученные данные обрабатывали при
лигандов использовали рабочую буферную смесь,
помощи программного обеспечения BIAevaluation
содержащую 150 мМ NaCl, 50 мМ CaCl2, 50 мМ
3 (BiaCore AB, GE Healthcare, Швеция).
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
176
ПЕТРОВ и др.
54
56
67
N
69
66
N
52
55
70
10"
N
O
51
39
53
68
71
9" OH
O
37
6"
38
40
AcHN
HN
7"
8" OH
35
15"
O
18
36
HO
41
HN
OH
34
5"
19
O
17
15
3"
OH
O
16
14
O 4"
88
9
NH
N 89
2"
13
25
11
86
N 90
O8
N
27
1" OH
5
12
24
N3
7
O
NH
85
AcHN
10
6
26
42
46
87
2
48
20"
HN1
4
28
45
O
O
O
O
50
3
20
44
43
10
47
49
21N22
30
O
32
29
23
NH
31
57
61
63
33
60
O
O
65
58
59
64
62
105
104
133
N
107
O
106
N
OH
N
109
132
108
129
OH
131
AcHN
130
21" OH
2
ЗАКЛЮЧЕНИЕ
ных с использованием триантеннарных лигандов
более сложного строения. Неожиданно лиганд 1
Разработаны и синтезированы новые высокоэф-
упрощенного строения продемонстрировал рав-
фективные триантеннарные лиганды, которые мо-
новесную константу диссоциации, близкую к та-
гут служить векторами для адресной доставки ле-
ковой лиганда 2, предложенного на основании
карственных средств в клетки печени. Полученные
литературных данных. Преимуществами лиган-
лиганды содержат концевую азидную группу, по-
зволяющую вводить их в реакцию [3+2]-азид-ал-
да 1 является более простая и короткая схема син-
теза, а также возможность выделения углеводного
кинового циклоприсоединения, которая является
ортогональной для большинства функциональных
фрагмента без использования колоночной хрома-
групп, присутствующих как в лиганде, так и в по-
тографии. Данные результаты в будущем можно
тенциальных препаратах для адресной доставки.
использовать для оптимизации получения триан-
Оба лиганда 1 и 2 демонстрируют высокую аф-
теннарных лигандов асиалогликопротеинового
финность к асиалогликопротеиновому рецептору;
рецептора за счет упрощения и удешевления их
значения равновесных констант диссоциации ком-
синтеза.
плекса «лиганд-рецептор» находятся в субнаномо-
БЛАГОДАРНОСТИ
лярном диапазоне, что на 6 порядков ниже равно-
весных констант диссоциации комплекса рецепто-
Авторы выражают благодарность сотрудникам
ра и природного лиганда N-ацетилгалактозамина,
лаборатории физико-химических методов анализа
и лишь на порядок больше значений KD, получен-
строения вещества химического факультета МГУ
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
177
имени М.В. Ломоносова доктору химических
6. Иваненков Я.А., Маклакова С.Ю., Белоглазкина Е.К.,
наук, профессору Лебедеву Альберту Тарасовичу,
Зык Н.В., Назаренко А.Г., Тоневицкий А.Г., Коте-
лянский В.Э., Мажуга А.Г. Усп. хим. 2017, 86, 750-
кандидату химических наук Ковалеву Сергею
776. [Ivanenkov Y.A., Maklakova S.Yu., Beloglazki-
Васильевичу, а также Белоглазкину Андрею
na E.K., Zyk N.V., Nazarenko A.G., Tonevitsky A.G.,
Александровичу за помощь в исследовании.
Kotelianski V.E., Majouga A.G. Russ. Chem. Rev.
ФОНДОВАЯ ПОДДЕРЖКА
2017, 86, 750-776.] doi 10.1070/rcr4707
7. Щегравина Е.С., Сачкова А.А., Усова С.Д., Ню-
Исследование выполнено при финансовой под-
чев А.В., Грачева Ю.А., Федоров А.Ю. Биоорг. хим.
держке Российского научного фонда, гранты РНФ
2021, 47, 71-98. [Shchegravina E.S., Sachkova A.A.,
№ 21-73-00106 (синтез поливалентных лиган-
Usova S.D., Nyuchev A.V., Gracheva Y.A., Fedo-
дов асиалогликопротеинового рецептора) и РНФ
rov A.Y. Russ. J. Bioorg. Chem. 2021, 47, 71-98.] doi
№ 20-63-46029 (исследование аффинности мето-
10.1134/S1068162021010222
дом спектроскопии поверхностного плазмонного
8. Brennan F.R., Shaw L., Wing M.G., Robinson C. Mol
резонанса), а также Программы развития МГУ.
Biotechnol. 2004, 27, 59-74. doi 10.1385/MB:27:1:59
9. Kang B.K., Chon S.K., Kim S.H., Jeong S.Y., Kim M.S.,
ИНФОРМАЦИЯ ОБ АВТОРАХ
Cho S.H., Lee H.B., Khang G. Int. J. Pharm. 2004, 286,
Петров Ростислав Александрович, ORCID:
147-156. doi 10.1016/j.ijpharm.2004.08.008
10. Sinha R., Kim G.J., Nie S., Shin D.M. Mol. Cancer
Ther. 2006, 5, 1909-1917. doi 10.1158/1535-7163.
Петров Станислав Александрович, ORCID:
mct-06-0141
11. Stern S.T., McNeil S.E. Toxicol. Sci. 2007, 101, 4-21.
Маклакова Светлана Юрьевна, ORCID: https://
doi 10.1093/toxsci/kfm169
orcid.org/0000-0001-5290-9353
12. Akbarzadeh A., Rezaei-Sadabady R., Davaran S.,
Joo S.W., Zarghami N., Hanifehpour Y., Samiei M.,
Лопухов Антон Владимирович, ORCID: https://
Kouhi M., Nejati-Koshki K. Nanoscale Res. Lett. 2013,
orcid.org/0000-0002-3517-505X
8, 102. doi 10.1186/1556-276X-8-102
13. Bildstein L., Dubernet C., Couvreur P. Adv. Drug Deliv.
orcid.org/0000-0002-9357-8236
Rev. 2011, 63, 3-23. doi 10.1016/j.addr.2010.12.005
Белоглазкина Елена Кимовна, ORCID: https://
14. Wang M., Li Z., Liu F., Yi Q., Pu C., Li Y., Luo T.,
Liang J., Wang J. J. Med. Chem. 2021, 64, 14793-
orcid.org/0000-0001-6796-8241
14808. doi 10.1021/acs.jmedchem.1c01365
КОНФЛИКТ ИНТЕРЕСОВ
15. Petrov R.A., Mefedova S.R., Yamansarov E.Y.,
Maklakova S.Y., Grishin D.A., Lopatukhina E.V.,
Авторы заявляют об отсутствии конфликта ин-
Burenina O.Y., Lopukhov A.V., Kovalev S.V.,
тересов.
Timchenko Y.V., Ondar E.E., Ivanenkov Y.A., Ev-
СПИСОК ЛИТЕРАТУРЫ
teev S.A., Vaneev A.N., Timoshenko R.V., Klyach-
ko N.L., Erofeev A.S., Gorelkin P.V., Beloglazki-
1. SchwartzA.L.,Ashwell G. Crit. Rev. Biochem. Mol. Biol.
na E.K., Majouga A.G. Mol. Pharm. 2020, 18, 461-
1984, 16, 207-233. doi 10.3109/10409238409108716
468. doi 10.1021/acs.molpharmaceut.0c00980
2. Spiess M. Biochemistry. 1990, 29, 10009-10018. doi
16. Rico L., Østergaard M.E., Bell M., Seth P.P., Hanes-
10.1021/bi00495a001
sian S. Bioorg. Med. Chem. Lett. 2018, 28, 2652-2654.
3. Stockert R.J. Physiol. Rev. 1995, 75, 591-609. doi
doi 10.1016/j.bmcl.2018.06.002
10.1152/physrev.1995.75.3.591
17. Petrov R.A., Maklakova S.Y., Ivanenkov Y.A., Pet-
4. Kunjiappan S., Pavadai P., Vellaichamy S., Ram Ku-
rov S.A., Sergeeva O.V., Yamansarov E.Y., Saltyko-
mar Pandian S., Ravishankar V., Palanisamy P., Go-
va I.V., Kireev I.I., Alieva I.B., Deyneka E.V., Sof-
vindaraj S., Srinivasan G., Premanand A., Sankarana-
ronova A.A., Aladinskaia A.V., Trofimenko A.V.,
rayanan M., Theivendren P. Drug Devel. Res. 2021, 82,
Yamidanov R.S., Kovalev S.V., Kotelianski V.E.,
309-340. doi 10.1002/ddr.21758
Zatsepin T.S., Beloglazkina E.K., Majouga A.G.
5. D’souza A.A., Devarajan P.V. J. Сontrol. Rel. 2015,
Bioorg. Med. Chem. Lett. 2018, 28, 382-387. doi
203, 126-139. doi 10.1016/j.jconrel.2015.02.022
10.1016/j.bmcl.2017.12.032
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
178
ПЕТРОВ и др.
18. Ivanenkov Y.A., Majouga A.G., Petrov R.A., Pet-
31. Meier M., Bider M.D., Malashkevich V.N., Spiess M.,
rov S.A., Kovalev S.V., Maklakova S.Y., Yamansa-
Burkhard P. J. Mol. Biol. 2000, 300, 857-865. doi
rov E.Y., Saltykova I.V., Deyneka E.V., Filkov G.I.,
10.1006/jmbi.2000.3853
Kotelianski V.E., Zatsepin T.S., Beloglazkina E.K.,
32. Kolatkar A.R., Leung A.K., Isecke R., Brossmer R.,
Bioorg. Med. Chem. Lett. 2018, 28, 503-508. doi
Drickamer K., Weis W.I. J. Biol. Chem. 1998, 273,
10.1016/j.bmcl.2017.12.004
19502-19508. doi 10.1074/jbc.273.31.19502
19. Prakash T.P., Graham M.J., Yu J., Carty R., Low A.,
33. Feinberg H., Torgersen D., Drickamer K., Weis W.I.
Chappell A., Schmidt K., Zhao C., Aghajan M., Mur-
J. Biol. Chem. 2000, 275, 35176-35184. doi 10.1074/
ray H.F., Riney S., Booten S.L., Murray S.F., Gaus H.,
jbc.M005557200
Crosby J., Lima W.F., Guo S., Monia B.P., Sway-
34. D’Souza A.A., Jain P., Galdhar C.N., Samad A., Dega-
ze E.E., Seth P.P. Nucleic Acids Res. 2014, 42, 8796-
ni M.S., Devarajan P.V. AAPS J. 2013, 15, 696-706.
8807. doi 10.1093/nar/gku531
doi 10.1208/s12248-013-9474-6
20. Springer A.D., Dowdy S.F. Nucleic Acid Ther. 2018,
35. Baenziger J.U., Maynard Y. J. Biol. Chem. 1980, 255,
28, 109-118. doi 10.1089/nat.2018.0736
4607-4613. doi 10.1016/s0021-9258(19)85538-2
21. Weng Y., Xiao H., Zhang J., Liang X.-J., Huang Y.
36. Iobst S.T., Drickamer K. J. Biol. Chem. 1996, 271,
Biotechnol. Adv. 2019, 37, 801-825. doi 10.1016/
6686-6693. doi 10.1074/jbc.271.12.6686
j.biotechadv.2019.04.012
37. Mamidyala S.K., Dutta S., Chrunyk B.A., Preville C.,
22. Hu B., Zhong L., Weng Y., Peng L., Huang Y., Zhao Y.,
Wang H., Withka J.M., McColl A., Subashi T.A.,
Liang X.-J. Sig Transduct. Target Ther. 2020, 5, 1-25.
doi 10.1038/s41392-020-0207-x
Hawrylik S.J., Griffor M.C., Kim S., Pfefferkorn J.A.,
Price D.A., Menhaji-Klotz E., Mascitti V., Finn M.G.
23. Craig K., Abrams M., Amiji M. Expert Opin.
J. Am. Chem. Soc. 2012, 134, 1978-1981. doi 10.1021/
Drug Deliv.
2018,
15,
629-640. doi
10.1080/
ja2104679
17425247.2018.1473375
38. Biessen E.A., Beuting D.M., Roelen H.C., van de Ma-
24. Zimmermann T.S., Karsten V., Chan A., Chiesa J.,
rel G.A., Van Boom J.H.,Van Berkel T.J. J. Med. Chem.
Boyce M., Bettencourt B.R., Hutabarat R., Nochur S.,
1995, 38, 1538-1546. doi 10.1021/jm00009a014
Vaishnaw A., Gollob J. Mol. Ther. 2017, 25, 71-78. doi
10.1016/j.ymthe.2016.10.019
39. Valentijn A.R.P., van der Marel G.A., Sliedregt L.A.,
25. Cedillo I., Chreng D., Engle E., Chen L., McPherson A.,
van Berkel T.J., Biessen E.A., van Boom J.H.
Rodriguez A. Molecules. 2017, 22, 1356. doi 10.3390/
Tetrahedron. 1997, 53, 759-770. doi 10.1016/S0040-
molecules22081356
4020(96)01018-6
26. Setten R.L., Rossi J.J., Han S. Nat. Rev. Drug Discov.
40. Khorev O., Stokmaier D., Schwardt O., Cutting B.,
2019, 18, 421-446. doi 10.1038/s41573-019-0017-4
Ernst B. Bioorg. Med. Chem. 2008, 16, 5216-5231. doi
10.1016/j.bmc.2008.03.017
27. Al Shaer D., Al Musaimi O., Albericio F., de la Tor-
re B.G. Pharmaceuticals. 2020, 13, 40. doi 10.3390/
41. Sliedregt L.A., Rensen P.C., Rump E.T., van Santb-
ph13030040
rink P.J., Bijsterbosch M.K., Valentijn A.R.P., van der
28. de Paula Brandão P.R., Titze-de-Almeida S.S., Titze-
Marel G.A., van Boom J.H., van Berkel T.J., Bies-
de-Almeida R. Mol. Diagn. Ther. 2020, 24, 61-68. doi
sen E.A. J. Med. Chem. 1999, 42, 609-618. doi
10.1007/s40291-019-00438-6
10.1021/jm981078h
29. Chong S., Agarwal S., Agarwal S., Aluri K.C., Arcipre-
42. Rajeev K.G., Nair J.K., Jayaraman M., Charisse K.,
te M., Brown C., Charisse K., Cichocki J, Fitzge-
Taneja N., O’Shea J., Willoughby J.L., Yucius K.,
rald K., Goel V., Gu Y., Guenther D., Habtemariam B.,
Nguyen T., Shulga-orskaya S., Milstein S.
Jadhav V., Janas M., Jayaraman M., Kurz J., Li J.,
Chembiochem.
2015,
16,
903-908. doi
10.1002/
Liou S., Liu J., Liu X., Maclauchlin C., Maier M.,
cbic.201500023
Manoharan M., McDougall R., Nair J., Ramsden D.,
43. Sanhueza C.A., Baksh M.M., Thuma B., Roy M.D.,
Robbie G., Schmidt K., Smith P., Theile C., Vaish-
Dutta S., Preville C., Chrunyk B.A., Beaumont K.,
naw A., Waldron S, Wu J.T., Xu Y., Zhang X., Zla-
Dullea R., Ammirati M., Liu S., Gebhard D., Fin-
tev I., Castellanos-Rizaldos E. Drug Metab. Dispos.
ley J.E., Salatto C.T., King-Ahmad A., Stock I.,
2021. doi 10.1124/dmd.121.000428
Atkinson K., Reidich B., Lin W., Rajesh K., Tu M.,
30. Onizuka T., Shimizu H., Moriwaki Y., Nakano T., Ka-
Menhaji-Klotz E., Price D.A., Liras S., Finn M.G.,
nai S., Shimada I., Takahashi H. The FEBS J. 2012, 279,
Mascitti V. J. Am. Chem. Soc. 2017, 139, 3528-3536.
2645-2656. doi 10.1111/j.1742-4658.2012.08643.x
doi 10.1021/jacs.6b12964
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023
СИНТЕЗ И АФФИННОСТЬ НОВЫХ ТРИАНТЕННАРНЫХ ЛИГАНДОВ
179
44. Lee Y.C., Townsend R.R., Hardy M.R., Lönngren J.,
49. Chen Y., Zhang Q., Flach C., Mendelsohn R., Galoppi-
Arnarp J., Haraldsson M., Lönn H. J. Biol. Chem. 1983,
ni E., Reyes P.I., Yang K., Li R., Li G., Lu Y. Anal.
258, 199-202. doi 10.1016/S0021-9258(18)33240-X
Bioanal. Chem. 2017, 409, 6379-6386. doi 10.1007/
45. Connolly D.T., Townsend R.R., Kawaguchi K.,
s00216-017-0577-2
Bell W.R., Lee Y.C. J. Biol. Chem. 1982, 257, 939-945.
doi 10.1016/S0021-9258(19)68290-6
50. Маклакова С.Ю., Кучеров Ф.А., Петров Р.А., Гоп-
46. Tae H.S., Sundberg T.B., Neklesa T.K., Noblin D.J.,
ко В.В., Шипулин Г.А., Зацепин Т.С., Белоглазки-
Gustafson J.L., Roth A.G., Raina K., Crews C.M.
на Е.К., Зык Н.В., Мажуга А.Г., Котелянский В.Э.
ChemBioChem.
2012,
13,
538-541. doi
10.1002/
Изв. АН. Сер. хим. 2015, 64, 1655-1655. [Maklako-
cbic.201100793
va S.Y., Kucherov F.A., Petrov R.A., Gopko V.V.,
47. Cardona C.M., Gawley R.E. J. Org. Chem. 2002, 67,
Zatsepin T.S., Beloglazkina E.K., Zyk N.V., Majou-
1411-1413. doi 10.1021/jo0161678
ga A.G., Koteliansky V.E., Shipulin G.A. Russ. Chem.
48. Chadwick J., Jones M., Mercer A.E., Stocks P.A.,
Bull. 2015, 64, 1655-1662.] doi 10.1007/s11172-015-
Ward S.A., Park B.K., O’Neill P.M. Bioorg. Med. Chem.
2010, 18, 2586-2597. doi 10.1016/j.bmc.2010.02.035
1056-6
Synthesis and Affinity of Novel Asialoglycoprotein
Receptor Triantennary Ligands
R. A. Petrov*, S. A. Petrov, D. A. Grishin, I. G. Kolmakov, D. S. Abramchuk,
V. T. Tkachenko, E. A. Vlasova, S. Yu. Maklakova, A. V. Lopukhov,
N. L. Klyachko, and E. K. Beloglazkina
Chemistry Department, Lomonosov Moscow State University, Leninskie gory, 1/3, Moscow, 119991 Russia
*e-mail: petrovrostaleks@gmail.com
Received May 22, 2022; revised April 11, 2022; accepted April 13, 2022
New triantennary N-acetylgalactosamine derivatives of tris(hydroxymethyl)aminomethane were synthesized
and used to form complexes with asialoglicoprotein receptor, originally found on hepatocytes. Equilibrium
dissociation constants (KD) of asialoglicoprotein receptor and obtained compounds were measured using surface
plasmon resonance spectroscopy technique. The KD values were in the subnanomolar range, being 6 orders
of magnitude lower than the KD of the complex of the receptor and N-acetylgalactosamine, its native ligand.
The synthesized ligands exhibit much stronger binding to receptor in comparison to the natural ligand. These
results suggest that synthesized ligands are promising agents for the targeted delivery of various therapeutic
agents in hepatocytes.
Keywords: targeted delivery, asialoglycoprotein receptor ligands, hepatocellular carcinoma, N-acetylgalac-
tosamine derivatives
ЖУРНАЛ ОРГАНИЧЕСКОЙ ХИМИИ том 59 № 2 2023