Журнал прикладной химии. 2020. Т. 93. Вып. 6
ОРГАНИЧЕСКИЙ СИНТЕЗ И ТЕХНОЛОГИЯ ОРГАНИЧЕСКИХ ПРОИЗВОДСТВ
УДК 541.64:539.(199+3)
ВЛИЯНИЕ ЗАМЕСТИТЕЛЕЙ НА ЭНЕРГЕТИЧЕСКИЙ БАРЬЕР
ВНУТРЕННЕГО ВРАЩЕНИЯ В АМИНОНИРОЭТИЛЕНАХ
© Б. Э. Крисюк1, Т. М. Сыпко1,2
1 Институт проблем химической физики РАН,
142432, Московская обл., г. Черноголовка, пр. Академика Семенова, д. 1
2 Московский государственный университет им. М. В. Ломоносова,
119991, г. Москва, ГСП-1, Ленинские горы, д. 1, стр. 51
E-mail: bkris@mail.ru
Поступила в Редакцию 23 октября 2019 г.
После доработки 8 февраля 2020 г.
Принята к публикации 8 февраля 2020 г.
Перспективные энергоемкие материалы 1,1-диамино-2,2-динитроэтилен (ДАДНЭ), а также его
производные, в которых один или оба атома водорода аминогруппы замещены на группу NH2 или ОН,
изучали методами квантовой химии с использованием гибридного функционала PBE0 c базисным набо-
ром cc-pVDZ и метода связанных кластеров на уровне CCSD/aug-cc-pVDZ. Термостабильность таких
веществ зависит от величины энергетического барьера внутреннего вращения Er вокруг связи С=С.
Показано, что введение указанных заместителей приводит к понижению Er . Величина Er в основном
определяется структурой внутримолекулярных водородных связей. Введение аминогруппы приводит
к более значительному уменьшению Er , чем введение гидроксильной группы.
Ключевые слова: квантово-химический расчет; FOX-7; внутримолекулярные водородные связи; энер-
гия активации вращения вокруг С=С-связи
DOI: 10.31857/S0044461820060158
Одним из самых перспективных соединений для
ется в том числе последними результатами исследо-
создания безопасных в эксплуатации взрывчатых
вания механизма термического разложения ДАДНЭ
веществ является 1,1-диамино-2,2-динитроэтилен
методом молекулярной динамики в конденсирован-
(ДАДНЭ, или FOX-7) из-за его высокой энергоемко-
ной фазе [6, 7]. Однако предположение об отщепле-
сти при одновременной термической стабильности
нии группы NO2 в лимитирующей стадии реакции
и низкой чувствительности к удару и трению [1-5].
противоречит данным об экспериментальной энергии
Несмотря на большое число работ, посвященных из-
активации термолиза Ea, равной 191 кДж·моль-1 [8],
учению термолиза ДАДНЭ, детальный механизм пер-
так как прочность связи C-NO2 составляет около
вичной стадии его термических превращений до сих
290 кДж·моль-1. Также в качестве начальной лими-
пор остается предметом активной дискуссии. В ка-
тирующей стадии термолиза ДАДНЭ предполагали
честве первичной реакции термолиза предполагали
нитро-нитритную изомеризацию (энергия активации
отщепление NO2, и это предположение подтвержда-
около 250 кДж·моль-1) [9, 10] и внутримолекуляр-
891
892
Крисюк Б. Э., Сыпко Т. М.
ный перенос атома водорода с аминогруппы на атом
углерода и перенос атома кислорода нитрогруппы
на другой атом углерода [11]. Величины энергии
активации для двух последних реакций составляют
205 и 226 кДж·моль-1 [11] соответственно, и обе эти
величины уже довольно близки к экспериментальной
величине Ea. Также в [11, 12] исследован механизм
первичной стадии термолиза через перенос атома
водорода с амино- на нитрогруппу с образовани-
ем нитроновой кислоты в качестве промежуточного
продукта. Этот процесс протекает с низкой энергией
активации, но дальнейшие превращения нитроновой
кислоты проходят через реакции с высокими энерге-
тическими барьерами, в результате чего эффектив-
ная энергия активации данного канала составляет
Для рассматриваемой задачи эти вещества явля-
284 кДж·моль-1 [11]. Таким образом, на настоящий
ются модельными, но при этом часть из них синте-
момент механизм лимитирующей стадии термолиза
зирована и исследована в качестве перспективных
ДАДНЭ остается дискуссионным.
энергоемких материалов. Достаточно активно из-
Важно отметить, что все рассмотренные меха-
учали соединение (2) и его соли [14-16]. Синтез и
низмы предполагают изменение гибридизации ато-
некоторые свойства соединения (3) описаны в работе
ма углерода, в результате чего двойная связь С=С
[17].
становится одинарной. При этом происходит пово-
Расчеты проводили на многопроцессорном кла-
рот плоскости нитрогрупп относительно плоскости
стере в вычислительном центре ИПХФ РАН с по-
аминогрупп. Таким образом, величина энергетиче-
мощью программ GAUSSIAN 09,* Revision C.01
ского барьера внутреннего вращения вокруг связи
и GAMESS [18]. Основные расчеты выполнены с
C=C является составной частью энергии активации
использованием метода DFT c гибридным функци-
лимитирующей стадии практически для любого ме-
оналом PBE0 [19] и базисным набором cc-pVDZ.
ханизма термолиза ДАДНЭ. Поэтому исследование
Выбор этого уровня расчета обусловлен тем, что
возможности внутреннего вращения в ДАДНЭ и его
он достаточно хорошо воспроизводит результаты,
замещенных помимо чисто теоретического интереса
полученные на значительно более высоком уровне
важно для анализа механизмов первичной стадии
расчета в работе [11] (расхождение наших данных
термолиза. В работе [13] выполнено теоретическое
с результатами цитируемой работы не превышает
исследование внутреннего вращения в молекулах
10 кДж·моль-1 [12]), и в то же время не так ресурсо-
ДАДНЭ и его циклических гомологов и показано, что
затратен. Многоконфигурационные расчеты MCSCF
на величину барьера вращения помимо собственно
и MRMP2 выполнены с помощью пакета GAMESS
структуры молекулы оказывают сильное влияние
внутри- и межмолекулярные взаимодействия.
* Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E.,
Цель настоящей работы — исследование влияния
Robb M. A., Cheeseman J. R., Scalmani G., Barone V.,
электронодонорных и акцепторных заместителей в
Mennucci B., Petersson G. A., Nakatsuji H., Caricato M.,
аминогруппе ДАДНЭ на энергетический барьер вра-
Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G.,
щения вокруг связи С=С.
Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R.,
Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O.,
Nakai H., Vreven T., Montgomery J. A., Jr., Peralta J. E.,
Экспериментальная часть
Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N.,
Staroverov V. N., Kobayashi R., Normand J., Raghavachari K.,
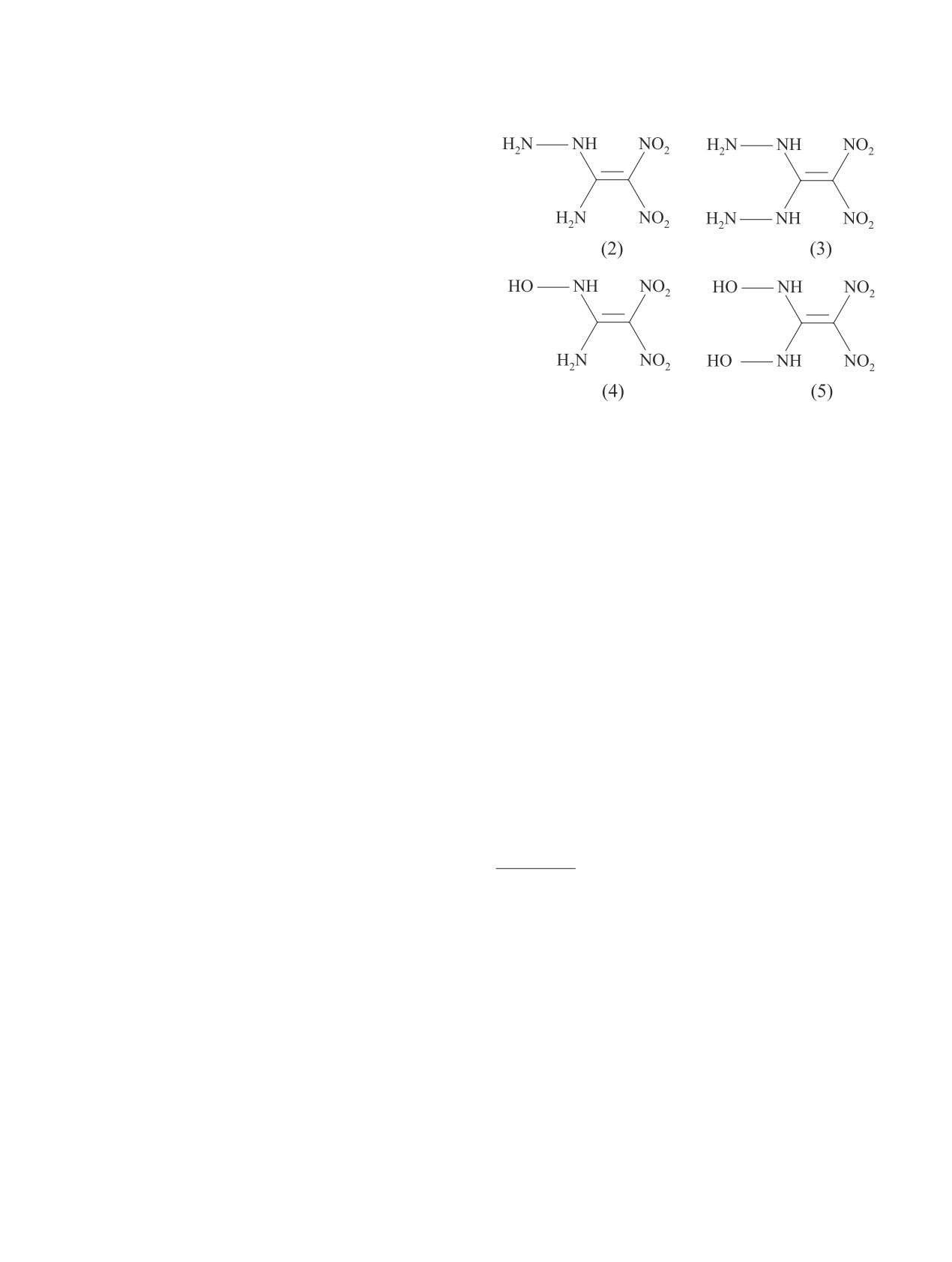
В качестве объектов исследования помимо са-
Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M.,
мого ДАДНЭ (1) выбраны производные ДАДНЭ,
Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B.,
в молекулах которых один или оба атома водоро-
Bakken V., Adamo C., Jaramillo J., Gomperts R.,
да аминогруппы замещены на группу NH2 [1-ами-
Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C.,
Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G.,
но-1-гидразино-2,2-динитроэтилен (2), 1,1-диги-
Voth G. A., Salvador P., Dannenberg J. J., Dapprich S.,
дразино-2,2-динитроэтилен (3)] или на -ОН-группу
Daniels A. D., Farkas Ö., Foresman J. B., Ortiz J. V.,
[1-амино-1-гидроксиламино-2,2-динитроэтилен (4),
Cioslowski J., Fox D. J. // Gaussian 09. Revision C.01.
1,1-дигидроксиламино-2,2-динитроэтилен (5)]:
Gaussian. Inc.: Wallingford CT, 2009.
Влияние заместителей на энергетический барьер внутреннего вращения в аминонироэтиленах
893
[18] и с использованием геометрии, оптимизирован-
щую вершине энергетического барьера вращения,
ной на уровне PBE/cc-pVDZ. Достоверность полу-
дополнительно оптимизировали методами TS и QST.
ченных DFT-результатов контролировали расчетом
Истинность положения барьера контролировали ска-
на уровне CCSD/aug-cc-pVDZ, выполненным для
нированием внутренней координаты реакции в обе
стационарных точек потенциальной поверхности.
стороны от барьера с помощью процедуры IRC (см.
Термодинамические расчеты выполняли с помощью
ссылку на с. 892).
программного пакета MOLTRAN с использованием
частот нормальных колебаний, рассчитанных кван-
Обсуждение результатов
тово-химически.
Внутреннее вращение исследовали сканировани-
Прежде всего отметим, что каждая из замещенных
ем торсионного угла между плоскостями амино- и
молекул имеет несколько ротамерных (таутомерных)
нитрогрупп. Сканирование осуществляли в оба на-
форм. В молекуле (2) их две: в первой аминогруппа
правления с шагом 5-10°. Геометрию, полученную
повернута от нитрогруппы (2а), во второй — к ни-
при сканировании и приблизительно соответствую-
трогруппе (2б):
В молекуле (3) таких форм три:
Аналогично для молекул (4) и (5) имеются две и
ных связей между амино- и нитрогруппами (которые
три формы — (4а), (4б) и (5а)-(5в) соответственно.
образуют энергетически выгодные шестичленные
Некоторые энергетические и структурные характе-
циклы с обеих сторон от связи углерод-углерод),
ристики ротамеров приведены в табл. 1.
так как при образовании переходного состояния в
Во всех молекулах наиболее выгодной является
любом механизме термолиза эти связи необходи-
конфигурация с индексом «а» [кроме соединения (5),
мо разорвать. Для замещенных молекул группы «а»
где энтальпии всех трех ротамеров близки]. В усло-
конфигурация водородных связей аналогична. В то
виях равновесия именно эта форма будет преобладать
же время в замещенных молекулах «б» водородные
в смеси ротамеров. Связано это с различной прочно-
связи наряду с такими же шестичленными циклами
стью водородных связей в этих ротамерах.
образуют с противоположной стороны связи С=С ме-
Термостабильность ДАДНЭ во многом обусловле-
нее выгодные семичленные циклы. В молекулах (3в)
на наличием сильных внутримолекулярных водород- и (5в) семичленные циклы образуются с обеих сторон
894
Крисюк Б. Э., Сыпко Т. М.
Таблица 1
Характеристики равновесных молекул
Торсионный угол между
Длина
Вид
Электронная энергия* Еtot,
Относительная энергия
плоскостями амино-
С=С-связи
молекулы
Хартри
ротамеров ΔН, кДж⋅моль-1
и нитрогрупп θ0, град
L, нм
(1)
-597.6423597
—
14.1
0.1425
(2а)
-652.8909151
0
8.4
0.1431
(2б)
-652.8718951
+50.0
14.3
0.1412
-652.8718949
(3а)
-708.1213688
0
17.9
0.1426
(3б)
-708.1199905
+3.6
25.5
0.1423
(3в)
-708.1107714
+27.8
42.1
0.1427
(4а)
-672.7285963
0
2.1
0.1423
(4б)
-672.7192314
+24.6
36.2
0.1422
(5а)
-747.8051409
0
20.5
0.1419
(5б)
-747.8062007
-2.8
43.0
0.1437
(5в)
-747.8024022
+7.2
43.9
0.1429
* С учетом энергии нулевых колебаний и термической поправки.
от связи С=С. В исходных равновесных молекулах
то в молекулах (1)-(5) величины L близки и состав-
плоскости амино- и нитрогрупп развернуты относи-
ляют 0.141-0.144 нм (табл. 1). Следовательно, связь
тельно друг друга на существенно различные углы θ0
С=С в ДАДНЭ и замещенных формально является
(табл. 1). Отметим, что значения θ0 минимальные у
промежуточной меду двойной и одинарной, т. е. «по-
ротамеров группы «а»: для ДАДНЭ эта величина со-
луторной». Влиянию заместителей, рассмотренных
ставляет 14°, для других — от 2 до 20°. Наибольший
в настоящей работе, эта связь подвержена незначи-
разворот плоскостей наблюдается в группе «в» (42-
тельно, поэтому можно ожидать, что вклад прочности
44°), что также обусловлено структурой боковых
π-связи в барьер вращения во всех изучаемых молеку-
циклов, замкнутых водородными связями.
лах будет примерно одинаков. Тогда решающий вклад
Вопрос о взаимных переходах и равновесии рота-
в изменение величины энергетического барьера вра-
меров выходит за рамки настоящей работы. Отметим
щения в ряду исследуемых изолированных молекул
лишь, что из-за делокализации электронов никакие
должны оказывать внутримолекулярные водородные
связи в рассматриваемых молекулах не являются
связи.
двойными, и вращение вокруг них возможно, т. е. в
Торсионный угол между плоскотями амино- и ни-
газовой или жидкой фазе эти ротамеры скорее всего
трогрупп в исследованных ротамерах имеет отличное
будут находиться в равновесии.
от нуля значение, в группах «б» и «в» он максимален
На барьер вращения в молекулах, подобных
и составляет величину от 15 до 40°. Это еще раз сви-
ДАДНЭ, влияют два взаимосвязанных фактора:
детельствует о сильном мезомерном эффекте и о том,
а) прочность π-связи, б) упомянутые выше водород-
что связь между углеродами в этих молекулах сильно
ные связи. И водородная, и π-связи при вращении раз-
отличается от классической двойной С=С-связи, вра-
рываются. Прочность π-связи сильно зависит от нали-
щение вокруг нее вполне возможно.
чия полярных заместителей и внутримолекулярного
Вращение вокруг кратной связи в общем случае
сопряжения. В исследуемых молекулах π-электроны
связано с разрывом и последующим образованием
частично делокализованы и наблюдается мезомерный
π-связи, и в этом приближении однодетерминант-
эффект. При этом степень делокализации электронов
ный метод PBE может описывать процесс некор-
зависит от внутримолекулярного взаимодействия.
ректно. Чтобы убедиться, что для данной задачи
По длине связи С=С (L) можно косвенно характери-
метод PBE дает адекватные результаты, были выпол-
зовать степень делокализации π-электронов. Если в
нены расчеты на уровне MCSCF и MRMP2 (базис
этилене и этане L = 0.134 и 0.154 нм соответственно,
cc-pVDZ). Для молекулы ДАДНЭ величина барье-
Влияние заместителей на энергетический барьер внутреннего вращения в аминонироэтиленах
895
ра внутреннего вращения составила 59 (MCSCF) и
ного максимума наблюдается еще один небольшой
92 кДж·моль-1 (MRMP2) . Первая величина получена
максимум, который связан с разворотом N-Н-связи
без учета корреляции электронов и потому не точна,
при нулевом угле между плоскостями.
а вторая достаточно хорошо соответствует данным
Наибольший барьер вращения наблюдается у не-
расчетов PBE (табл. 2). Отметим также, что данные
замещенного ДАДНЭ. Введение одного заместите-
РВЕ хорошо соответствуют результатам расчетов на
ля -OH или -NH2 практически в одинаковой мере
более высоком уровне теории CCSD. Энергия акти-
снижает этот барьер (на 6-8 кДж·моль-1). Введение
вации вращения по результатам РВЕ примерно на
второго заместителя еще больше понижает его, при-
10 кДж·моль-1 выше, но в ряду изучаемых молекул
чем аминогруппа в этом смысле заметно эффективнее
оба метода изменение энергетического барьера вра-
гидроксильной.
щения описывают одинаково. Следовательно, вы-
Еще более ярко этот результат виден на примере
бранный DFT-метод применим для данной задачи,
молекул группы «б» (см. рисунок, б). Здесь введение
и поэтому остальные результаты получены на этом
гидроксильных групп приводит к аналогичному с «а»
уровне расчета.
результату, барьеры для (4а) и (4б), для (5а) и (5б)
Расчетные данные на рисунке и в табл. 2 сгруп-
практически одинаковы и в парах, и между собой.
пированы по принципу подобия конфигурации водо-
Это говорит о том, что прочности водородных связей
родных связей в молекулах (см. выше). Рассмотрим с
в шестичленном цикле с группировками N…H…O и
этой точки зрения полученные результаты.
в семичленном цикле с O…H…O близки. Иная кар-
За ноль на всех графиках принят угол θ, соот-
тина наблюдается для аминозамещенных молекул.
ветствующий положению максимума энергии при
В конфигурации «б» барьер вращения резко сни-
вращении, так как минимумы энергии у всех молекул
жается: в молекуле (2б) (одна аминогруппа) — до
соответствуют различным межплоскостным углам,
35 кДж·моль-1; введение второй аминогруппы
и если отсчитывать угол вращения от нулевого угла
(3б) снижает барьер вращения еще больше — до
или от минимума, то кривые пересекались бы, что
29 кДж·моль-1, т. е. при прочих равных условиях
затруднило бы их представление.
замена шестичленного цикла с водородными связями
Для всех молекул с индексом «а» кривые сканиро-
на семичленный со связями N…H…O резко понижает
вания торсионного угла подобны, для них характерен
барьер вращения в изучаемых системах.
достаточно высокий барьер вращения (см. рисунок, а)
В молекулах группы «в» (семичленные циклы с
70-80 кДж·моль-1 (табл. 2). Исключение составляет
обеих сторон С С-связи) отмеченные выше законо-
лишь молекула (3), где барьер ниже примерно на
мерности также проявляются: для гидроксильных
10 кДж·моль-1. На расстоянии около 90° от основ-
заместителей барьер существенно выше, чем для
Таблица 2
Длина C=C-связи и значения энергий активации вращения, рассчитанные на уровнях PBE0/cc-pVDZ
и CCSD/aug-cc-pVDZ
Энергетический барьер вращения, кДж⋅моль-1
Вид молекулы
Длина С=С-связи, нм
PBE0
CCSD
(1)
0.1425
81.3
72.9
(2а)
0.1431
72.9
55.9
(3а)
0.1426
61.4
49.2
(4а)
0.1423
74.7
61.6
(5а)
0.1419
72.4
60.0
(2б)
0.1412
35.4
26.9
(3б)
0.1423
28.8
19.1
(4б)
0.1422
61.7
(69.3)
—
(5б)
0.1437
61.7
(71.2)
—
(3в)
0.1427
23.3
16.0
(5в)
0.1429
56.0
—
896
Крисюк Б. Э., Сыпко Т. М.
Изменение электронной энергии молекул в зависимости от величины угла между плоскостями амино- и нитрогрупп
для молекул группы «а» (а), «б» (б) и «в» (в).
аминогрупп (см. рисунок, в). В целом для этих рота-
дит достаточно гладко. Там же, где есть семичленные
меров характерно минимальное из всех форм значе-
циклы, перестроение водородных связей происходит
ние барьера вращения.
скачкообразно. При этом поиск и оптимизацию пе-
Сканирование торсионного угла мы осуществля-
реходного состояния осуществляли во всех случаях
ли в обе стороны. При этом для молекул группы «а»
с использованием геометрии двух состояний: с кри-
кривые сканирования «вперед» и «назад» совпадают,
вой сканирования «вперед» и «назад» вблизи точки
тогда как для молекул групп «б» и «в» наблюдается
пересечения этих кривых. И в большинстве случаев
их пересечение. Сканирование «вперед» после точки
полученная при такой оптимизации точка ложилась
пересечения приводит к дальнейшему росту энергии,
точно на пересечение кривых сканирования. Именно
и перескок на кривую сканирования «назад» проис-
для таких состояний приведены значения барьера
ходит при углах, больших, чем точка пересечения
вращения в табл. 2. Но в двух случаях (4б) и (5б)
кривых. Это связано с тем, что происходит разрыв
применение такой процедуры привело к некоторо-
имеющейся в исходном состоянии водородной связи
му повышению энергии. Эти значения приведены
и переориентация водорода на противоположный
в табл. 2 в скобках. Для этих переходных состояний
гетероатом. Этот процесс происходит во всех изу-
также характерно наличие одной мнимой частоты,
ченных молекулах, однако если водородные связи
и сканирование IRC также приводило к исходному
замкнуты в шестичленный цикл, то процесс происхо-
и конечному состояниям. Однако эти состояния по
Влияние заместителей на энергетический барьер внутреннего вращения в аминонироэтиленах
897
величине торсионного угла θ смещены на 10-15° от-
них же характерен наиболее высокий барьер враще-
носительно точки пересечения кривых сканирования
ния. Самый низкий барьер наблюдается у молекул
«вперед» и «назад» и находятся вблизи продолжения
группы «в», причем в ротамере (3в) величина этого
одной из этих кривых. Поэтому мы пришли к выводу,
барьера снижена примерно до 20 кДж·моль-1.
что эти переходные состояния не являются оптималь-
На основании этих результатов можно ожидать,
ными, они находятся на гребне поверхности потенци-
что термостабильность замещенных ДАДНЭ будет
альной энергии (одна мнимая частота), но не в сед-
ниже, чем у незамещенной молекулы, а самым неу-
ловой точке. И за величину барьера в этом случае мы
стойчивым соединением будет молекула (3).
принимаем точку пересечения кривых сканирования,
а геометрию оптимального переходного состояния с
Благодарности
помощью стандартных процедур найти не удалось.
Еще более выражен этот эффект для молекулы
Авторы признательны И. Н. Зюзину за ценные
(5в). В этом случае даже неоптимальное переходное
советы при постановке задачи и полезное обсуж-
состояние найти не удалось, и энергию барьера мы
дение.
получили только по точке пересечения кривых ска-
нирования.
Финансирование работы
Как упоминалось выше, достоверность полу-
ченных значений энергетических барьеров враще-
Работа выполнена на средства Института проблем
ния проверили расчетом на более высоком уровне
химической физики РАН по теме 0089-2019-0005
CCSD/aug-cc-pVDZ. Результаты таких расчетов
«Фундаментальные и проблемно-ориентированные
приведены в табл. 2 для тех молекул, где на уровне
исследования в области создания энергетических
PBE/cc-pVDZ удалось в явном виде определить ге-
конденсированных систем (ЭКС) различного назна-
ометрию переходных состояний. В целом расчеты
чения».
CCSD/aug-cc-pVDZ подтвердили сделанный ра-
нее вывод, что для рассматриваемых задач метод
Конфликт интересов
PBE/cc-pVDZ дает адекватные результаты. Значения
барьера вращения при CCSD расчете закономерно
Авторы заявляют об отсутствии конфликта инте-
меньше на 8-15 кДж·моль-1, чем дает расчет PBE.
ресов, требующего раскрытия в данной статье.
Но все закономерности изменения величины барье-
ра вращения оба метода описывают одинаково, да и
Информация об авторах
различие в 10 кДж·моль-1 не является критическим.
Таким образом, расчет на более высоком уровне под-
Крисюк Борис Эдуардович, д.х.н., проф., ORCID:
тверждает описанные выше закономерности.
В заключение отметим, что согласно нашим дан-
Сыпко Тимофей Михайлович,
ным и работе [13] значение барьера внутреннего
вращения в ДАДНЭ составляет величину порядка
80 кДж·моль-1, а энергия активации термолиза по
Список литературы
данным литературы [8] — 191 кДж·моль-1. Сравнивая
эти величины, можно заключить, что барьер враще-
[1] Latypov N. V., Bergman J., Langlet A., Wellmar U.,
ния составляет почти половину энергии активации
Bemm U. Synthesis and reactions of 1,1-diamino-
термолиза, что еще раз подтверждает актуальность
2,2-dinitroethylene // Tetrahedron. 1998. V. 54. N 38.
настоящего исследования.
P. 11525-11536.
[2] Bellamy A. J. FOX-7 (1,1-diamino-2,2-dinitroethene)
Выводы
// High Energy Density Materials / Ed. T. M. Klapötke.
Ser. Structure and Bonding / Ed. D. M. P. Mingos.
На основании квантово-химических расчетов по-
V. 125. P. 1-33. Berlin, Springer, 2007.
казано, что введение OH- и NH2-групп в качестве
заместителей в молекулу ДАДНЭ (FOX-7) приводит
[3] Sikder A. K., Sikder N. A review of advanced high
к снижению барьера внутреннего вращения, причем
performance, insensitive and thermally stable energetic
наиболее существенно барьер снижает введение двух
materials emerging for military and space applications //
аминогрупп [молекула (3)]. Из всех ротамерных форм
J. Hazard. Mater. 2004. V. 112. N 1-2. P. 1-15. https://
в равновесии преобладают молекулы группы «а», для
doi.org/:10.1016/j.jhazmat.2004.04.003
898
Крисюк Б. Э., Сыпко Т. М.
[4] Trzciński W. A., Cudziło S ., Chyłek Z., Szymańczyk L.
[12]
Крисюк Б. Э., Веретин В. С. О некоторых пер-
Detonation properties of 1,1-diamino-2,2-
вичных реакциях термолиза 1,1-диамино-2,2-ди-
dinitroethene (dadne) // J. Hazard. Mater. 2008.
нитроэтилена (FOX-7) по данным DFT расчетов.
Часть 1. Прямой перенос водорода на нитрогруп-
jhazmat.2008.01.026
пу // Бутлеровские сообщ. 2017. Т. 49. № 2. С. 31-
[5] Nair U. R., Asthana S. N., Subhananda Rao A.,
35. ROI: jbc-01/17-49-2-31
Gandhe B. R. Advances in high energy materials //
[13]
Крисюк Б. Э. Влияние внутри- и межмолекуляр-
Defence Sci. J. 2010. V. 60. N 2. P. 137-151. https://
ных взаимодействий на возможность внутреннего
doi.org/10.14429/dsj.60.327
вращения вокруг С=С связи в молекулах FOX-7 и
[6] Liu Y., Li F., Sun H. Thermal decomposition of FOX-7
его производных // Хим. физика. 2020. Т. 39. № 1.
studied by ab initio molecular dynamics simulations //
C. 3-6 [Krisyuk B. E. Influence of intramolecular and
Teor. Chem. Acc. 2014. V. 133. N 10. ID 1557. https://
intermolecular interactions on internal rotation around
doi.org/10.1007/s00214-014-1567-5
c=c bond in fox-7 molecules and its derivatives //
[7] Jiang H., Jiao Q., Zhang C. Early events when heating
Russ. J. Phys. Chem. B. 2020. V. 14. N 1. P. 1-4.
1,1-diamino-2,2-dinitroethylene: self-consistent
charge density-functional tight-binding molecular
[14]
Bellamy A. J., Latypov N. V., Goede P. Transamination
dynamics simulations // J. Phys. Chem. C. 2018.
reactions of 1,1-diamino-2,2-dinitroethene (FOX-7)
V. 122. N 27. P. 15125-15132.
// J. Chem. Res. (S). 2002. N 7. P. 257-257. https://
doi.org/10.3184/030823402103172103
[8] Burnham A. K., Weese R. K., Wang R., Kwok Q. S. M.,
[15]
Axthammer Q. J., Krumm B., Klapötke T. M. The
Jones D. E. G. Thermal properties of FOX-7. 35; Int.
exciting chemistry of 1,1-diamino-2,2-dinitroethene
annual conference of ICT. Karlsruhe, Germany, 2005.
and 1-amino-1-hydrazino-2,2-dinitroethene // J. Phys.
Publ. 70.
Chem. A. 2017. V. 121. N 18. P. 3567-3579. https://
[9] Gindulyte A., Massa L., Huang L., Karle J.
doi.org/10.1021/acs.jpca.7b01742
Proposed mechanism of 1,1-diamino-dinitroethylene
[16]
Katritzky A. R., Sommen G. L., Gromova A. V.,
decomposition: A density functional theory study // J.
Witek1 R. M., Steel P. J., Damavarapu R. Synthetic
Phys. Chem. A. 1999. V. 103. N 50. P. 11045-11051.
routes towards tetrazolium and triazolium dinitro-
methylides // Chem. Heterocyclic Comp. 2005. V. 41.
[10] Храпковский Г. М., Николаева Е. В., Чачков Д. В.,
N 1. P. 127-134.
Шамов А. Г. Теоретическое изучение механизма
[17]
Frumkin A. E., Yudin N. V., Suponitsky K. Yu.,
нитро-нитритной перегруппировки и ее роли в
Sheremetev A. B. 1-Amino-1-hydroxyamino-2,2-
процессах газофазного мономолекулярного рас-
dinitroethene: Novel insights in chemistry of FOX-7
пада с-нитросоединений // ЖПХ. 2004. Т. 74. № 6.
// Mendeleev Commun. 2018. V. 28. N 2. P. 135-137.
С. 983-996 [Khrapkovskii G. M., Nikolaeva E. V.,
Chachkov D. V., Shamov A. G. Theoretical study of
[18]
Schmidt M. W., Baldridge K. K., Boatz J. A,.
the mechanism of the nitro-nitrite rearrangement and
Elbert S. T., Gordon M. S., Jensen J. H., Koseki S.,
its role in gas-phase monomolecular decomposition
Matsunaga N., Nguyen K. A., Su S., Windus T. L.,
of C-nitro compounds // Russ. J. Gen. Chem. 2004.
Dupuis M., Montgomery J. A. General atomic and
molecular electronic-structure system // J. Comput.
B:RUGC.0000042427.28020.77].
Chem. 1993. V. 14. N 11. P. 1347-1363.
[11] Kiselev V. G.. Gritsan N. P. Unexpected primary
reactions for thermolysis of 1,1-diamino-2,2-
[19]
Adamo C., Barone V. Toward reliable density
dinitroethylene (FOX-7) revealed by ab initio
functional methods without adjustable parameters:
calculations // J. Phys. Chem. 2014. V. 118. N 36.
The PBE0 model // J. Chem. Phys. 1999. V. 110. N 13.