

Журнал аналитической химии, 2019, T. 74, № 1, стр. 48-66
Оптические сенсорные системы на основе полиэлектролитного комплекса пероксидазы с хитозаном для определения биологически активных веществИ. А. Веселова, Т. Н. Шеховцова
И. А. Веселова 1, *, Т. Н. Шеховцова 1
1 Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, ГСП-2, Ленинские горы, 1, стр. 3, Россия
* E-mail: irina.veselova@mail.ru
Поступила в редакцию 02.04.2018
После доработки 16.07.2018
Принята к публикации 16.07.2018
Аннотация
В обзоре обобщены и систематизированы работы авторов за последние 15 лет, характеризующие возможности, достоинства и перспективы применения оптических сенсорных систем на основе пероксидазы для определения широкого круга биологически активных веществ – фенольных соединений и гидропероксидов различного строения, фенотиазинов, катехоламинов и их метаболитов – в целях контроля качества лекарственных препаратов, пищевых продуктов, биомедицинских исследований, клинической диагностики.
Одной из актуальных задач современной аналитической химии является создание простых, надежных и чувствительных сенсорных систем для контроля качества лекарственных и косметологических препаратов, пищевых продуктов, биохимического мониторинга, клинической диагностики. Перспективность применения сенсорных устройств в практике химического анализа обусловлена рядом их преимуществ по сравнению с традиционными аналитическими методиками: экспрессностью и экономичностью анализа, технологической и методической простотой, отсутствием необходимости привлечения высококвалифицированного персонала. Включение в такие аналитические средства биологических компонентов – ферментов, антител, ДНК – позволяет повысить чувствительность и селективность анализа и расширяет его возможности за счет высокой избирательности биохимических взаимодействий.
В последние 20–25 лет во всем мире активно проводятся исследования, направленные на создание ферментативных сенсорных систем для определения биологически активных веществ в объектах окружающей среды, продуктах питания, фармацевтических препаратах, физиологических жидкостях. Большинство существующих ферментативных сенсоров традиционно основано на электрохимической регистрации аналитического сигнала, и такие биосенсоры позволяют успешно определять органические соединения многих классов, в то время как созданию и развитию биосенсоров с оптическим (спектрофотометрическим, флуориметрическим и хемилюминесцентным детектированием) уделяется меньшее внимание.
Для создания сенсорных устройств используют такие ферменты, которые катализируют превращение либо только одного соединения (примеры таких узко специфических ферментов крайне малочисленны), либо группы близких по свойствам веществ. Особый интерес представляют ферменты, катализирующие превращение разных по свойствам и строению соединений, например субстратов-восстановителей и субстратов-окислителей.
В этой связи пероксидаза из корней хрена занимает особую нишу в биохимических методах анализа. Этот хорошо изученный высокоактивный коммерческий фермент катализирует превращение разных групп органических веществ, определение которых представляет значительный интерес. В частности, субстратами-восстановителями пероксидазы являются фенольные соединения различного строения, а в роли субстратов-окислителей могут выступать органические пероксиды [1–3]. К первым относятся многие важнейшие экотоксиканты, антиоксиданты природного и промышленного назначения, вторые могут исполнять роль маркеров качества пищевых, лекарственных и косметологических продуктов, а также являются важным техническим и фармацевтическим сырьем. Кроме того, как субстраты-восстановители, так и субстраты-окислители пероксидазы являются важнейшими маркерами различных социально-значимых заболеваний. Определение всех перечисленных выше классов соединений − важная задача химического анализа.
Основное требование, предъявляемое к любым химическим сенсорным системам, заключается в том, что отклик их чувствительного слоя должен быть пропорционален концентрации только определяемого соединения (или нескольких соединений при их групповом определении). При этом компоненты матрицы анализируемого образца не должны влиять на результаты измерения аналитического сигнала. Однако, как правило, реальные объекты, представляющие наибольший аналитический интерес, имеют матрицы сложного состава (например, биологические жидкости), в том числе нерастворимые в воде (многие косметические и лекарственные препараты, пищевые продукты). Компоненты этих матриц могут не только вносить погрешности в результаты измерения, но в ряде случаев значительно затрудняют процедуру анализа. По этой причине при анализе реальных объектов часто возникает потребность дополнительной пробоподготовки анализируемого образца с использованием токсичных или агрессивных органических растворителей, фильтрования или разделения, что существенно увеличивает погрешность результатов измерений, продолжительность анализа, а также осложняет его выполнение.
Перспективный подход к решению перечисленных проблем, по мнению авторов, заключается в создании твердофазных сенсорных систем, основанных на формировании и измерении оптического (спектрофотометрического или флуоресцентного) сигнала не в анализируемом растворе, а непосредственно на поверхности сенсора в его чувствительном слое. Есть все основания полагать, что внедрение таких индикаторных систем и приемов, расширяющих возможности ферментативных методов, в дальнейшем позволит упрочить место оптических биосенсоров среди современных средств анализа.
Настоящий обзор посвящен проблеме совершенствования существующих и создания новых оптических сенсорных устройств на основе иммобилизованной пероксидазы для определения биологически активных веществ.
ПЕРОКСИДАЗА В САМОСОБИРАЮЩЕМСЯ КОМПЛЕКСЕ С ХИТОЗАНОМ КАК ОСНОВА БИОРАСПОЗНАЮЩЕГО СЛОЯ ОПТИЧЕСКОЙ СЕНСОРНОЙ СИСТЕМЫ
Пероксидаза из корней хрена, как упомянуто выше, − наиболее часто используемый в химическом анализе фермент [2, 3]. Однако расширение ее аналитического применения ограничено недостаточной специфичностью и чувствительностью к ряду субстратов и эффекторов, а также малой стабильностью нативного фермента и низкой эффективностью биокатализа в органических средах при определении биологически активных соединений в мало- или нерастворимых в воде объектах [4].
В течение трех последних десятилетий интерес исследователей к поиску подходов к осуществлению ферментативного катализа в средах органических растворителей неуклонно растет [5–8]. Следует отметить, что в неполярных (гидрофобных) растворителях, практически не влияющих на структуру белков, ферменты, как правило, сохраняют каталитически активную конформацию [9, 10] и стабильны [11, 12]. В присутствии полярных органических растворителей вследствие денатурации белковой глобулы биокатализаторы частично или полностью теряют каталитическую активность [13, 14]. Среди приемов, рекомендуемых для повышения устойчивости биокатализаторов к этому типу инактивации, можно отметить следующие: иммобилизация ферментов на твердых носителях [15], замена аминокислотных остатков белка [16, 17], его ковалентная модификация низкомолекулярными реагентами [18, 19] и полимерами [20], молекулярный импринтинг ферментов с нерастворимыми в воде лигандами с последующей лиофилизацией [21]. Однако ни один из этих способов стабилизации биокатализаторов не позволяет сохранить их высокую каталитическую активность в широком концентрационном диапазоне полярных органических растворителей.
Перспективным подходом (ранее не использовавшимся в химическом анализе) к преодолению указанных проблем является включение биокатализаторов в самособирающиеся структуры полиэлектролитов. Достоинством комплексов фермент–полиэлектролит, образованных за счет неспецифических электростатических взаимодействий, является возможность моделирования физико-химических свойств комплекса и кинетических параметров катализируемых им реакций варьированием природы и молекулярной массы полимеров, изменением их соотношения в реакционной смеси в процессе иммобилизации, условий формирования комплекса (природы буферных растворов, рН, ионной силы и т.д.) [22]. Таким образом, появляются возможности создания высокоактивных и стабильных ферментативных систем с заданными свойствами (чувствительностью и селективностью) для решения конкретных аналитических задач.
Следует отметить, что способ включения фермента в полиэлектролитный комплекс отличается простотой применяемых методик, обеспечивает равномерное распределение биокатализатора в объеме носителя, что позволяет получать стабильные иммобилизованные препараты с хорошо воспроизводимыми характеристиками [7, 21]. Кроме того, такие системы легко формируются, в ряде случаев оптически прозрачны и удобны для дальнейшего применения в химическом анализе. Образование полиэлектролитных комплексов между полиионами является на сегодняшний день наиболее простым путем формирования структур как нано- (водорастворимые коньюгаты, наночастицы), так и микроскопических (физические гели, пленки) размеров [22]. Последующее их закрепление на твердых носителях (оптических стеклах, тест-полосках, пластинках и т.д.) приводит к значительному улучшению механических свойств ферментных препаратов и служит основой для конструирования биосенсоров [23–25].
Выбор природы полиэлектролита для образования комплекса с пероксидазой. Для формирования нековалентных полиэлектролитных ферментных комплексов перспективно применение природных полисахаридов. Повышенный интерес к этому типу полимеров обусловлен их высокой биологической активностью и способностью образовывать интерполиэлектролитные комплексы с белками. Варьируя химическую природу полисахаридов и используя их различные производные, можно получать матрицы с оптимальными для пероксидазы характеристиками, в том числе обладающие распознающими способностями, необходимыми для дальнейшего применения ее иммобилизованных препаратов, в частности, в составе биосенсоров. При выяснении влияния природы полиэлектролитов на физико-химические свойства пероксидазы нами были изучены природные полисахариды и их синтетические производные трех различных типов: анионные – карбоксиметилцеллюлоза и альгинат кальция, неионные – агароза, β-циклодекстрин и крахмал, а также катионный – хитозан [26]. Активность и стабильность пероксидазы в их присутствии контролировали по скорости индикаторной реакции окисления пероксидом водорода о-дианизидина [7, 27]. Несмотря на различную природу и тип полисахаридов, все они оказывали активирующее действие (22 ± 4%) на скорость пероксидазного превращения органического субстрата. При этом чрезвычайно важно для дальнейшего аналитического применения ферментных препаратов, что наиболее стабильной оказалась пероксидаза в присутствии хитозана [7]. Хитозан – полисахарид, обладающий высоким сродством к белкам, особенно к ферментам, и хорошей сорбционной способностью. Его часто применяют в качестве носителя для иммобилизации биомолекул разными способами: ковалентным и нековалентным связыванием, капсулированием, включением в гели и т.д. Этому способствуют такие его свойства, как биосовместимость, низкая токсичность, химическая инертность, способность образовывать пленки, гели, мембраны; механическая прочность, высокая проницаемость по отношению к воде, гидрофильность. Хитозан является перспективной матрицей для создания оптических сенсоров, поскольку не поглощает в ближней УФ и видимой областях спектра [27, 28]. Кроме того, наличие реакционноспособных аминогрупп в составе полимера, как это будет показано ниже, позволяет регулировать чувствительность и селективность пероксидазы к ряду органических субстратов, например фенолам различного строения [28].
Влияние на каталитическую активность пероксидазы хитозанов с различными молекулярными массами (со степенью деацилирования 85%) изучено с использованием той же индикаторной реакции окисления пероксидом водорода о-дианизидина. Установлено, что все хитозаны – низкомолекулярные (со средними молекулярными массами (ММ) 5, 10, 25 кДа) и высокомолекулярный (со средней ММ 150 кДа) при их различных содержаниях в реакционной смеси (0.01−10 и 0.001−0.05 мас. % соответственно) оказывают на пероксидазу (0.1 нМ) активирующее действие, степень которого зависит как от молекулярной массы полимера, так и от его концентрации. В наибольшей степени активирует пероксидазу 0.1%-ный (по массе) раствор хитозана со средней ММ 10 кДа (рис. 1) [7].
Рис. 1.
Зависимость степени активирования (А, %) пероксидазы от молекулярной массы (ММ, кДа) и содержания (схитозана, мас. %) хитозана в реакционной смеси (0.05 М фталатный буферный раствор, рН 5.0). ММ хитозана, кДа: 1 – 5; 2 – 10; 3 – 25; 4 – 150.
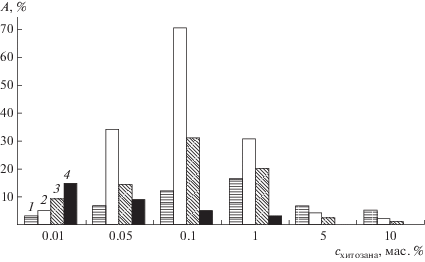
Активирующее действие хитозанов на пероксидазу обусловлено, очевидно, образованием ими самособирающегося комплекса {фермент–полисахарид}, более каталитически активного, чем нативный биокатализатор. Аналогичное повышение активности таких ферментов, как ксантиноксидаза и хитиназа, за счет образования ими полиэлектролитных комплексов с хитозаном отмечено в работе [29].
Наличие электростатических взаимодействий между пероксидазой и хитозаном (на примере полимера с ММ 150 кДа) доказано нами при изучении влияния хлорида калия (0.001−0.1 М) на скорость пероксидазного окисления о-дианизидина в присутствии полисахарида (известно, что сильные электролиты разрушают электростатические взаимодействия между полиионами). Показано, что степень активирующего действия хитозана на пероксидазу понижается по мере увеличения концентрации KCl в реакционной смеси. При этом в отсутствие полисахарида хлорид калия в изученном интервале концентраций не влияет на скорость индикаторной реакции [7].
Влияние хитозанов с различными молекулярными массами на стабильность пероксидазы. Для изучения стабильности препаратов иммобилизованной пероксидазы ее полиэлектролитные комплексы с хитозанами со средними ММ 5, 10, 25 и 150 кДа предварительно закрепляли физической сорбцией на твердом сорбенте [7] и затем хранили в герметичной упаковке при 4°С. Активность полученных ферментных препаратов контролировали до тех пор, пока она сохранялась на уровне не менее 50% от первоначальной. Стабильность полиэлектролитных комплексов с низкомолекулярными хитозанами возрастала с увеличением ММ полисахарида. Наиболее стабильным (активность на уровне 50% от первоначальной сохранялась в течение 120 сут) оказался комплекс в присутствии 10 мас. % хитозана с ММ 25 кДа. Однако максимальную стабильность проявил полиэлектролитный комплекс, образованный высокомолекулярным (ММ 150 кДа) хитозаном с содержанием 0.001 мас. %: он сохранял не менее 50% от первоначальной активности в течение 550 сут (рис. 2).
Рис. 2.
Продолжительность (t, сут) сохранения не менее 50% активности (от начальной) препаратами пероксидазы, иммобилизованными на пенополиуретане, в присутствии хитозанов с различными молекулярными массами и содержаниями в смеси для иммобилизации. ММ хитозана, кДа: 1 – 5, 2 – 10, 3 – 25, 4 – 150.
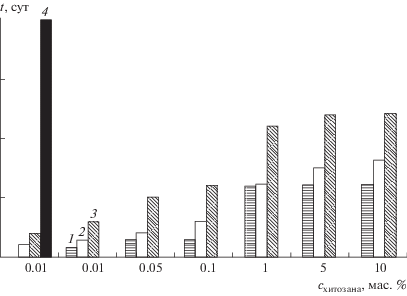
В связи с тем, что стабильность биокатализатора в составе комплекса является одной из наиболее важных аналитических характеристик сенсорных систем, для дальнейших исследований выбрали хитозан со средней ММ 150 кДа.
Влияние рН, ионной силы и природы буферного раствора на каталитическую активность комплекса {пероксидаза–хитозан} детально изучено в оптимальных условиях формирования нековалентных комплексов, образующихся в результате электростатических взаимодействий между полиэлектролитами. Такое исследование необходимо, прежде всего, в тех случаях, когда значения рКа полимера (рКа хитозана составляет 6.3−6.5, он растворим только в протонированной форме [29, 30]) и изоэлектрической точки фермента (pI пероксидазы составляет 7.2 [31]) различаются незначительно.
Каталитическая активность пероксидазы в реакции окисления о-дианизидина в присутствии 0.0005–0.01 мас. % высокомолекулярного хитозана оказалась максимальной в 0.05 М фталатном буферном растворе при рН 5.9–6.2 [7]. Таким образом, активность комплекса максимальна в области рН, в которой, судя по значениям рКа хитозана и точки электронейтральности пероксидазы, предположительно имеется наибольшее количество депротонированных групп хитозана и еще достаточное количество протонированных групп пероксидазы, необходимых для формирования полиэлектролитного комплекса. Не меньшее значение имеет и содержание хитозана в системе. Наибольшую каталитическую активность (в два раза выше, чем у нативного фермента) комплекс {фермент−полисахарид} проявляет в 0.05 М фталатном буферном растворе при содержании высокомолекулярного хитозана в реакции 0.006−0.009 мас. %.
При изучении влияния природы буферного раствора на свойства и условия формирования комплекса использовали помимо фталатного 0.05 М фосфатный и цитратный буферные растворы с рН 5.9−6.2. В цитратном буферном растворе хитозан при различных его содержаниях (0.0005−0.01 мас. %) в реакционной смеси не изменяет каталитическую активность пероксидазы. В фосфатном буферном растворе хитозан активирует пероксидазу, но слабее, чем во фталатном. Изменение характера и степени влияния полисахарида на фермент может быть обусловлено либо значительным увеличением ионной силы буферного раствора, либо изменением заряда аниона, либо совместным действием этих двух факторов [7].
Для выяснения причин влияния природы буферного раствора на условия образования комплекса изучены диаграммы распределения форм фталевой, фосфорной и лимонной кислот и рассчитаны значения ионной силы соответствующих буферных растворов при рН 5.9. При этом значении рН ионная сила возрастает в следующем ряду 0.05 М буферных растворов: фосфатный (I = 0.03 M), фталатный (I = 0.09 M), цитратный (I = 0.18 M). Причем ионная сила, при которой степень активирования пероксидазы хитозаном составляет более 60%, в 0.05 М цитратном буферном растворе всего в 1.5 раза ниже, чем в 0.2 М фталатном буферном растворе с I = 0.27 M. Следовательно, природа буферного раствора незначительно влияет на ионную силу раствора, от которой могли бы зависеть условия формирования комплекса {пероксидаза–хитозан}.
Следует отметить, что согласно распределительным диаграммам при рН 5.9 около 7% фосфорной и 80% лимонной кислот находятся в виде двухзарядных ионов; в случае фталевой кислоты при данном рН такие ионы отсутствуют. По данным [32] присутствие двухзарядных анионов кислот способствует сшиванию молекул хитозана между собой, что и происходит при рН 5.9 в цитратном буферном растворе. В фосфатном буферном растворе протекают два конкурирующих процесса: взаимодействие пероксидазы с хитозаном и взаимодействие хитозан–хитозан. И если в первом случае исключается возможность образования комплекса между молекулами белка и полисахарида, то во втором содержание полиэлектролитного комплекса в системе мало.
Распределение по размерам частиц комплекса {пероксидаза–хитозан}. При исследовании методом фотонно-корреляционной спектроскопии однородности получаемых частиц полисахаридного комплекса и их среднего гидродинамического радиуса установлено, что в оптимальных условиях формирования комплекса частицы пероксидазы с хитозаном представляют собой упорядочную структуру со средним размером частиц 22 ± 3 нм [4]. Столь высокая однородность получаемых частиц на наноуровне свидетельствует о перспективности их использования в дальнейшем для получения гомогенных и воспроизводимых биочувствительных пленок как основы оптических сенсоров.
Влияние диметилсульфоксида на каталитическую активность комплекса {пероксидаза–хитозан}. В смесях полярных органических растворителей ферменты часто теряют каталитическую активность вследствие денатурации биомолекулы, степень которой зависит как от полярности органического растворителя, так и от его содержания в реакционной среде [33, 34]. При этом комплексы белков с полиэлектролитами устойчивы в присутствии органических растворителей [35–39].
Нами изучена активность и стабильность комплекса {пероксидаза–хитозан} в среде полярного органического растворителя − диметилсульфоксида (ДМСО). Выбор ДМСО обусловлен его довольно частым применением в медицинской практике для обеспечения направленного транспорта лекарственных веществ через кожный покров и клеточные мембраны. Как следствие, он входит в состав многих фармацевтических препаратов. Кроме того, ДМСО используют для подготовки проб к анализу биохимическими методами. Например, его применяют в качестве экстрагента физиологически активных веществ из клеточных мембран различной структуры, биологических жидкостей и других биообъектов, растворителя фармацевтических препаратов, лекарственных средств и т.д. [40]. Кроме того, этот растворитель смешивается с водой в любых соотношениях, что позволяет исследовать его влияние на активность пероксидазы в широком интервале содержаний в реакционной смеси.
Нами установлено, что при содержании ДМСО 30 об. % в реакции окисления о-дианизидина комплекс {пероксидаза–хитозан} в два раза активнее, чем нативный фермент; при содержании органического растворителя 60 об. % они характеризуются одинаковой каталитической активностью, а в присутствии 70 об. % ДМСО каталитические свойства проявляет только комплекс [7].
Сравнение стабильности пероксидазы нативной и в составе комплекса с хитозаном в присутствии 30 об. % ДМСО показало, что фермент сохраняет половину первоначальной активности в среде органического растворителя в течение 7 и 28 ч соответственно.
Формирование оптически прозрачных пленок на основе комплекса {пероксидаза–хитозан}. Для создания сенсоров на основе комплекса {пероксидаза–хитозан} со спектрофотометрической и флуориметрической регистрацией аналитического сигнала нами разработана простая технология формирования биораспознающих пленок на оптических стеклах и в ячейках полистирольного планшета. При сравнительном изучении пропускания пленок после их выдерживания в течение часа в водном растворе (0.05 M фосфатный буферный раствор с рН 6.5) и в водно-органической среде (в присутствии 30 об. % ДМСО) установлено, что после выдерживания в присутствии органического растворителя пленки более прозрачны (Т = 96 ± 2%, n = 5), чем пленки, выдержанные в водном растворе (Т = 81 ± 4%, n = 5). Для изучения морфологии и топографии пленок, полученных из различных сред, использовали метод атомно-силовой микроскопии (АСМ). Рис. 3 свидетельствует о том, что пленки после их выдерживания в 30%-ном (по объему) растворе ДМСО более однородные и гладкие, чем пленки, полученные из водного раствора (флуктуации по высоте на 1 кв. микрон поверхности составили 30−35 и 140−150 нм соответственно). Толщина оптических пленок, равная 5 микрон, установлена методом оптической спектроскопии [4, 41].
Рис. 3.
АСМ-профили и изображения поверхности биочувствительного слоя на основе самособирающегося комплекса {пероксидаза–хитозан} после выдерживания в 0.05 М фосфатном буферном растворе с рН 6.5 (рис. справа) и среде вода (0.05 М фосфатный буферный раствор с рН 6.5)–ДМСО (30 об. %) (рис. слева).
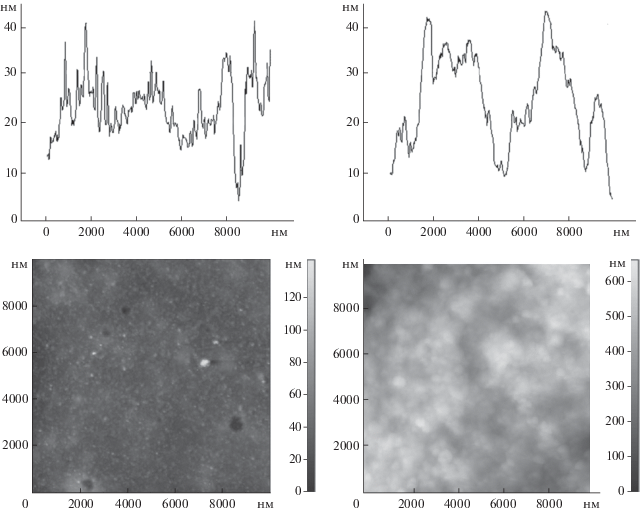
Таким образом, нами показано, что включение пероксидазы в комплекс с хитозаном позволяет получать высокоактивные и стабильные в водных и водно-органических средах ферментные препараты, применение которых открывает широкие перспективы для создания оптических сенсорных систем и устройств, способных функционировать как в водных, так и в водно-органических средах.
КОНСТРУКЦИЯ ТВЕРДОФАЗНЫХ СПЕКТРОФОТОМЕТРИЧЕСКИХ И ФЛУОРИМЕТРИЧЕСКИХ СЕНСОРНЫХ УСТРОЙСТВ, СХЕМЫ РЕГИСТРАЦИИ АНАЛИТИЧЕСКОГО СИГНАЛА
Использование твердофазных оптических сенсорных систем, основанных на формировании и измерении аналитического сигнала вне раствора, позволяет расширить аналитические возможности сенсоров за счет определения биологически активных веществ в объектах на основе сложных матриц (мутных, окрашенных, нерастворимых в воде, неизвестного состава) при их минимальной пробоподготовке. Однако следует отметить, что чувствительность, селективность и особенно экспрессность описанных в литературе твердофазных сенсоров недостаточны для решения многих аналитических задач. Подход, основанный на формировании и измерении аналитического сигнала на твердой поверхности, ранее не использовали при создании спектрофотометрических и флуоресцентных сенсорных систем для определения фенольных соединений, гидропероксидов, фенотиазинов, катехоламинов и их метаболитов.
В качестве простейшей конструкции твердофазных сенсорных устройств, основанных на формировании и измерении аналитического оптического сигнала не в анализируемом растворе, а непосредственно на поверхности сенсора (в его чувствительном слое), нами предложена пластина, по размерам адаптированная под кюветные отделения стандартных спектрофотометров и флуориметров. Поверхность пластины покрыта чувствительным слоем, содержащим фермент и компоненты индикаторной реакции, иммобилизованные в хитозане, для того, чтобы индикаторное соединение оставалось после проведения реакции в этом чувствительном слое [4, 28, 41]. Смесь наносили капельным методом и равномерно распределяли по поверхности пластины, расположенной на строго горизонтальной поверхности, и высушивали на воздухе при комнатной температуре [4, 28].
Для проведения индикаторной реакции сенсорное устройство погружали в раствор, содержащий определяемое соединение, выдерживали в нем необходимое время (непроточный вариант проведения индикаторной реакции), извлекали и высушивали на воздухе при комнатной температуре [4, 28, 41].
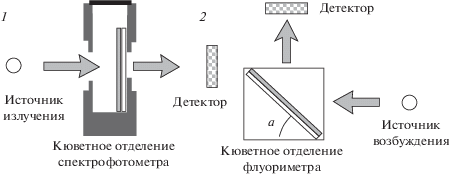
Аналитический сигнал регистрируют в режиме поглощения (спектрофотометрическое детектирование) или отражения (флуориметрическое детектирование) непосредственно в чувствительном слое на поверхности пластины (подложки). Подложку устанавливают на фронтальной поверхности кюветного отделения спектрофотометра или в кюветном отделении флуориметра перед отражающей поверхностью−зеркалом соответственно (рис. 4). Такая конструкция позволяет при необходимости сочетать два способа измерения аналитического сигнала. При использовании отражающей поверхности в случае флуориметрической регистрации сигнала значительно повышается чувствительность определения, поскольку при этом в детектор попадает больше возбужденного излучения.
Рис. 4.
Схемы измерения поглощения (1) и флуоресценции (2) чувствительного слоя твердофазного оптического биосенсорного устройства.
Однако серьезной проблемой при регистрации флуоресцентного сигнала с использованием твердофазных сенсорных устройств предложенной конструкции является высокое значение фонового сигнала в области 300–500 нм (λex = 300–400 нм и λem = = 400–500 нм). Наличие интенсивного фонового сигнала делает фактически невозможным измерение полезного аналитического сигнала. С ц-елью выявления природы фонового сигнала, уменьшения его величины, а также установления рабочей области спектра (а, следовательно, и выбора подходящих индикаторных систем) использован подход, заключающийся в изменении угла отражения возбуждающего излучения. Установлено, что отношение интенсивности флуоресценции фона к интенсивности флуоресценции индикатора пиронина Б минимально при угле 75°. Рекомендуемая ширина входной и выходной щелей составляет 5.0 нм [31].
Таким образом, нами предложены новые типы конструкций и устройств для регистрации аналитического сигнала на твердой поверхности с уче-том их дальнейшего использования в универсальных серийных спектрофотометрах и флуориметрах.
СПЕКТРОФОТОМЕТРИЧЕСКИЕ СЕНСОРНЫЕ СИСТЕМЫ ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ И ИХ ПРИМЕНЕНИЕ В ХИМИЧЕСКОМ АНАЛИЗЕ
Определение ряда фенольных соединений и пероксидов различного строения. Наличие реакционноспособных аминогрупп в составе хитозана обусловливает его способность вступать в некоторые реакции, характерные для аминов. Известно [42], например, что он реагирует с хинонами. Предложено несколько путей практического применения такого взаимодействия, включающих удаление фенольных соединений из сточных вод после их ферментативного окисления и получение модифицированных полимеров на основе хитозана. До сих пор, однако, этот подход не нашел применения в аналитической практике. В то же время аддукт хитозана и хинона сильно поглощает свет в УФ и видимой областях спектра и, таким образом, эта реакция может быть использована для определения фенольных соединений после их предварительного окисления, т.е. возможно создание оптической биосенсорной системы для определения фенольных соединений, которая не требует использования дополнительных хромогенных агентов. Работа предложенного сенсорного устройства основана на двух последовательных процессах: ферментативном окислении фенольного соединения с образованием хинонного продукта и взаимодействии образовавшегося хинонного продукта с аминогруппами хитозана с образованием сильно поглощающего свет аддукта [4, 41]. Последний процесс детально исследован и описан в работах Пейна с соавт. [42, 43], который предположил, что реакция образования аддукта хитозан–хинон протекает по механизму присоединения Михаэля [44].
При разработке спектрофотометрического биосенсорного устройства модельным водорастворимым фенольным соединением служил гидрохинон, который является субстратом пероксидазы. Гидрохинон применяют как антиоксидант при производстве полимеров и в качестве отбеливающего кожу компонента в некоторых фармацевтических препаратах наружного применения.
При оптимальных условиях формирования биораспознающей матрицы на поверхности оптического стекла и регистрации аналитического сигнала возможно определение гидрохинона со следующими характеристиками: диапазон определяемых концентраций 20–200 мкМ, sr = 0.02 при концентрации гидрохинона 20 мкМ (n = 4), предел обнаружения 3 мкМ (табл. 1) [4].
Таблица 1.
Аналитические характеристики спектрофотометрической сенсорной системы для определения различных фенольных соединений и гидропероксидов
| Аналит | Диапазон определяемых концентраций, мкM* | Предел обнаружения, мкM | Чувствительность, M–1 | Органический растворитель, об. % |
|---|---|---|---|---|
| Фенольные соединения | ||||
| Фенол | 20–100 | 13 | 2 × 103 | – |
| Гидрохинон | 20–200 | 3 | 7 × 103 | – |
| Пирокатехин | 20–250 | 7 | 3 × 103 | – |
| Кверцетин | 10–150 | 3 | 12 × 103 | ДМСО (10) |
| Рутин | 10–150 | 10 | 4 × 103 | ДМСО (10) |
| Эскулетин | 10–200 | 10 | 4 × 103 | ДМСО (10) |
| Гидропероксиды | ||||
| Пероксид водорода | 100–1000 | 45 | 25 × 102 | – |
| 2-Бутанолпероксид | 50–1000 | 32 | 12 × 102 | ДМСО (20) |
| Бензоилпероксид | 50–250 | 45 | 9 × 102 | ДМСО (20) |
| трет-Бутилгидропероксид | 150–2500 | 60 | 10 × 102 | Ацетон (50) |
Следует отметить, что аддукты продуктов ферментативного окисления некоторых фенольных соединений и хитозана поглощают свет в УФ или видимой областях спектра при 300 нм или меньших длинах волн [4, 41] Однако в большинстве случаев их влияние на определение гидрохинона при 345 нм невелико и может быть устранено при необходимости методами хемометрики. Поглощение самой пленки хитозана при 345 нм невелико и хорошо воспроизводимо. Другие вещества, присутствующие в системе, также не влияют на аналитический сигнал: продукт ферментативного окисления гидрохинона (хинон) – единственное вещество в системе, которое взаимодействует с хитозаном и таким образом внедряется в пленку, поглощение которой и служит аналитическим сигналом.
Биораспознающий слой обладает высокой стабильностью (последнюю изучали при концентрации гидрохинона 100 мкМ). Перед использованием пластину с ним можно хранить в течение по меньшей мере 3 мес. в холодильнике. Начальный отклик биосенсорного устройства был принят равным 100%. Отклик биосенсорного устройства после 5 дней хранения при комнатной температуре (25°C) составил 69 ± 7% (n = 4, P = 0.95), а после 90 сут хранения этого же сенсора при 4°C – 98 ± 8% (n = 4, P = 0.95). Более того, аддукт, используемый для определения, также очень стабилен во времени, и его поглощение практически не изменяется при хранении биосенсора в течение, по крайней мере, 6 недель после определения. Важной особенностью разработанного биосенсорного устройства является то, что аналитическим сигналом является поглощение самой пластинки, а не реакционного раствора. За счет этого возможен анализ эмульсий и непрозрачных растворов без предварительного отделения матрицы. Если какие-либо частицы образца (например, крема) остаются на поверхности пластинки, то их можно смыть с пленки водой. Вследствие того, что определяемое соединение связано с хитозаном ковалентно, вымывание его из пленки невозможно. Более того, в случае разработанного нами оптического биосенсорного устройства значения оптической плотности при концентрациях определяемого соединения 20–200 мкМ находятся в диапазоне 0.4–1.6, и, следовательно, нет необходимости в использовании дорогих высокочувствительных приборов. Это выгодно отличает предложенное сенсорное устройство от сенсора аналогичной конструкции, разработанного Абдуллой и соавт. [41], отклик которого в присутствии значительно бóльших концентраций определяемого соединения (500–9000 мкМ пирокатехина) меняется в диапазоне 0.01–0.06 ед. опт. плотн. соответственно.
В качестве объектов для апробации методик определения гидрохинона часто выбирают содержащие его фармацевтические мази и гели для отбеливания кожи. Известно, что долговременное употребление гидрохинонсодержащей продукции опасно для здоровья, поэтому использование гидрохинона ограничено или запрещено во многих странах [45]. Кроме того, такие образцы представляют интерес с точки зрения подготовки их проб к анализу. Для демонстрации эффективности сенсора в анализе фармацевтической продукции его применили для определения гидрохинона в креме для депигментации кожи (“Ахромин” производства Ален Мак, Болгария), где он является единственным активным компонентом (табл. 2). Результат 1.9 ± 0.1% (n = 4, P = = 0.95), полученный методом стандартных добавок при использовании предложенного биосенсорного устройства, не отличается от результата, полученного методом ВЭЖХ с амперометрическим детектированием (1.9 ± 0.1%, n = 3, P = 0.95), и соответствует значению, заявленному производителем (1.9%). Следует отметить, что использование предложенного нами устройства в анализе реального образца на содержание гидрохинона значительно удобнее, чем анализ методами ВЭЖХ и спектрофотометрии в УФ-диапазоне, поскольку не нужно добиваться прозрачности образца; достаточно гомогенизации навески образца в воде или ДМСО.
Таблица 2.
Результаты определения фенольных соединений и органических пероксидов в реальных объектах с использованием спектрофотометрической сенсорной системы
| Объект/пробоподготовка | Определяемое вещество/сопутствующие компоненты по данным на упаковке | Найдено с использованием биосенсорного устройства | Заявлено производителем |
|---|---|---|---|
| Крем “Ахромин”/cуспендирование образца в воде, спирте или ацетонитриле | Гидрохинон/парафин, протегин Х, глицерин, ланолин, хлорид натрия, ютанол G, гидрохинон, витконол АРМ, парсол МСХ, отдушка розы, метабисульфит натрия, трилон ВД, трилон В, молочная кислота | 1.9 ± 0.1% | 1.9% |
| Витамины “Аскорутин”/cуспендирование таблетки в ДМСО | Рутин/аскорбиновая кислота, сахар; крахмал картофельный; кальция стеарат; тальк | 0.051 ± 0.003 г/табл. | 0.05 г/табл. |
| Порошок для инъекций “Корвитин”/pастворение образца в ДМСО | Кверцетин/поливинилпирролидон, натрия гидроксид | 0.052 ± 0.004 г/0.5 г | 0.05 г/0.5 г |
| Гель “Базирон”/cуспендирование образца в воде | Бензоил пероксид/акрилат сополимер, полоксамер 182, карбомер 940 (карбополь 980), глицерин, динатрия эдетат, натрия диоктил сульфосукцинат, пропиленгликоль, кремний коллоидный безводный, натрия гидроксид | 5.0 ± 0.4% | 5% |
Отклик сенсорной системы по отношению к фенольным соединениям понижается в ряду кверцетин, гидрохинон, катехол, рутин, эскулетин, фенол. В случае таких фенолов, как резорцин, пирогаллол и флуороглюцин отклик биосенсорной системы отсутствует. По-видимому, это обусловлено тем, что продукты окисления этих фенольных соединений не взаимодействуют с хитозаном с образованием аддукта Михаэля, поскольку, как известно, все изученные фенолы окисляются пероксидом водорода в присутствии пероксидазы [1–3]. Использование в качестве аналитической системы двух последовательных реакций позволяет устранить мешающее влияние многих компонентов нефенольной структуры (за исключением хинонов), которые могут присутствовать в реальных объектах.
При разработке методик определения органических гидропероксидов в качестве индикаторного (хромогенного) вещества использовали пирокатехин. Определение проводили в условиях, оптимальных для определения фенольных соединений, с концентрацией пирокатехина 1 мМ. Чувствительность биосенсорного устройства к органическим гидропероксидам снижается в ряду: 2-бутанон пероксид, бензоилпероксид, трет-бутилпероксид. Аналитические характеристики всех разработанных методик представлены в табл. 1.
Аналитические возможности спектрофотометрической сенсорной системы апробированы при определении девяти фенольных соединений различного строения, а также органических гидропероксидов. При определении фенольных соединений (кверцетина, рутина, эскулетина) и органических пероксидов (2-бутанонпероксида и бензоилпероксида), мало растворимых в водных растворах, реакцию проводили в присутствии ДМСО (10 и 20 об. % соответственно) (табл. 2) [46].
Определение фенотиазинов в водно-органических средах. Фенотиазины – класс искусственно синтезированных гетероциклических соединений, используемых в медицине в качестве транквилизаторов, антидепрессантов, нейролептиков, антиаллергенов и т.д. [47]. В основе химической структуры этой группы препаратов лежит гетероциклическая система, состоящая из шестичленного гетероцикла тиазина, конденсированного с двумя ядрами бензола [48].
Вследствие своей амфифильной природы производные фенотиазинового ряда при попадании в организм взаимодействуют с клеточной мембраной и распределяются между двумя фазами − водным раствором и гидрофобным липидным бислоем (клеточной мембраной) [49]. Для эффективного направленного переноса производных фенотиазина в клетки (например, хлорпромазина в клетки эритроцитов) в состав фармацевтических препаратов в качестве эффективного транспортера сквозь мембранные структуры часто добавляют полярный органический растворитель ДМСО. Этот же растворитель применяют для экстракции фенотиазинов из биологических жидкостей, тканей, а также для растворения лекарственных средств при подготовке проб различных объектов к анализу [50]. Определению фенотиазинов в биологической матрице обычно предшествует сложная пробоподготовка ─ экстракция и предконцентрирование фенотиазинов. Необходимость проведения стадии пробоподготовки связана, во-первых, с малыми концентрациями препаратов в биологических объектах (порядка ppm или ppb), а, во-вторых, со способностью фенотиазинов связываться с молекулами белков, присутствующими в анализируемых объектах.
Наиболее распространенным и удобным способом выделения фенотиазинов из матрицы является их жидкостно–жидкостная экстракция в подходящий органический растворитель, не смешивающийся с водой [51]. В качестве растворителя для экстракции фенотиазинов из плазмы крови чаще всего используют смесь неполярного (гексана) и небольшого объема (5 об. %) полярного (в том числе и ДМСО) растворителей [52, 53].
Разработанное сенсорное устройство использовано для определения следующих фенотиазинов: хлорпромазина (или аминазина), промазина и трифторперазина (или трифтазина). Эти препараты коммерчески доступны, их широко применяют в медицинской практике в качестве лекарственных препаратов; механизм их пероксидазного окисления частично изучен и описан [54, 55].
Ранее установлено [47], что промазин, хлорпромазин и трифторперазин в нейтральной среде активируют пероксидазное окисление таких субстратов пероксидазы, как никотинамидадениндинуклеотид (НАДН) и аскорбат-ион. Этот же эффект так называемой “субстрат-субстратной” активации мы обнаружили в реакции окисления о-дианизидина. Увеличение скорости окисления о-дианизидина пропорционально концентрации фенотиазинов в реакционной смеси, что положено в основу определения промазина, хлорпромазина и трифторперазина в водных растворах в присутствии 30 об. % ДМСО (табл. 3) [4].
Таблица 3.
Метрологические характеристики методик определения фенотиазинов по их активирующему действию на реакцию окисления о-дианизидина, катализируемого нативным ферментом (I) и комплексом {пероксидаза–хитозан} (II) в водных растворах и в присутствии 30% (по объему) ДМСО (n = 5)
| Определяемое соединение | Диапазон определяемых концентраций, мкМ | sr при сн (n = 5) | |
|---|---|---|---|
| 0.05 М фталатный буферный раствор с рН 5.8 | |||
| Промазин | I | 0.03–0.2 | 0.03 |
| II | 0.02–01 | 0.05 | |
| Хлорпромазин | I | 0.07–0.5 | 0.07 |
| II | 0.05–0.4 | 0.04 | |
| Трифторперазин | I | 0.1–1 | 0.05 |
| II | 0.09–0.7 | 0.08 | |
| 0.05 М фталатный буферный раствор с рН 5.8 + ДМСО (30 об. %) | |||
| Промазин | I | 0.1–0.8 | 0.07 |
| II | 0.02–0.1 | 0.05 | |
| Хлорпромазин | I | 0.5–2 | 0.06 |
| II | 0.05–0.4 | 0.04 | |
| Трифторперазин | I | – | – |
| II | 0.09–0.7 | 0.08 | |
Как видно из табл. 3, метрологические характеристики методик определения фенотиазинов в водно-органическом растворе в присутствии комплекса {пероксидаза–хитозан} гораздо лучше, чем методик в присутствии нативного биокатализатора. Коэффициент чувствительности определения промазина и хлорпромазина в водно-органической среде в присутствии комплекса больше в 6 и 11.5 раз соответственно по сравнению с тем же параметром аналогичных методик в отсутствие хитозана. Нижние границы определяемых концентраций фенотиазинов в водно-органической среде в присутствии комплекса на порядок ниже, чем в случае использования нативной пероксидазы в водных растворах. Следует отметить, что в отсутствие хитозана разработать методику определения самого трудноокисляемого из изученных субстратов пероксидазы – трифторперазина – в водно-органической среде не удалось. Предложенная методика определения промазина применена для анализа органических экстрактов гексан–ДМСО из плазмы венозной крови человека.
ФЛУОРЕСЦЕНТНЫЕ СЕНСОРНЫЕ СИСТЕМЫ ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ И ИХ ПРИМЕНЕНИЕ В ХИМИЧЕСКОМ АНАЛИЗЕ
Предложены два типа новых индикаторных систем для определения фенольных соединений и пероксидов различного строения, включающие следующие процессы: 1) взаимодействие продуктов ферментативного окисления фенолов с хитозаном, меченным флуоресцентной меткой, сопровождающееся уменьшением интенсивности флуоресценции; 2) пероксидазная дериватизация катехоламинов и их метаболитов с ароматическими диаминами – о-фенилендиамином или этилендиамином – с образованием флуоресцирующих производных [28].
Флуоресцентная сенсорная система для определения биологически активных веществ на основе хитозана, меченного изотиоцианатом родамина Б. Распространенный подход при создании флуоресцентных сенсоров заключается во введении в их чувствительный слой соединений, обладающих сильной собственной флуоресценцией, и последующем определении компонентов индикаторной системы по тушению флуоресценции [56]. Биочувствительный слой в этом случае представляет собой смесь хитозана, меченного флуоресцентной меткой (изотиоцианатом родамина Б), и пероксидазы. При ферментативном окислении фенольных соединений интенсивность флуоресценции биочувствительного слоя уменьшается пропорционально содержанию аналита.
Определение фенольных соединений. Оптимальные условия функционирования сенсорной системы на основе пероксидазы в комплексе с хитозаном, меченным изотиоцианатом родамина Б (концентрации хитозана, флуоресцентной метки, фермента, пероксида водорода, толщины пленки, концентрации и рН буферного раствора, длин волн возбуждения и испускания) приведены в работе [28]. С использованием этой системы разработаны методики определения пирокатехина, резорцина, гидрохинона, допамина и адреналина (табл. 4).
Таблица 4.
Метрологические характеристики определения дигидроксифенолов, катехоламинов и гидропероксидов в водной среде с использованием биосенсорной системы на основе хитозана, меченного флуоресцентной меткой
| Определяемое соединение | Диапазон линейности, мкМ | сmin, мкМ | sr (при сн, n = 4, P = 0.95) |
|---|---|---|---|
| Фенольные соединения | |||
| Пирокатехин | 0.5–5.0 | 0.10 | 0.04 |
| Резорцин | 0.5–7.5 | 0.15 | 0.03 |
| Гидрохинон | 0.5–7.5 | 0.20 | 0.03 |
| Допамин | 5.0–50.0 | 2.0 | 0.05 |
| Адреналин | 5.0–50.0 | 1.5 | 0.06 |
| Гидропероксиды | |||
| Пероксид водорода | 100–1000 | 45 | 0.08 |
| Пероксид мочевины | 25–250 | 10 | 0.08 |
| 2-Бутанонпероксид | 100–1000 | 40 | 0.08 |
| Бензоилпероксид | 50–750 | 45 | 0.07 |
| трет-Бутилгидропероксид | 250–25 000 | 230 | 0.07 |
Отклик чувствительного слоя сенсорного устройства аддитивен при концентрациях аналитов, входящих в диапазоны линейности градуировочных графиков. Поскольку коэффициенты чувствительности определения различаются по величине, информация, полученная при анализе объекта, одновременно содержащего смесь фенолов, позволяет сделать вывод лишь об их суммарном содержании в образце.
Таким образом, разработанные биосенсорные устройства на основе хитозана, меченного флуоресцентной меткой, могут быть использованы для анализа объектов, содержащих только одно фенольное соединение (например, фармацевтических и косметических препаратов), либо для оценки суммарного содержания фенольных соединений в анализируемом образце (например, в промышленных и сточных водах), а также при анализе с использованием совокупности сенсоров на основе разных индикаторных реакций. Анализ более сложных смесей (биологических жидкостей) требует создания более чувствительных, селективных и стабильных индикаторных систем.
Определение пероксидов различного строения. Флуоресцентный отклик биочувствительного слоя на основе пероксидазы в комплексе с хитозаном, меченным изотиоцианатом родамина Б, пропорционален концентрации не только восстановителя – фенольного соединения, но и окислителя – пероксида. Вследствие этого перспективно его применение для определения пероксидов различного строения, в том числе органических. В качестве субстрата-восстановителя использовали пирокатехин (фенольное соединение с наибольшим коэффициентом чувствительности определения) при концентрации 2.5 мкМ, соответствующей середине диапазона линейности. Метрологические характеристики методик определения ряда пероксидов приведены в табл. 4.
По чувствительности и воспроизводимости результатов определения органических гидропероксидов описанная флуоресцентная биосенсорная система сопоставима с известными аналогами, в том числе твердофазным спектрофотометрическим биосенсорным устройством на основе хитозана (несколько превосходя последний). При этом существенное преимущество твердофазного флуоресцентного биосенсорного устройства по сравнению со спектрофотометрическим аналогом заключается в меньшей продолжительности анализа при комнатной температуре (10–15 мин вместо 24 ч) [4, 28, 41].
С целью повышения чувствительности определения гидропероксидов индикаторную реакцию проводили в среде прямых и обращенных мицелл ПАВ, которые должны способствовать повышению активности фермента за счет изменения его конформации и доступности активного центра. Кроме того, в мицеллярных средах возможно увеличение аналитического сигнала вследствие повышения растворимости малорастворимых аналитов. Введение молекул ПАВ в раствор флуорофора может сопровождаться увеличением интенсивности флуоресценции в результате его включения в мицеллу и уменьшения числа его вращательных степеней свободы, что понижает долю безызлучательных переходов. Поскольку действие биочувствительного слоя на основе меченого хитозана основано на тушении его собственной флуоресценции, введение молекул ПАВ может как повысить, так и понизить чувствительность определения (вследствие наличия в системе двух конкурентных процессов). Следовательно, необходимо выделить вклад каждого процесса в формирование аналитического сигнала. По этой причине изучили возможность использования мицеллярных сред параллельно для описанного выше флуоресцентного и спектрофотометрического сенсоров [28, 41].
Установлено, что проведение реакции в мицеллярных средах сопровождается существенным по сравнению с немицеллярной системой (до 2 раз в случае мицелл на основе додецилсульфата натрия, ДДС) увеличением интенсивности собственной флуоресценции хитозановой пленки (в том числе при проведении контрольного опыта). При этом значительно ухудшается воспроизводимость результатов измерений (sr ~ ~ 0.2–0.3 при n = 4, P = 0.95). Метрологические характеристики методик определения пероксидов в среде прямых мицелл ПАВ с использованием спектрофотометрического биосенсора позволили выявить следующие закономерности: чувствительность определения всех пероксидов, кроме трет-бутилгидропероксида, возрастает в ряду сред цетилтриметиламмония бромид (ЦТАБ) < немицеллярная среда < ТВИН 80 < ДДС, причем повышение чувствительности закономерно сопровождается сужением диапазона линейности [41].
Таким образом, аналитические характеристики предложенной нами флуоресцентной биосенсорной системы на основе хитозана, меченного изотиоцианатом родамина Б, сопоставимы с характеристиками сенсоров, описанных в литературе [57–60]. При этом значительное преимущество предложенной сенсорной системы заключается в том, что использованная при его создании методика формирования и измерения аналитического сигнала позволяет анализировать негомогенные и непрозрачные среды. Однако достигаемые чувствительность и селективность определения недостаточны для решения некоторых важных аналитических задач, например анализа биологических жидкостей и тканей на содержание катехоламинов, таких как допамин, адреналин, и их метаболитов (гомованилиновой и ванилилминдальной кислот).
ФЛУОРЕСЦЕНТНАЯ СЕНСОРНАЯ СИСТЕМА ДЛЯ ОПРЕДЕЛЕНИЯ БИОЛОГИЧЕСКИ АКТИВНЫХ ВЕЩЕСТВ НА ОСНОВЕ ВЗАИМОДЕЙСТВИЯ ПРОДУКТОВ ФЕРМЕНТАТИВНОГО ОКИСЛЕНИЯ ФЕНОЛЬНЫХ СОЕДИНЕНИЙ С ДЕРИВАТИЗИРУЮЩИМИ АГЕНТАМИ
Перспективным для определения катехоламинов (допамин, адреналин) и их метаболитов (гомованилиновая и ванилилминдальные кислоты) представляется подход, основанный на измерении флуоресценции их производных. Дериватизирующими агентами, обеспечивающими чувствительное и селективное определение фенольных соединений по флуоресценции их производных, являются ароматические и алифатические амины.
Реакция дериватизации катехоламинов и их метаболитов ароматическими и алифатическими аминами осуществляется по следующей схеме: на первой стадии определяемое соединение окисляется до соответствующего хинона с открытой цепью, который далее вступает в реакцию внутримолекулярного присоединения по Михаэлю. В щелочной среде образовавшийся аддукт Михаэля мгновенно восстанавливается до гидроксииндола, который далее окисляется с образованием соответствующего о-хинона с закрытой цепью. Конечный продукт окисления взаимодействует с дериватизирующими агентами с образованием производных бензоксазола или хиноксалина. Идентичность продуктов ферментативной и неферментативной дериватизации доказана в работе [61] методами спектрофотометрии и жидкостной хромато-масс-спектрометрии на примере взаимодействия адреналина и бензиламина.
Определение фенольных соединений. Для разработки флуоресцентных методик определения фенольных соединений в сенсорном варианте в качестве дериватизирующих агентов использовали о-фенилендиамин (о-ФДА) и этилендиамин (ЭДА), дериватизаты которых флуоресцируют в области 500–600 нм. Интенсивность флуоресценции пропорциональна концентрации фенольных соединений. Спектры возбуждения и флуоресценции симметричны, а положение максимумов флуоресценции не зависит от длины волны возбуждающего излучения [28].
Для получения наибольшего количества флуоресцирующего аддукта в биочувствительном слое оптимизировали условия формирования аналитического сигнала (концентрацию фермента, пероксида водорода, дериватизирующего агента, время реакции, рН и концентрацию буферного раствора). При использовании иммобилизованного о-ФДА в качестве модельных фенольных соединений использовали простейший изомерный дигидроксифенол – пирокатехин, катехоламины (допамин и адреналин) и их метаболиты (гомованилиновую и ванилилминдальные кислоты). При применении ЭДА ограничились разработкой методик определения пирокатехина, допамина и адреналина (табл. 5).
Таблица 5.
Метрологические характеристики определения фенольных соединений с использованием флуоресцентной биосенсорной системы на основе дериватизации с о-фенилендиамином и этилендиамином
| Определяемое соединение | Диапазон линейности, нМ | сmin, нМ | sr (при сн, n = 5, P = 0.95) |
|---|---|---|---|
| Дериватизирующий агент – о-фенилендиамин | |||
| Пирокатехин | 50–1000 | 20 | 0.03 |
| Допамин | 50–500 | 18 | 0.03 |
| Адреналин | 10–2500 | 5 | 0.04 |
| Гомованилиновая кислота | 10–250 | 5 | 0.03 |
| Ванилилминдальная кислота | 5–100 | 3 | 0.03 |
| Дериватизирующий агент – этилендиамин | |||
| Пирокатехин | 500–5000 | 180 | 0.04 |
| Допамин | 250–5000 | 75 | 0.06 |
| Адреналин | 250–1000 | 150 | 0.08 |
Чувствительность и воспроизводимость результатов измерений, а также экспрессность анализа существенно возрастают при переходе от измерения сигнала в растворе к измерению сигнала на поверхности, а также при замене ЭДА на о-ФДА. Так, биосенсор на основе о-ФДА позволяет определять катехоламины и их метаболиты на наномолярном уровне концентраций, что достаточно для определения этих соединений в биологических жидкостях человека. При этом время, необходимое для достижения 90%-ной величины аналитического сигнала, не превышает 30 с. Биосенсорное устройство на основе ЭДА позволяет определять фенольные соединения на микромолярном уровне концентраций, а продолжительность реакции составляет от 2 до 15 мин. Отклик чувствительного слоя биосенсоров на основе о-ФДА и ЭДА сохраняется на уровне 90–95% от первоначального в течение двух дней при 25°С и не менее 4 недель при –20°С. При переходе от измерения аналитического сигнала в растворе к использованию биосенсоров сужается диапазон линейности, что свидетельствует о концентрировании продуктов окисления фенольных соединений в полимерной пленке. Как и в случае описанной ранее твердофазной флуоресцентной биосенсорной системы на основе меченого хитозана, увеличение объема реакционной системы сопровождается понижением чувствительности определения и расширением диапазона определяемых концентраций.
На примере анализа смесей, содержащих одновременно попарно допамин и адреналин, гомованилиновую кислоту и адреналин, а также ванилилминдальную кислоту и адреналин, установлено, что как в растворе, так и при использовании биосенсорного утройства, адреналин значительно превосходит остальные модельные фенольные соединения по скорости образования его флуоресцентных производных в реакции дериватизации с о-ФДА, вследствие чего величина аналитического сигнала в перечисленных выше системах неаддитивна. Так, определение допамина, гомованилиновой и ванилилминдальной кислот в присутствии адреналина возможно только при их многократном (в 100–1000 раз) избытке. При одновременном введении в реакционную систему допамина и гомованилиновой кислоты, допамина и ванилилминдальной кислоты, а также гомованилиновой и ванилилминдальной кислот сенсорная система дает аддитивный отклик. Следует отметить, что при рН 10 (оптимальном для определения гомованилиновой кислоты) скорость реакции ферментативной дериватизации ванилилминдальной кислоты с о-ФДА настолько мала, что даже при двукратном избытке ванилилминдальная кислота не мешает определению гомованилиновой кислоты. В норме содержание гомованилиновой кислоты в биологических жидкостях превышает содержание ванилилминдальной кислоты. Следовательно, с учетом возможного разбавления анализируемого образца, а также требуемой и достигаемой чувствительности определения катехоламинов и их метаболитов биосенсор на основе о-ФДА может быть использован для индивидуального определения гомованилиновой кислоты в крови и моче (при рН 10), а также для совместного определения гомованилиновой и ванилилминдальной кислот в моче (при рН 9.5).
Таким образом, разработанная нами флуоресцентная биосенсорная система на основе реакции пероксидазной дериватизации с о-ФДА превосходит описанные в литературе аналоги по чувствительности определения фенольных соединений и сопоставима с ними по селективности определения и воспроизводимости результатов измерения, а также стабильности аналитического сигнала [60, 61]. Достигаемые чувствительность и селективность определения, а также небольшое время отклика (30 с) позволяют использовать предложенную сенсорную систему для индивидуального определения катехоламинов и их метаболитов в фармацевтических препаратах и для определения гомованилиновой и ванилилминдальной кислот в биологических жидкостях.
Определение пероксидов. Описанные выше биосенсорные системы также использованы для разработки методик определения пероксидов различного строения. В качестве дериватизирующего агента использовали о-ФДА, поскольку биосенсор на его основе имеет лучшие метрологические характеристики, чем биосенсорное устройство на основе ЭДА. Метрологические характеристики определения пероксидов с помощью флуоресцентной биосенсорной системы на основе о-ФДА приведены в табл. 6.
Таблица 6.
Метрологические характеристики определения пероксидов в водной и мицеллярной средах с помощью флуоресцентной биосенсорной системы на основе дериватизации с о-фенилендиамином
| Определяемое соединение | Диапазон линейности, мкМ | сmin, мкМ | sr (при сн, n = 4, P = 0.95) |
|---|---|---|---|
| Пероксид водорода (вода) | 25–500 | 10 | 0.03 |
| Пероксид водорода (ЦТАБ) | 100–2500 | 80 | 0.15 |
| Пероксид водорода (ТВИН) | 50–500 | 30 | 0.15 |
| Пероксид мочевины | 25–500 | 15 | 0.05 |
| 2-Бутанонпероксид | 100–1000 | 80 | 0.06 |
| Бензоилпероксид | 50–500 | 30 | 0.07 |
| трет-Бутилгидропероксид | 50–2500 | 40 | 0.13 |
Следует отметить, что имеется относительно небольшое число работ [62–69], посвященных оптическим сенсорным системам для определения пероксида водорода и органических пероксидов. Существующие оптические сенсоры позволяют определять пероксиды преимущественно на микро/миллимолярном уровне, т.е. по чувствительности сравнимы (в единичных случаях превосходят [62, 63]) со спектрофотометрическими и флуоресцентными сенсорными системами на основе полиэлектролитного комплекса {пероксидаза–хитозан}. При этом только единичные оптические сенсоры (согласно литературным сведениям) апробированы в анализе реальных объектов – оптически прозрачных сред (дождевой воды и раствора для дезинфекции контактных линз), не требующих дополнительной пробоподготовки [67, 68]. Важным преимуществом регистрации аналитического сигнала непосредственно в биораспознающем слое сенсорных систем, предложенных нами, является возможность анализа окрашенных растворов, а также образцов, нерастворимых в воде и мицеллярных средах (растворах, суспензиях разной плотности и полярности), без предварительной пробоподготовки.
ПРИМЕНЕНИЕ ФЛУОРЕСЦЕНТНЫХ БИОСЕНСОРНЫХ СИСТЕМ В АНАЛИЗЕ РЕАЛЬНЫХ ОБЪЕКТОВ
Флуоресцентные биосенсорные системы, основанные на тушении флуоресценции (с меченым хитозаном) и возрастании сигнала (дериватизация с о-ФДА), а также спектрофотометрическая система на основе комплекса {пероксидаза–хитозан} апробированы при определении фенольных соединений (гидрохинона, допамина, адреналина) и органических пероксидов (бензоилпероксида, пероксида мочевины) в составе косметических и лекарственных препаратов различной природы (растворимых и нерастворимых в воде). Следует подчеркнуть, что пробоподготовка была ограничена гомогенизацией (в случае водонерастворимых препаратов) или разбавлением (в случае инъекционных растворов) анализируемого образца в воде. Результаты определения фенольных соединений в фармацевтических препаратах хорошо согласуются с содержаниями, указанными производителем (табл. 7).
Таблица 7.
Результаты определения гидрохинона, допамина, адреналина, бензоилпероксида и пероксида мочевины в фармацевтических и лекарственных препаратах в водной среде(I) и в среде прямых мицелл ПАВ(II)
| Тип биосенсорной системы | Фармацевтический препарат | Определяемое соединение | Найдено, % | Заявлено произво- дителем, % | |
|---|---|---|---|---|---|
| I | II | ||||
| Спектрофотометрическая сенсорная система | Ахромин | Гидрохинон | 2.1 ± 0.2 | – | 1.9 |
| Базирон | Бензоилпероксид | 4.8 ± 0.7 | 4.8 ± 0.7 | 5.0 | |
| Пероксидерм | 2.8 ± 0.4 | 2.5 ± 0.3 | 2.5 | ||
| Splat Exreem White | Пероксид мочевины | 0.09 ± 0.01 | 0.09 ± 0.01 | 0.1 | |
| Флуоресцентная сенсорная система на основе хитозана, меченного флуоресцентной меткой | Ахромин | Гидрохинон | 2.1 ± 0.2 | – | 1.9 |
| Допамин – Ферейн | Допамин | 0.53 ± 0.04 | 0.5 | ||
| Адреналина гидрохлорид | Адреналин | 0.1 ± 0.01 | 0.1 | ||
| Ксилокаин – Адреналин | (5.4 ± 0.6) × 10–4 | 5 × 10–4 | |||
| Базирон | Бензоилпероксид | 4.7 ± 0.6 | 5.0 | ||
| Пероксидерм | 2.5 ± 0.4 | 2.5 | |||
| Splat Exreem White | Пероксид мочевины | 0.11 ± 0.02 | 0.1 | ||
| Флуоресцентная сенсорная система на основе дериватизации с о-ФДА | Допамин – Ферейн | Допамин | 0.54 ± 0.07 | 0.5 | |
| Адреналина гидрохлорид | Адреналин | 0.11 ± 0.02 | 0.1 | ||
| Ксилокаин – Адреналин | (4.9 ± 0.6) × 10–4 | 5 × 10–4 | |||
| Базирон | Бензоилпероксид | 4.7 ± 0.4 | 5.0 | ||
| Пероксидерм | 2.4 ± 0.4 | 2.5 | |||
| Splat Exreem White | Пероксид мочевины | 0.11 ± 0.01 | 0.1 | ||
Флуоресцентную биосенсорную систему на основе о-ФДА применили для определения конечных продуктов метаболизма катехоламинов (гомованилиновой и ванилилминдальной кислот) в моче. Анализ проводили способом стандартных добавок при условиях, оптимальных для определения гомованилиновой кислоты (селективное определение последней) и ванилилминдальной кислоты (определение суммарного содержания гомованилиновой и ванилилминдальной кислот). Результаты определения гомованилиновой кислоты хорошо согласуются с данными, полученными при определении этого соединения методом ВЭЖХ с амперометрическим детектированием [28].
* * *
Таким образом, наши исследования по созданию и совершенствованию существующих оптических (спектрофотометрических и флуоресцентных) сенсорных систем позволили предложить оригинальные подходы к иммобилизации широко используемого в анализе фермента пероксидазы, обеспечивающие его активность и стабильность при хранении и протекании каталитических процессов в неблагоприятных для белка условиях и средах. Разработаны новые типы конструкций и устройств для регистрации аналитического сигнала твердой фазы (биочувствительного слоя) на основе универсальных серийных спектрофотометров и флуориметров. Предложены новые индикаторные системы для определения фенольных соединений – фенотиазинов, катехоламинов и их метаболитов, пероксидов различного строения для обеспечения контроля качества лекарственных препаратов, биомедицинских исследований, клинической диагностики. Достоинством разработанных оптических сенсорных систем является простота их изготовления и эксплуатации. Чувствительность, селективность и быстродействие предложенных устройств достаточны для определения перечисленных выше аналитов в фармацевтической продукции, а в ряде случаев и в биологических жидкостях человека. Формирование и измерение аналитического сигнала вне раствора, непосредственно в чувствительном слое сенсора, позволяет анализировать непрозрачные и мутные среды и, следовательно, объекты на основе матриц сложного состава без дополнительной пробоподготовки. Последнее достоинство выгодно отличает предложенные биосенсоры от описанных в литературе аналогов, основанных на формировании и измерении аналитического сигнала в растворе.
Авторы выражают благодарность к.х.н. A.B. Кирейко, к.х.н. П.В. Родионову и к.х.н. Л.И. Малининой за участие в постановке задач, проведении экспериментов и обсуждении результатов исследований.
Работа выполнена при финансовой поддержке Российского Научного фонда (проект № 14-23-00012).
Список литературы
Захарова Г.С., Упоров В.И., В.И. Тишков. Пероксидаза из корней хрена: Модилирование свойств химической модификацией химической глобулы // Успехи биол. химии. 2011. Т. 51. С. 37.
That T. Ngo. Peroxidase in chemical and biochemical analysis // Anal. Lett. 2010. V. 43. № 10–11. P. 1572.
Shivakumar A. Role of peroxidase in clinical assays: A short review // J. Clin. Nutr. Diet. 2017. V. 3. №. 2. P. 1.
Veselova I.A., Malinina L.I., Rodionov P.V., Shekhovtsova T.N. Properties and analytical applications of the self-assembled complex {peroxidase–chitosan} // Talanta. 2012. V. 102. P. 101.
Minteer Sh.D. Enzyme Stabilization and Immobilization. Methods and Protocols. Series: Methods in Molecular Biology. Heidelberg: Springer, 2010. 679 p.
Veselova I.A., Kireiko A.V., Shekhovtsova T.N. Catalytic activity and the stability of horseradish peroxidase increase as a result of its incorporation into a polyelectrolyte complex with chitosan // Prikl. Biokhim. Mikrobiol. 2009. V. 45. P. 125.
Klibanov AM. Improving enzymes by using them in organic solvents // Nature. 2001. V. 409. P. 241.
Martinek K., Levashov A.V., Khmelnitsky Y.L., Klyachko N.L., Berezin I.V. Colloidal solution of water in organic solvents: a microgeterogeneous medium for enzymatic reactions // Science. 1982. V. 218. P. 889.
Ramirez-Corredores M., Borole A. Biocatalysis in Oil Refining. Elsevier: London, 2007. 416 p.
Fitzpatrick P.A., Steinmetz A.C.U., Ringe D., Klibanov A.M. Enzyme crystal structure in a neat organic solvent // Proc. Natl. Acad. Sci. USA. 1993. V. 90. P. 8653.
Yennawar H.P., Yennawar N.H., Farber G.K. A structural explanation for enzyme memory in nonaqueous solvents // J. Am. Chem. Soc. 1995.V. 117. P. 577.
Mozhaev V.V., Poltevsky K.G., Slepnev V.I., Badun G.A., Levashov A.V. Homogeneous solutions of hydrophilic enzymes in nonpolar organic solvents. New systems for fundamental studies and biocatalytic transformations // FEBS Lett. 1991. V. 292. P. 159.
Wu X.J., Choi M.M.F., Wu X.M. An organic-phase optical phenol biosensor coupling enzymatic oxidation with chemical reduction // Analyst. 2004. V. 129. P. 1143.
Khmelnitsky Yu. L., Mozhaev V.V., Belova A.B., Sergeeva M.V., Martinek K. Denaturation capacity: a new quantitative criterion for selection of organic solvents as reaction media in biocatalysis // Eur. J. Biochem. 1991. V. 198. P. 31.
Zaks A., Klibanov A.M. Enzymatic catalysis in nonaqueous solvents // J. Biol. Chem. 1988. V. 263. P. 3194.
Vakurov A.V., Gladilin A.K., Levashov A.V., Khmelnitsky, Y.L. Dry enzyme-polymer complexes − stable organosoluble biocatalysts for nonaqueous enzymology // Biotechnol. Lett. 1994. V. 16. P. 175.
Dordick J.S. Designing enzymes for use in organic solvents // Biotechnol. Prog. 1992. V. 8. P. 259.
Sears P., Schuster M., Wang P., Witte K., Wong C.-H. Engineering subtilisin for peptide coupling: studies on the effects of counterions and site-specific modifications on the stability and specificity of the enzyme // J. Am. Chem. Soc. 1994. V. 116. P. 6521.
Khmelnitsky Yu. L., Belova A.B., Levashov A.V., Mozhaev V.V. Relationship between surface hydrophilicity of a protein and its stability against denaturation by organic solvents // FEBS Lett. 1991. V. 284. P. 267.
Kudryashova E.V., Artemova T.M., Vinogradov A.A., Gladilin A.K., Mozhaev V.V., Levashov A.V. Stabilization and activation of alpha-chymotrypsin in water-organic solvent systems by complex formation with oligoamines // Protein Eng. 2003. V. 16. P. 303.
Stepankova V., Bidmanova S., Koudelakova T., Prokop Z., Chaloupkova R. Strategies for stabilization and activation of biocatalysts in organic solvents // ACS Catal. 2013. V. 3. №. 12. P. 2823.
Rich J.O., Mozhaev V.V., Dordick J.S., Clark D.S., Khmelnitsky Yu. L. Molecular imprinting of enzymes with water-insoluble ligands for nonaqueous biocatalysis // J. Am. Chem. Soc. 2002. V. 124. P. 5254.
Лукачева Л.В., Закемовская А.А., Карякина Е.Е., Зоров И.Н., Синицын А.П., Сухачева М.В., Нетрусов А.И., Карякин А.А. Определение глюкозы и лактозы в продуктах питания с помощью биосенсоров на основе берлинской лазури // Журн. аналит. химии. 2007. Т. 62. № 4. С. 429. (Lukacheva L.V., Zakemovskaya A.A., Karyakina E.E., Zorov I.N., Sinitsyn A.P., Sukhacheva M.V., Netrusov A.I., Karyakin A.A. Determination of glucose and lactose in food products with the use of biosensors based on Berlin blue // J. Analyt. Chem. 2007. V. 62. № 4. Р. 388.)
Karyakin A.A., Kotel’nikova E.A., Lukacheva L.V., Karyakina E.E. Optimal environment for glucose oxidase in perfluorosulfonated ionomer membranes: Improvement of first-generation biosensors // Anal. Chem. 2002. № 74. P. 1593.
Kazakova L.I., Shabarchina L.I., Anastasova S., Pavlov A.M., Vadgama P. Skirtach A.G., Sukhorukov G.B. Chemosensors and biosensors based on polyelectrolyte microcapsules containing fluorescent dyes and enzymes // Anal. Bioanal. Chem. 2013. V. 405. P. 1559.
Chaniotakis N.A. Enzyme stabilization strategies based on electrolytes and polyelectrolytes for biosensor applications // Anal. Bioanal. Chem. 2004. V. 378. P. 89.
Veselova I.A., Shekhovtsova T.N. Visual determination of mercury(II) using horseradish peroxidase immobilized on polyurethane foam // Anal. Chim. Acta. 1999. V. 392. P. 151.
Byeon J.H. Multifunctional metal-polymer nanoagglomerates from single-pass aerosol self-assembly // Sci. Rep. 2016. V. 6. № 27006 (open access).
Rodionov P.V., Veselova I.A., Shekhovtsova T.N. A solid-phase fluorescent biosensor for the determination of phenolic compounds and peroxides in samples with complex matrices // Anal. Bioanal. Chem. 2014. V. 406. P. 1531.
Zikakis J.P. Chitin, Chitosan and Related Enzymes. Orlando: Academic press, 1984. 1066 p.
Balesteros A., Plou F.J., Iborra J.L., Holling P.I. Stability and Stabilization of Biocatalysts. Series: Progress of Biotechnology. Elsevier: Amsterdam, 1998. 756 p.
Decher G. Fuzzy nonoassemblies: toward layered polymeric multicomposites // Science. 1997. V. 277. P. 1232.
Jayakumar R., Prabaharan M., Muzzarelly R. Chitosan for Biomaterials. Advances in Polymer Science. Springer. Inc., 2011. 236 p.
Khmelnitsky Yu. L., Levashov A.V., Klyachko N.L., Martinek K. Engineering biocatalytic systems in organic media with low water content // Enzyme Microb. Technol. 1988. V. 10. P. 710.
Halling P.J. Thermodynamic predictions for biocatalysis in nonconvential media: theory, tests and recommendations for experimental design and analysis // Enzyme Microb. Technol. 1994. V. 16. P. 178.
Levitsky V., Lozano P., Gladilin A., Iborra J.L. Stability of immobilized enzyme−polyelectrolyte complex against irreversible inactivation by organic solvents // Prog. Biotechnol. 1998. V. 5. P. 417.
Mozhaev V.V., Kudryashova E.V., Efremova N.V., Topchieva I.N. Stability of α-chymotrypsin conjugated with poly(ethylene glycols) and proxanols at high temperature and in water-cosolvent mixtures // Biotechnol. Techn. 1996. V. 10. P. 849.
Kudryashova E.V., Gladilin A.K., Vakurov A.V., Heitz F., Levashov A.V., Mozhaev V.V. Enzyme−polyelectrolyte complexes in water-ethanol mixtures: negatively charged groups artificially introduced into α-chymotrypsin provide additional activation and stabilization effects // Biotechnol. Bioeng. 1997. V. 55. P. 267.
Shipovskov S., Levashov A. Tyrosinase: Polybrene noncovalent complexes in water-ethanol mixtures // Biotechnol. Bioeng. 2003. V. 84. P. 258.
Xu K., Griebenov K., Klibanov A.M. Correlation between catalytic activity and secondary structure subtilisin dissolved in organic solvents // Biotechnol. Bioeng. 1997. V. 56. P. 485.
Griebenov K., Klibanov A.M. Can conformational changes be responsible for solvent and excipient effects on the catalytic behavior of subtilisin Carlsberg in organic solvents? // Biotechnol. Bioeng. 1997. V. 53. P. 351.
Олейник Л.И., Веселова И.А., Родионов П.В., Будашов И.А., Шеховцова Т.Н. Оптический биосенсор на основе комплекса {пероксидаза–хитозан} для определения гидрохинона // Заводск. лаборатория. Диагностика материалов. 2011. Т. 77. № 4. С. 23.
Payne, G. F., M. V. Chaubal, T. A. Barbari. Enzyme-catalyzed polymer modification: reaction of phenolic compounds with chitosan films // Polymer. 1996. V. 37. P. 4643.
Kumar G., Bristow J.F., Smith P.J., Payne G.F. Enzymatic gelation of the natural polymer chitosan // Polymer. 2000. V. 41. P. 2157.
Abdullah J., Ahmad M., Karuppiah N., Heng L.Y., Sidek H. Chitosan-based tyrosinase optical phenol biosensor employing hybrid nafion/sol-gel silicate for MBTH immobilization // Talanta. 2006. V. 40. P. 527.
Erdem A., Pabuccuoglu A., Meric B., Kerman K., Osnoz M. Electrochemical biosensor based on horseradish peroxidase for the determination of oxidizable drugs // Turk. J. Med. Sci. 2000. V. 30. P. 349.
Родионов П.В., Алиева Е.А., Сергеева Е.А., Павлова М.Е., Веселова И.А., Шеховцова Т.Н. Определение пероксида водорода и органических пероксидов в мицеллярных и водно-органических средах с использованием спектрофотометрического биосенсора на основе пероксидазы из корней хрена // Журн. аналит. химии. 2016. Т.71. № 9. С. 971.
Рогожина Т.В., Рогожин В.В. Стационарная кинетика индивидуального и совместного окисления фенотиазинов в присутствии пероксидазы хрена // Исследовано в России (электронный журнал). 2002. С. 767.
Madej K., Kala M., Wozniakievicz M. LC and non-aqueous CE determination of phenothiazines in autopsy samples // Chromatographia. 2005. V. 62. P. 533.
Pola A., Michalak K., Burliga A., Motohashi N., Kawase M. Determination of lipid bilayer/water partition coefficient of new phenothiazines using the second derivative of absorption spectra method // Eur. J. Pharm. Sci. 2004. V. 21. P. 421.
Gordeliy V.I., Kiselev M.A., Pole A.V., Teixeira J. Lipid membrane structure and interactions in DMSO/water mixture // Biophys. J. 1998. V. 75. P. 2343.
Escribano J., Garcia-Canovas F., Garcia-Carmona F., Losano J.A. Kinetic study of the transient phase of a second-order chemical reaction coupled to an enzymic step: application to the oxidation of chlorpromazine by peroxidase– hydrogen peroxide // Biochim. Biophys. Acta. 1985. V. 831. P. 313.
Ashton Action Q. J. Phenothiazines – Advances in Research and Application. Atlanta: Schlolarly Editions, 2012. 107 p.
Yamini Y., Faraji M. Extraction and determination of trace amounts of chlorpromazine in biological fluids using magnetic solid phase extraction followed by HPLC // J. Pharm. Anal. 2014. V. 4. P. 279.
Nakano M., Sugoika K., Nakano H., Takyu C., Inaba H. Generation of electronically exited species during enzymatic oxidation of chlorpromazine and related compound // Biochem. Biophys. Res. Commun. 1985. V. 3. P. 952.
Kelder P.P., de Mol N.J., Fischer M.J.E., Janssen L.H.M. Kinetic evaluation of the oxidation of phenothiazine derivatives by methemoglobin and horseradish peroxidase in the presence of hydrogen peroxide. Implications for the reaction mechanisms // Biochim. Biophys. Acta. 1994. V. 1205. P. 230.
Ma O., Lavertu M., Sun J., Nguyen S., Buschmann M.D., Winnik F.M., Hoemann C.D. Precise derivatization of structurally distinct chitosans with rhodamine B isothiocyanate // Carbohydr. Polym. 2008. V. 72. P. 616.
Drexhage K.H. Fluorescence efficiency of laser dyes // J. Res. 1976. V. 80. P. 421.
Park S.A., Jang E., Koh W.G., Kim B. Fabrication and characterization of optical biosensors using polymer hydrogel microparticles and enzyme-quantum dot conjugates. // Sens. Actuators B. 2010. V. 150. P. 120.
Родионов П.В., Веселова И.А., Шеховцова Т.Н. Оптические сенсоры для определения фенольных соединений различного строения. // Журн. аналит. химии. 2013. Т. 68. № 11. С. 1044.
Veselova I.A., Malinina L.I., Barsukova M.E., Tokareva A.I., Buslova T.S., Pirogov A.V., Shekhovtsova T.N. A novel multi-purpose enzymatic system and procedures for the rapid fluorescent determination of flavonoids in herbal pharmaceuticals and plant materials. // Talanta. 2017. V. 171. P. 108.
Demiyanova A.S., Sakharov I.Yu. High chemiluminescent activity of Fe III-TAML activator in aqueous-organic media and its use in determination of organic peroxides // Analyst. 2015. V. 140. P. 2964.
Vdovenko M.M., Demiyanova A.S., Kopylov K.E., Sakharov I.Yu. FeIII–TAML activator: A potent peroxidase mimic for chemiluminescent determination of hydrogen peroxide // Talanta. 2014. V. 125. P. 361.
Sakuragawa A., Taniai T., Okutani T. Fluorometric determination of microamounts of hydrogen peroxide with an immobilized enzyme prepared by coupling horseradish peroxidase to chitosan beads // Anal. Chim. Acta. 1998. V. 374. P. 191.
Rubtsova M.Y., Kovba G.V., Egorov A.M. Chemiluminescent biosensors based on porous supports with immobilized peroxidase // Biosens. Bioelectron. 1998. V. 12. P. 75.
Collaudin A.B., Blum L.J. Enhanced luminescent response of a fiber-optic sensor for H2O2 by a high-salt-concentration medium // Sens. Actuators B. 1997. V. 38–39. P. 189.
Porsh H.E., Wolfbeis O.S. Optical sensor for hydrogen peroxide // Mikrochim. Acta. 1989. V. 1. P. 41.
Lobnik A., Cajlakovic M. Sol-gel based optical sensor for continuous determination of dissolved hydrogen peroxide // Sens. Actuators B. 2001. V. 74. P. 194.
Hanko M., Bruns N., Tiller J.C., Heinze J. Optical biochemical sensor for determining hydroperoxides in nonpolar organic liquids as archetype for sensors consisting of amphiphilic conetworks as immobilization matrices // Anal. Bioanal Chem. 2006. V. 386. P. 1273.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии