

Журнал аналитической химии, 2019, T. 74, № 10, стр. 729-751
От жидкостно-газовой хроматографии к хроматомембранному массообменному процессу
Л. Н. Москвин a, О. В. Родинков a, *
Санкт-Петербургский государственный университет, Институт химии
198504 Санкт-Петербург, Петродворец, Университетский просп., 26, Россия,
* E-mail: o.rodinkov@spbu.ru
Поступила в редакцию 20.12.2018
После доработки 22.04.2019
Принята к публикации 22.04.2019
Аннотация
Рассмотрены физико-химические закономерности и аналитические возможности методов разделения, основанных на проявлении капиллярных эффектов в гидрофобных микропористых средах: жидкостно-газовой хроматографии со стационарной газовой фазой в порах твердофазного носителя и осуществляемого в бипористой матрице хроматомембранного массообменного процесса в системах жидкость–газ и жидкость–жидкость. Показаны преимущества этих методов по сравнению с традиционными схемами осуществления массообменных процессов в тех же системах с точки зрения более высокой эффективности и возможности разделения веществ в непрерывном режиме.
ЖИДКОСТНО-ГАЗОВАЯ ХРОМАТОГРАФИЯ
Открытие жидкостно-газовой хроматографии. После появления жидкостно-жидкостной и газожидкостной хроматографии стало очевидным, что открытая М.С. Цветом хроматография – это не просто новый метод ”адсорбционного анализа”, как он характеризовал ее в одной из своих первых работ, а универсальная схема разделения веществ, применимая практически к любому из возможных сочетаний фаз. Однако, несмотря на разнообразие вариантов хроматографии, реализованных к началу 80-х гг., трудно было представить осуществимость хроматографического процесса со стационарной газовой фазой (СГФ). До этого времени практически даже не возникал вопрос: возможно ли с физико-химических позиций обращение фаз в газожидкостной хроматографии, т.е. может ли газовая фаза выполнять роль стационарной фазы и соответственно имеет ли право на существование жидкостно-газовая хроматография (ЖГХ).
Открытие этого метода произошло, как это часто бывает в науке, случайно [1, 2]. В процессе поиска методов анализа газов, растворенных в воде, заметили необычный эффект: при пропускании воды, содержащей растворенные газы, через колонку с пористым политетрафторэтиленом (ПТФЭ), предварительно продутую газом-носителем, в первых порциях фильтрата, значительно превосходящих свободный объем колонки, не обнаруживались исходные газообразные примеси. Причем характер изменения концентраций газов в фильтрате в зависимости от объема пропущенной воды соответствовал закономерностям фронтального хроматографического процесса (рис. 1).
Рис. 1.
Зависимость отношения концентраций растворенных в воде газов на выходе (с) и входе (с0) колонки, заполненной пористым политетрафторэтиленом и продутой аргоном (свободный объем колонки 8 см3): 1 – кислород, 2 – водород, 3 – азот.

Были предложены и проверены три гипотезы, которые могли бы объяснить эффект удерживания в колонке с ПТФЭ растворенных в воде газов. Первая гипотеза предполагала адсорбционное удерживание газов на ПТФЭ, вторая – удерживание за счет распределения в системе жидкость–жидкость, и третья – распределение в системе жидкость–газ. Второе предположение о распределении в системе жидкость–жидкость основывалось на известном факте изменения физико-химических свойств воды в узких капиллярах, и соответственно изменении растворимости газов в такой воде, находящейся в порах носителя.
Отбросить две первые гипотезы, как несостоятельные, позволили данные о поглощении азота и кислорода из воды на колонках с различными сорбентами, отличающимися по удельной поверхности и способу предварительной подготовки. Как видно из табл. 1, отсутствуют какие-либо корреляции между удельной поверхностью адсорбентов и количеством удерживаемых газов, что позволило отвергнуть гипотезу об адсорбционном механизме их удерживания. Снижение же количества удерживаемых из водных растворов газов при заполнении порового пространства ПТФЭ гептаном, в котором растворимость газов в несколько раз больше, чем в воде, подтвердило несостоятельность предположения о жидкостно–жидкостном механизме удерживания газов. Оставалось признать, что справедлива последняя из гипотез – удерживание газов в неподвижной газовой фазе.
Таблица 1.
Объем газов (V), поглощаемых из насыщенной воздухом воды в колонке, заполненной различными сорбентами и предварительно продутой аргоном (Sуд – удельная поверхность; Vс – объем колонки, 8 cм3)
| Сорбент | Sуд, м2/г | V/Vс | |
|---|---|---|---|
| кислород | азот | ||
| Оксид алюминия | 270 | 0.012 | 0.035 |
| Силикагель КСС-2,5 | 400 | 0.012 | 0.033 |
| Активный уголь СКТ | 1200 | 0.021 | 0.045 |
| Пористый ПТФЭ | 0.5–1.0 | 0.105 | 0.202 |
| Стружка из ПТФЭ | 0.01 | 0.034 | 0.094 |
| Пористый ПТФЭ, насыщенный гептаном | – | 0.033 | 0.090 |
Были определены объемы удерживания в колонке с ПТФЭ нескольких газов, существенно различающихся по растворимости в воде при различных температурах. Установлено, что чем меньше растворимость газа в воде и соответственно чем больше его коэффициент распределения между СГФ и подвижной водной фазой, тем больше объем удерживания. Это подтвердило третью гипотезу о жидкостно-газовом механизме удерживания растворенных в воде газов в колонке с пористым ПТФЭ.
Первые работы по ЖГХ были выполнены с использованием широко распространенных в настоящее время монолитных сорбентов [3, 4], впервые предложенных в 60-х гг. прошлого века [5] для осуществления жидкостно–жидкостной хроматографии и названные в те годы компактными, а позднее блочными [6]. Эти бипористые сорбенты (матрицы) состоят из связанных при спекании в единый монолит пористых гранул ПТФЭ. Пространство между гранулами образует систему открытых макропор, размеры которых составляют десятые доли мм и зависят от размеров гранул ПТФЭ, взятых для повторного спекания. По макропорам перемещается полярная подвижная жидкая фаза (ПЖФ), а сами гранулы содержат открытые поры микронных размеров, в которых находится стационарная неполярная жидкая или газовая фаза. Первый случай соответствует экстракционной хроматографии, а второй – ЖГХ. Учитывая достаточно большие размеры исходных гранул, ожидать от подобных сорбентов высокой эффективности массообмена нельзя, однако бипористые сорбенты и носители нашли применение в экспрессном радиохимическом хроматографическом анализе [7] и, как будет показано ниже, могут служить для осуществления хроматомембранных массообменных процессов.
Через год после выхода статьи об открытии ЖГХ с экспериментальными доказательствами возможности осуществления подобного процесса появилась статья Гиддингса и Майерса [8] с предсказанием этого метода на основании теоретических соображений. Американские исследователи априори предположили, что благодаря исключительно большой скорости диффузии разделяемых веществ в газовой фазе, где она примерно в 105 раз выше, чем в жидкостях, по эффективности ЖГХ будет значительно превосходить другие варианты жидкостной хроматографии. Однако для этого необходимо, чтобы СГФ находилась в изолированных друг от друга порах носителя, во избежание продольного диффузионного размывания зон разделяемых компонентов.
Физико-химические закономерности жидкостно-газовой хроматографии. Принципиальная возможность такого процесса закономерно следует из известного факта возникновения в порах гидрофобных не смачиваемых водой материалов отрицательного капиллярного давления, препятствующего их заполнению водой. Величина капиллярного давления определяется следующим выражением [9]:
(1)
${{P}_{{\text{c}}}} = {{\left| {2\sigma \cos \theta } \right|} \mathord{\left/ {\vphantom {{\left| {2\sigma \cos \theta } \right|} r}} \right. \kern-0em} r},$При всей кажущейся очевидности приведенных аргументов, убедить в достоинствах нового метода мог только эксперимент. Полученные зависимости высоты, эквивалентной теоретической тарелке (ВЭТТ), от скорости потока элюента в ЖГХ имеют тот же характер, что и для традиционных видов жидкостной хроматографии. Однако значения ВЭТТ в сопоставимых условиях оказываются в 3–4 раза меньше. Так, при размерах частиц сорбентов 150–200 мкм величина ВЭТТ составляет всего 1–2 мм при скорости элюирования до 3 см/с, что хорошо согласуется с теоретическими предсказаниями [8].
Аналитические возможности жидкостно-газовой хроматографии. Отсутствие детекторов, высоко чувствительных к растворенным газам, существенно ограничивает область аналитического применения ЖГХ в элюентном варианте. Более рациональным является сочетание фронтального варианта ЖГХ как метода суммарного концентрирования и выделения из жидкой пробы в газовую фазу летучих веществ или веществ, способных образовывать летучие продукты, с их последующим газохроматографическим определением в потоке газа-носителя [14, 15]. На первой стадии анализа пробу жидкости пропускают через колонку для ЖГХ, при этом происходит извлечение растворенных газов в СГФ. На второй стадии колонку для ЖГХ продувают потоком газа-носителя, который переносит извлеченные компоненты через влагоотделительный сосуд и осушитель в аналитическую газохроматографическую колонку, а затем в детектор.
С точки зрения количественного анализа наиболее рациональны два режима извлечения газообразных аналитов [16]: режим полного (количественного) извлечения определяемых компонентов из всего объема пробы, который должен быть меньше объема до проскока наименее удерживаемого из определяемых компонентов, и режим предельного равновесного насыщения определяемыми компонентами всего объема газовой фазы в колонке для ЖГХ, когда через нее пропускают объем пробы, заведомо больший, чем удерживаемый объем наиболее удерживаемого из определяемых компонентов. Режим равновесного насыщения обеспечивает более высокую (в 1.5–2 раза) чувствительность определения, однако для его реализации требуется больший объем пробы (приблизительно в 3–5 раз), а также соблюдение условий постоянства объема СГФ в колонке.
Разработанные на основе предложенной схемы анализа газохроматографические методики позволяют с относительной погрешностью менее 20% проводить экспрессное (5–10 мин) определение растворенных в воде неорганических газов (водород, кислород, азот, оксиды углерода), начиная с концентраций 5–20 мкг/л, при использовании детектора по теплопроводности, и органических газов (метан, этан, этилен, пропан, бутан), начиная с 20–60 нг/л, при использовании пламенно-ионизационного детектора. Предел обнаружения зависит от чувствительности детектора и коэффициента распределения определяемого компонента между жидкой и газовой фазами.
Фронтальный вариант ЖГХ позволяет извлекать растворенные газы не только из водных растворов, но и из других полярных жидкостей [17]. В том случае, когда анализируемая жидкость в силу своего низкого поверхностного натяжения смачивает носитель, но неограниченно смешивается с водой (низшие спирты, кетоны, альдегиды и др.), ее разбавляют водой до наступления инверсии смачиваемости и пропускают через колонку для ЖГХ.
ЖИДКОСТНО-ГАЗОАДСОРБЦИОННАЯ ХРОМАТОГРАФИЯ
Физико-химическая модель жидкостно-газоадсорбционной хроматографии. В соответствии с теорией капиллярности СГФ находится в микро- и мезопорах не смачиваемых жидкостью носителей, в то время как макропоры и пустоты между частицами носителя, где капиллярное давление пренебрежимо мало, заполнены жидкой фазой (рис. 2). Учитывая высокую удельную поверхность носителей, удерживание компонентов в рассматриваемой хроматографической системе в общем случае наряду с распределением между ПЖФ и СГФ обусловлено адсорбцией компонентов на межфазных границах жидкость–твердое тело и газ–твердое тело [18]. Согласно равновесной аддитивной теории удерживания [19] в рассматриваемой трехфазной системе приведенный объем удерживания компонента VR представляет собой сумму трех слагаемых [18]:
(3)
${{V}_{{\text{R}}}} = {{К }_{{{\text{GL}}}}}{{V}_{{\text{G}}}} + {{К }_{{{\text{GL}}}}}{{К }_{{{\text{GS}}}}}{{S}_{{{\text{GS}}}}} + {{К }_{{{\text{LS}}}}}{{S}_{{{\text{LS}}}}},$Рис. 2.
Схема расположения фаз в жидкостно-газовой хроматографии: 1 – стационарная газовая фаза, 2 – подвижная жидкая фаза, 3 – твердофазный носитель.

Таблица 2.
Вклад различных процессов в удерживание из водных растворов на неполярном полимерном сорбенте порапаке QS (20°С, концентрация компонентов 3 × 10–5 М) [20]
| Компонент | K | KGS, см3/м2 | Объем колонки, см3 | КGLVG, см3 | ККGSSGS, см3 | VR, см3 |
|---|---|---|---|---|---|---|
| Азот | 65 | 0.00071 | 3.8 | 72 | 33 | 112 |
| Кислород | 32 | 0.00072 | 3.8 | 35 | 16 | 54 |
| Бутан | 6.7 | 3.9 | 0.05 | 0.27 | 260 | 250 |
| Гексан | 20 | 22 | 0.05 | 0.8 | 4350 | 4500 |
| Бензол | 0.20 | 38 | 0.05 | <0.1 | 75 | 79 |
| 1,2-Дихлорэтан | 0.038 | 25 | 0.28 | <0.1 | 65 | 68 |
| Этилацетат | 0.0048 | 59 | 0.28 | <0.1 | 17 | 16 |
| Ацетон | 0.0017 | 3.3 | 3.8 | <0.1 | 4.4 | 4.0 |
| Метанол | 0.00014 | 2.2 | 3.8 | <0.1 | 2.4 | <0.1 |
Распределение в системе жидкость–газ (первое слагаемое уравнения (3)) вносит основной вклад в удерживание только для так называемых “постоянных газов” (гелий, водород, кислород, азот и др.), практически не адсорбирующихся на мезопористых адсорбентах. Неожиданным с точки зрения общепринятых представлений выглядит отсутствие значимого удерживания ЛОВ за счет адсорбции на носителе непосредственно из жидкой фазы (третье слагаемое пренебрежимо мало). Это, однако, находит удовлетворительное объяснение, исходя из сопоставления энергетических параметров адсорбции из газовой и жидкой фаз и существенно большей величины SGS по сравнению с SLS. Как показали результаты исследований, те же закономерности распространяются и на другие мезопористые адсорбенты.
Принимая во внимание, что при фильтрации жидкой фазы через не смачиваемый ею мезопористый сорбент адсорбция летучих веществ происходит в основном из газовой фазы, заполняющей микро- и мезопоры, данный хроматографический процесс может рассматриваться как жидкостно-газоадсорбционная хроматография (ЖГАХ) [21].
Жидкостно-газоадсорбционная хроматография в процессах разделения и концентрирования летучих органических веществ. Представление о процессе ЖГАХ имеет принципиальное значение для объяснения закономерностей удерживания веществ в обращенно-фазовой жидкостно-адсорбционной (ОФ ЖАХ) и твердофазной экстракции (ТФЭ) ЛОВ из водных растворов и для поиска способов оптимизации этих процессов.
До недавнего времени в ОФ ЖАХ и особенно в ТФЭ в случае выделения ЛОВ из водных растворов общепринятой являлась процедура предварительного смачивания гидрофобных сорбентов полярным органическим растворителем, смешивающимся с водой, для увеличения площади контакта сорбента с водным раствором SLS. Как показали проведенные нами исследования [22, 23], данная процедура не способствует более эффективному удерживанию выделяемых веществ сорбентом и менее эффективна, чем продувка сорбента газом перед фильтрацией пробы. Это находит логичное объяснение в рамках предложенной физико-химической модели процесса ЖГАХ, согласно которой преимущественный вклад в удерживание ЛОВ вносит адсорбция не из жидкой, а из газовой фазы, находящейся в микро- и мезопорах сорбента. Предварительное смачивание уменьшает величину SGS, т.е. площадь контакта СГФ и сорбента. Особенно негативно предварительное смачивание влияет на объем до проскока Vb, из которого происходит 95%-ное извлечение аналитов.
Другим важным следствием механизма ЖГАХ является возможность температурной оптимизации ТФЭ [24, 25]. В рамках модели ЖГАХ, когда основной вклад в удерживание вносит адсорбция из стационарной газовой фазы, можно принять:
При этом становится неоднозначным влияние температуры на эффективность сорбционного концентрирования, поскольку с повышением температуры происходит, с одной стороны, увеличение коэффициента распределения KGL, а с другой – уменьшение коэффициента адсорбции KGS. Сопоставление температурных зависимостей величин KGL и KGS для неполярных полимерных сорбентов показывает, что при повышении температуры от 20 до 40°С для некоторых классов ЛОВ (простые и сложные эфиры, спирты, кетоны) KGL увеличивается сильнее, чем уменьшается KGS. В результате, как показали результаты проведенных исследований, повышение температуры в указанном диапазоне приводит к увеличению объемов удерживания этих ЛОВ из водных рас-творов в 1.5–2 раза в соответствии с уравнением (4). В еще гораздо большей степени, чем VR, для полярных ЛОВ c повышением температуры возрастают объемы до проскока Vb (рис. 3), поскольку повышение температуры сопровождается также значительным уменьшением вязкости подвижной фазы и, как следствие, увеличением эффективности массообмена [25].
Рис. 3.
Температурная зависимость относительных объемов до проскока при динамической сорбции из водных растворов на полисорбе-1 (концентрация летучих органических веществ в растворе 100 мг/л): 1 – бензол, 2 – метилэтилкетон, 3 – метилацетат, 4 – диэтиловый эфир, 5 – бутанол-1, 6 – триэтиламин.

Результаты проведенных исследований свидетельствуют о присутствии СГФ в порах сорбентов не только в случае водных, но и в случае типичных для ОФ ВЭЖХ водно-органических элюентов [26]. При этом величина объема СГФ влияет как на параметры удерживания, так и на эффективность процесса (табл. 3). Наблюдаемое уменьшение ВЭТТ при переходе от водных к водно-органическим элюентам с концентрацией органического компонента, еще не достаточной для смачивания сорбента (σ > 48 мДж/м2), можно объяснить уменьшением вязкости и соответствующим увеличением коэффициентов диффузии в ПЖФ. Однако наиболее существенным эффектом является увеличение ВЭТТ при уменьшении объема СГФ до значений, когда адсорбция начинает происходить непосредственно из ПЖФ. В целом независимо от величины объема СГФ эффективность ОФ ВЭЖХ в условиях, когда межфазный обмен в хроматографической системе осуществляется по механизму, одной из стадий которого является ЖГАХ, оказывается выше, чем в случае создания условий, при которых происходит межфазный обмен между жидкой фазой и поверхностью адсорбента.
Таблица 3.
Влияние присутствия стационарной газовой фазы на объем удерживания фенола и эффективность процесса ВЭЖХ на колонке 6 × 0.5 см с силасорбом С18 (10–15 мкм) при расходе элюента 3 мл/мин
| Элюент | σ, мДж/м2 | VR/V0 | VG/Vp, % | ВЭТТ, мм |
|---|---|---|---|---|
| Вода | 73 | 9.0 | 76 | 0.11 |
| 2.5% изопропанола в воде | 57 | 7.2 | 39 | 0.09 |
| 5% изопропанола в воде | 52 | 6.3 | 12 | 0.08 |
| 10% изопропанола в воде | 46 | 4.9 | <1 | 0.14 |
| 15% изопропанола в воде | 42 | 3.3 | <1 | 0.14 |
| 20% изопропанола в воде | 39 | 2.5 | <1 | 0.15 |
| 5% диоксана в воде | 58 | 5.7 | 30 | 0.10 |
| 10% диоксана в воде | 54 | 3.4 | 18 | 0.09 |
| 15% диоксана в воде | 51 | 2.1 | 5 | 0.07 |
| 20% диоксана в воде | 50 | 1.5 | <1 | 0.09 |
| 30% диоксана в воде | 47 | 1.2 | <1 | 0.14 |
Из полученных результатов следует возможность оптимизации состава водно-органического элюента с точки зрения повышения эффективности хроматографического процесса за счет применения органического компонента с более высоким поверхностным натяжением, обеспечивающего при прочих равных условиях создание больших объемов СГФ. Например, как следует из табл. 3, 15 %-ный раствор диоксана, обладая приблизительно одинаковой элюирующей силой с 20%-ным раствором изопропанола (об этом можно судить на основании близости величин VR/V0 для обоих растворов), обеспечивает приблизительно вдвое меньшую величину ВЭТТ.
ХРОМАТОМЕМБРАННЫЙ МАССООБМЕННЫЙ ПРОЦЕСС
Физико-химическая модель хроматомембранного массообменного процесса. Открытие ЖГХ и реализация жидкостно-жидкостной хроматографии в гидрофобных бипористых сорбентах (матрицах) послужили отправной точкой для возникновения идеи о новом массообменном процессе [27, 28]. Если гидрофобную бипористую матрицу с двух противоположных сторон ограничить гидрофобными микропористыми мембранами (рис. 4), то возникает система сообщающихся между собой открытых микропор мембран и матрицы. Эти микропоры, размеры которых составляют несколько микрометров, с одной стороны, недоступны для полярной жидкой фазы в силу несмачиваемости материала и возникающего вследствие этого отрицательного капиллярного давления, а с другой стороны, могут служить в качестве транспортных каналов для прохождения неполярной жидкой или газовой фазы. Пространство между гранулами матрицы образует систему открытых макропор (с размерами в несколько десятков микрометров), в которых капиллярное давление ничтожно мало, и по которым способна перемещаться полярная жидкая фаза.
Рис. 4.
Схема хроматомембранного массообменного процесса: 1, 2 – вход и выход полярной жидкой фазы; 3, 4 – вход и выход неполярной жидкой или газовой фазы; 5 – массообменная матрица; 6 – мембраны; 7 – корпус хроматомембранной ячейки.

Благодаря полной аналогии структур хроматомембранной массообменной матрицы и монолитного сорбента, межфазный обмен в ней подчиняется закономерностям хроматографического процесса, а возможности непрерывного выделения веществ из потока одной фазы в поток другой идентичны возможностям мембранных методов. Поэтому подобный массообменный процесс получил название хроматомембранного. Для осуществления непрерывного хроматомембранного массообменного процесса (ХМП) в системе жидкость–газ с одновременным движением жидкости по макропорам, а газа – по микропорам необходимо, чтобы в любой точке массообменной матрицы, в частности, на входе и на выходе из нее, выполнялось следующее условие [29]:
(5)
${{P}_{{{\text{N(G)}}}}} < {{P}_{{\text{P}}}} < {{P}_{{{\text{N(G)}}}}} + \left| {{{P}_{{\text{с }}}}} \right|,$Одновременное движение потоков обменивающихся фаз при осуществлении ХМП возможно в произвольных направлениях, определяемых расположением ограничивающих бипористую матрицу микропористых мембран. Однако практическое значение имеют три схемы относительного перемещения фаз: прямоточная (при движении фаз в одном направлении), противоточная (при движении фаз навстречу друг другу) и двухмерная схема (при движении фаз во взаимно перпендикулярных направлениях, рис. 4). В прямоточной схеме одна из фаз на входе в массообменный слой, где ее давление максимально, и на выходе из этого слоя, где ее давление минимально, контактирует с другой фазой, имеющей ту же направленность градиента давления. В соответствии с условием (5) допустимые градиенты давлений фаз ΔР (разность давлений на входе и выходе) определяются неравенством:
(6)
$\left| {\Delta {{Р }_{{\text{P}}}}--\Delta {{Р }_{{{\text{N(G)}}}}}} \right| \leqslant \left| {{{Р }_{{\text{c}}}}} \right|,$В случае противоточной и двухмерной схем одна из фаз на входе в массообменную матрицу контактирует с другой фазой, находящейся на выходе из него, и допустимые градиенты давлений должны отвечать более жесткому условию:
(7)
$\left| {\Delta {{Р }_{{\text{P}}}} + \Delta {{Р }_{{{\text{N(G)}}}}}} \right| \leqslant \left| {{{Р }_{{\text{c}}}}} \right|.$Таким образом, с точки зрения ширины диапазона возможных потоков фаз через массообменную матрицу прямоточная схема ХМП имеет преимущества по сравнению с двухмерной и противоточной схемами [31]. Это является причиной более высокого быстродействия прямоточной схемы, в результате которого изменение концентрации аналита в принимающей фазе после изменения ее концентрации в отдающей фазе происходит в прямоточной схеме значительно быстрее и не превышает 1–2 мин для ЛОВ с температурами кипения до 100°С [32].
Наряду с непрерывным вариантом возможен и дискретный вариант осуществления ХМП, при котором потоки двух фаз пропускаются через массообменное пространство не одновременно, а последовательно. В случае дискретного варианта градиент давления неподвижной фазы равен нулю, соответственно допустимый градиент давления подвижной фазы, как следует из неравенств (6) и (7), должен отвечать условию:
Влияние природы фаз и пористой структуры матрицы на параметры хроматомембранного массообменного процесса. Как следует из выраже-ний (6)–(8) важнейшей физико-химической характеристикой, от которой зависят возможности ХМП, является величина капиллярного давления Рc. Экспериментально найденные значения Рc для систем ПТФЭ–водная фаза–органическая фаза для различных органических фаз (система А) и ПТФЭ–полярная жидкая фаза–газовая фаза для различных полярных фаз (система B) представлены в табл. 4. В этих системах использованы мембраны из ПТФЭ со средним радиусом пор 0.9 мкм. В первой системе максимальные значения капиллярного давления для одной и той же пористой матрицы из ПТФЭ реализуются для наименее растворимых в воде углеводородов. В системах ПТФЭ–полярная жидкая фаза–газовая фаза величина капиллярного давления увеличивается с увеличением поверхностного натяжения жидкости σ и максимально для воды [31].
Таблица 4.
Экспериментально найденные значения капиллярного давления Pc в различных системах (пояснение см. в тексте)
| Система А | Система B | ||
|---|---|---|---|
| органическая фаза | Рc, МПа | полярная жидкая фаза | Рc, МПа |
| Трибутилфосфат | 0.031 | Иодбензол | 0.058 |
| Бутилацетат | 0.048 | Бензиловый спирт | 0.080 |
| Хлороформ | 0.084 | Нитробензол | 0.086 |
| Бензол | 0.086 | Этиленгликоль | 0.095 |
| Тетрахлорметан | 0.108 | Формамид | 0.134 |
| Октан | 0.123 | Вода | 0.171 |
Исходя из условий осуществления хроматомембранного процесса, можно сформулировать основные требования к материалу бипористой матрицы и к размерам макро- и микропор в ней. Материал матрицы, во-первых, должен быть химически инертным по отношению к обеим фазам, во-вторых, должен иметь максимальную величину краевого угла смачивания полярной жидкой фазой, т.е. быть максимально гидрофобным, а в-третьих, должен обеспечивать возможность получения бипористой матрицы с заданной структурой и воспроизводимыми свойствами поверхности. В максимальной степени указанным требованиям отвечает ПТФЭ. Бипористая матрица из ПТФЭ получается путем спекания исходного полимеризационного порошка-сырца, его размола, фракционирования и повторного спекания. Размеры микропор в ней соответствуют размерам частиц исходного полимеризационного порошка, а размеры макропор, в качестве которых служат зазоры между повторно спеченными частицами, сопоставимы с размерами этих частиц.
Критерии выбора размеров макропор в ХМП те же, что и при выборе размеров частиц сорбентов и носителей в хроматографии. В обоих случаях приходится искать компромисс между увеличением эффективности массообмена и уменьшением проницаемости массообменного слоя по отношению к полярной жидкой фазе при уменьшении размеров частиц. Как показали результаты проведенных исследований, для решения большинства аналитических задач диапазон размеров макропор от 0.1 до 1.0 мм можно принять в качестве рабочего диапазона, в пределах которого осуществляется выбор оптимума для решения конкретных задач [33].
В ХМП межфазный обмен происходит на границе микро- и макропор. Соответственно в случае системы жидкость–газ микропоры выступают лишь в роли каналов для прохождения газовой фазы и их радиус не оказывает значимого влияния на эффективность массообмена. Поэтому оптимальный радиус микропор соответствует условию достижения максимального расхода газовой фазы. Увеличение радиуса микропор, с одной стороны, ведет к увеличению проницаемости, а с другой, – к уменьшению Рс и, следовательно, к уменьшению допустимой величины ΔРN(G). Как показал расчет [29], зависимость расхода неполярной жидкой фазы от радиуса микропор бипористой матрицы и мембран проходит через максимум в диапазоне от 2 до 20 мкм, а газовой фазы – в диапазоне от 1 до 10 мкм. На основании результатов ртутной порометрии образцов ПТФЭ, полученных из различных марок исходного полимеризационного порошка, установлено [34], что для реализации ХМП в системе жидкость–газ оптимальным является ПТФЭ марки Ф-4ПН, а для системы жидкость–жидкость – марки Ф-4ТН. Указанные марки ПТФЭ являются продуктами суспензионной полимеризации тетрафторэтилена с различными размерами частиц [35].
Применение бипористых матриц и мембран с оптимальными размерами макро- и микропор позволяет реализовывать ХМП в достаточно широких диапазонах скоростей потоков обменивающихся фаз. Максимальные удельные расходы (мл/(мин см2)) фаз через хроматомембранную ячейку (ХМЯ) составляют для водной фазы 4–8, для органической фазы 0.3–0.6 и 10–20 для газовой фазы. В случае прямоточной схемы они возрастают в 4–5 раз [31, 32]. Увеличение абсолютных объемных скоростей потоков фаз в ХМП возможно за счет рационального выбора конфигурации бипористой матрицы. Например, увеличив площадь поперечного сечения матрицы перпендикулярно потоку газовой фазы и сократив протяженность матрицы по направлению движения этой фазы, можно достичь объемных скоростей потока до 0.5–1.0 л/мин. Подобная возможность, в частности, реализована при жидкостно-абсорбционном концентрировании аналитов из потока анализируемого воздуха [36].
Закономерности непрерывного хроматомембранного массообменного процесса и его описание в рамках тарелочной теории хроматографии. Хроматографический принцип межфазного обмена в ХМП делает целесообразным применение для описания его закономерностей тарелочной теории. В случае противоточной схемы процесса массообменный слой в ХМЯ можно рассматривать по аналогии с обычной хроматографической колонкой как ряд последовательно соединенных эквивалентных теоретических тарелок (в дальнейшем тарелок), характеризующихся ВЭТТ [37]. Для описания двухмерной схемы нами предложено [38] исходить из представления о массообменном слое как о прямоугольной матрице, состоящей из совокупности двухмерных тарелок, которые характеризуются ВЭТТ по направлению движения потока отдающей фазы (h0) и ВЭТТ по направлению движения потока принимающей фазы – экстрагента (hЕ). В рамках тарелочной теории на основании уравнений материального баланса и условия равновесия в пределах теоретической тарелки нами получены уравнения, связывающие концентрацию компонента в потоке принимающей фазы (экстрагента) на выходе из ХМЯ (cЕ) с его концентрацией в потоке отдающей фазы (пробы) на входе в ХМЯ (c0), отражающие закономерности прямоточной [31], противоточной [37] и двухмерной [28] схем ХМП. Установлено, что при осуществлении прямоточной и противоточных схем ХМП величина ВЭТТ составляет 5–15 мм. В случае двухмерной схемы величины h0 и hЕ зависят от агрегатного состояния соответствующей фазы. По направлению движения жидкой фазы ВЭТТ составляет те же 5–15 мм, а по направлению движения газовой фазы – 1–3 мм. Полученные значения ВЭТТ позволяют оценить число тарелок для массообменного слоя с известной протяженностью.
Общие закономерности ХМП, независимо от абсолютных значений коэффициентов распределения выделяемых аналитов, отражают зависимости величины cE/(c0K) от соотношения W/(WEK), где W и WE– объемные скорости потока отдающей и принимающей фаз, соответственно, а K – коэффициент распределения аналита, равный отношению его равновесных концентраций между принимающей и отдающей фазами. На подобных зависимостях независимо от схемы относительного перемещения обменивающихся фаз через ХМЯ можно выделить три области осуществления ХМП (рис. 5): область полного извлечения компонента из потока пробы в поток экстрагнена, когда с достаточной для практики точностью, можно принять
(9)
${{c}_{{\text{Е }}}} = {{{{c}_{0}}W} \mathord{\left/ {\vphantom {{{{c}_{0}}W} {{{W}_{{\text{E}}}}}}} \right. \kern-0em} {{{W}_{{\text{E}}}}}};$Рис. 5.
Зависимости отношения концентраций компонентов в принимающей и отдающей фазах от соотношения их потоков через хроматомембранную ячейку, рассчитанные [39] для числа тарелок N = 1 (1), N = 3 (2) и N = 10 (3), и экспериментальные, полученные для бензола (4) и хлороформа (5). I – область полного извлечения, II – область частичного извлечения и насыщения, III – область равновесного насыщения.
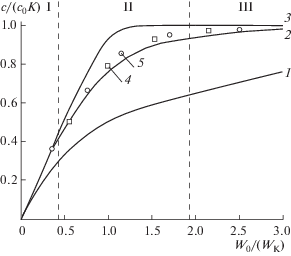
область частичного извлечения компонента; и область равновесного насыщения потока экстрагента выделяемым компонентом, когда можно принять
С точки зрения аналитического применения наибольший интерес представляют режимы полного извлечения и равновесного насыщения, которые обеспечивают минимальную погрешность анализа, поскольку в этом случае результаты анализа в минимальной степени подвержены влиянию изменений условий эксперимента. Область осуществления этих режимов зависит от выбранной схемы ХМП. В прямоточной схеме реализуется не более одной тарелки, поэтому если длина массообменного слоя больше ВЭТТ, то ее изменение не влияет на эффективность ХМП. В отличие от традиционного барботирования, для которого эффективность массообмена также характеризуется N = 1, прямоточная схема ХМП позволяет приблизительно на порядок увеличить допустимые для установления межфазного равновесия скорости потоков обменивающихся фаз и тем самым резко сократить продолжительность стадии выделения. Недостатком прямоточной схемы ХМП являются узкие диапазоны соотношения расходов фаз, при которых реализуются режимы полного извлечения и равновесного насыщения. Однако положительный эффект от увеличения числа тарелок в противоточной схеме постепенно ослабевает по мере увеличения N. Поскольку увеличение длины массообменного слоя приводит к пропорциональному увеличению инерционности экстракционной системы, применение громоздких ХМЯ становится нерациональным. С учетом величины ВЭТТ оптимальная длина массообменного слоя ХМЯ, при которой обеспечивается N ≈ 3–4, для противоточной схемы составляет 20–30 мм.
Аналогичные тенденции выявлены и для двухмерной схемы ХМП. Установлено, что к повышению концентрации выделяемого компонента в экстрагенте приводит увеличение протяженности массообменного слоя как по направлению движения отдающей, так и по направлению движения принимающей фазы. При прочих равных условиях, в частности, при одном и том же объеме массообменного слоя, максимальные значения сЕ достигаются при равном числе тарелок в направлении движения потоков отдающей и принимающей фаз, что делает оптимальным соотношение протяженности массообменного слоя в этих направлениях 5 : 1.
Классификация хроматомембранных методов. Хроматомембранные методы подобно хроматографическим многоварианты с точки зрения как агрегатного состояния фаз, участвующих в массообменном процессе, так и способов его осуществления. Однако при этом число возможных вариантов методов существенно меньше, так как ХМП, в отличие от хроматографии, осуществим только в системах с двумя флюидными фазами. Основные различия между возможными вариантами сводятся к схемным решениям организации межфазного контакта. Варианты различных хроматомембранных методов приведены в табл. 5.
Таблица 5.
Классификация хроматомембранных методов разделения веществ
| Классификационный признак | Варианты методов разделения | Сущность метода |
|---|---|---|
| Агрегатное состояние отдающей и извлекающей фаз | Жидкостная экстракция | Выделение веществ из одной жидкой фазы в другую |
| Газовая экстракция | Выделение веществ из полярной жидкой фазы в газовую | |
| Жидкостная абсорбция | Выделение веществ из газовой фазы в полярную жидкую | |
| Направление относительного перемещения фаз | Прямоточный | Движение фаз в одном направлении |
| Противоточный | Движение фаз в противоположных направлениях | |
| Двухмерный | Движение фаз во взаимно перпендикулярных направлениях | |
| Характер массообменного процесса | Непрерывный | Одновременное движение обеих фаз через ХМЯ |
| Дискретный | Попеременное движение через ХМЯ | |
| Режим выделения целевых компонентов | Полное извлечение | Количественное извлечение целевого компонента из пробы в принимающую фазу |
| Частичное извлечение | Частичное извлечение целевого компонента из пробы или частичное им насыщение принимающей фазы | |
| Равновесное насыщение | Равновесное насыщение принимающей фазы целевым компонентом |
АНАЛИТИЧЕСКОЕ ПРИМЕНЕНИЕ ХРОМАТОМЕМБРАННЫХ МЕТОДОВ РАЗДЕЛЕНИЯ ВЕЩЕСТВ
Основными областями аналитического применения хроматомембранных методов являются процессы выделения и концентрирования аналитов при анализе водных и газовых сред [29, 33, 39]. При этом проявляются основные достоинства ХМП – высокая эффективность массообмена, присущая хроматографическим методам, и непрерывный характер процесса, свойственный мембранным методам. Аналитическое применение находят все 3 метода, упомянутые в табл. 5: хроматомембранная жидкостная экстракция (ХЖЭ), хроматомембранная газовая экстракция (ХГЭ) и хроматомембранная жидкостная абсорбция (ХЖА). Первый из перечисленных методов прежде всего находит применение в проточных методах: в проточно-инжекционном (ПИА), в непрерывном проточном анализе (НПА) водных сред и циклическом инжекционном анализе (ЦИА), второй – в парофазном хроматографическом анализе жидких сред, и, наконец, третий – в анализе газовых сред и отличается большим разнообразием способов конечного определения выделенных аналитов [40, 41].
Аналитическое применение хроматомембранной жидкостной экстракции. В проточных методах ХЖЭ является альтернативой жидкостной экстракции в сегментированных потоках [42, 43], которая не позволяет достичь существенных коэффициентов концентрирования аналитов из-за ограничений в возможности варьирования соотношений объемов отдающей и принимающей фаз. В качестве иллюстрации аналитических возможностей непрерывной ХЖЭ можно привести осуществляемое в рамках НПА определение меди(II) в водных растворах с использованием экстрагента – раствора дитизона в СCl4 [44]. Выбранному соотношению объемных скоростей потоков водной пробы и экстрагента (20 : 1) через ХМЯ соответствует коэффициент концентрирования, равный 20. Сигнал проточного фотометрического детектора, установленного в линии потока экстрагента, прямо пропорционален концентрации меди в потоке водной пробы (рис. 6).
Рис. 6.
Изменение аналитического сигнала при определении меди (II) в водных растворах с использованием непрерывной хроматомембранной жидкостной экстракции. Анализируемая проба: водный раствор (рН 2) с содержанием меди, мкг/л: 10 (1), 20 (2), 30 (3). Экстрагент: CCl4 + 5 × 10–6 М дитизона. Скорость потока пробы 3 мл/мин, экстрагента – 0.15 мл/мин.

Возможность непрерывного выделения аналитов из потока водной пробы в поток экстрагента с непрерывным получением информации о концентрации аналитов в анализируемой среде выгодно отличает ХЖЭ от различных вариантов сорбционного концентрирования, предполагающих, как правило, чередование циклов сорбция–десорбция с периодическим определением аналитов в десорбирующем растворе. Примером практической реализации дискретного варианта ХЖЭ является определение нитрит-ионов [45] и анионных ПАВ [46]. При ХЖЭ анионных поверхностно-активных веществ в форме ионных ассоциатов с метиленовым голубым в хлороформ (продолжительность концентрирования 5 мин) с последующим фотометрическим детектированием нижняя граница диапазона определяемых концентраций составила 8 мкг/л. Увеличивая продолжительность стадии хроматомембранного концентрирования, предел обнаружения может быть снижен по крайней мере в 4 раза. В этом состоит принципиальные преимущество хроматомембранного экстракционного концентрирования по сравнению с сегментными схемами экстракции в ПИА.
Существенным недостатком ХЖЭ с непосредственной подачей анализируемой жидкости в ХМЯ является засорение бипористой гидрофобной матрицы взвешенными частицами, присутствующими в анализируемых водных пробах, например, в природной воде. При этом предварительная фильтрация пробы неприемлема из-за возможных потерь определяемых веществ. В этом случае более предпочтительным оказывается сочетание экстракционного концентрирования аналитов в экстракционно-хроматографической колонке из потока водной пробы с последующим их элюированием из колонки потоком экстрагента и хроматомембранным отделением экстракта от водной фазы. В этой схеме миниатюрная экстракционно-хроматографическая колонка является одновременно механическим фильтром для отделения взвесей, который периодически легко заменять, а хроматомембранная ячейка выступает в качестве высокоэффективного сепаратора двухфазных водно-органических потоков.
Рассмотренная двухстадийная схема концентрирования, основанная на сочетании экстракционно-хроматографического концентрирования аналитов с хроматомембранным отделением органического экстракта от водной фазы, может рассматриваться как общее решение проблемы жидкостной экстракции в потоке. Эта схема позволяeт, например, осуществлять проточное определение в природных водах нефтепродуктов [47, 48], фенолов [48, 49]. При этом их пределы обнаружения могут быть снижены пропорционально увеличению продолжительности стадии экстракционного концентрирования. Характеристики методик проточного анализа с ХЖЭ приведены в табл. 6.
Таблица 6.
Характеристики методик проточного и лабораторного анализа природных вод с хроматомембранной жидкостной экстракцией
| Определяемые вещества |
t, мин | Метод определения | сmin, мкг/л | Литература |
|---|---|---|---|---|
| Медь(II) | 5 | Фотометрический | 0.5 | [44] |
| Нитрит-ионы | Тот же | [45] | ||
| Анионные ПАВ | 5 | » | 8 | [46] |
| Нефтепродукты | 5 | Люминесцентный | 1 | [47, 48] |
| Сумма фенолов | 10 | Тот же | 1 | [48, 49] |
| Фосфат-ионы | 10 | Фотометрический | 3 | [50] |
| Силикат-ионы | 10 | Тот же | 3 | [50] |
| Медь(II) | 6 | » | 40 | [51] |
| Цинк(II) | 6 | » | 40 | ]51] |
| ПАУ | 4 | Газовая хроматография | 0.1 | [52] |
Аналитическое применение хроматомембранной газовой экстракции. Основная область применения газовой экстракции – извлечение летучих веществ из жидкой в газовую фазу для последующего газохроматографического определения. Как известно [53], газовая экстракция позволяет снизить пределы обнаружения по сравнению с непосредственным вводом анализируемого водного раствора в испаритель хроматографа, если коэффициент распределения KGL >10–3. Этому условию отвечают многие классы веществ, за исключением наиболее полярных, хорошо растворимых в воде (табл. 7).
Таблица 7.
Диапазоны коэффициентов распределения (KGL = сG/cL) различных классов веществ между водной и газовой фазой при комнатной температуре [33]
| Класс веществ | KGL | Класс соединений | KGL |
|---|---|---|---|
| Инертные газы, О2, N2, Н2 | 10–100 | Простые эфиры С2–С8 | 0.02–0.2 |
| Фторуглеводороды С1–С4 | 20–200 | Алкилацетаты и формиаты С3–С7 | 0.002–0.01 |
| Алканы С1–С6 | 5–30 | Кетоны С3–С6 | 0.001–0.02 |
| Алкены С2–С6 | 5–20 | Альдегиды С1–С5 | 2 × 10–4–3 × 10–3 |
| Алкины С2–С6 | 1–10 | Спирты С1–С5 | 10–4–10–3 |
| СО2, Н2S, N2O, NO, AsH3 | 0.2–4 | Амины С1–С4 | 10–5–10–4 |
| Алкилбензолы С6–С8 | 0.2–0.5 | Фенолы С6–С8 | 10–5–10–4 |
| Хлоругдеводороды С1–С3 | 0.04–1.0 | Карбоновые кислоты С1–С5 | 10–6–10–5 |
Общеметодические вопросы непрерывной ХГЭ рассмотрены в работах [29, 54]. Критериями выбора наиболее рациональных вариантов, схем и режимов ХГЭ являются значения КGL определяемых веществ, диапазон определяемых концентраций и режим проведения анализа (off-line или on-line) [37]. Дискретный вариант ХГЭ подчиняется тем же закономерностям, что и фронтальный вариант ЖГХ, но сложнее в аппаратурном отношении. Непрерывный вариант ХГЭ пригоден как для анализа в режиме on-line, так и для лабораторного анализа предварительно отобранных проб. В последнем случае пробу отбирают в сосуд емк. 50–100 мл, снабженный двумя отводными трубками для подачи в сосуд газа-носителя, который за счет избыточного давления вытесняет пробу из сосуда в ХМЯ. Выходящий из ХМЯ поток газа-носителя, управляемый с помощью регулятора расхода, направляют в дозирующую петлю крана дозатора и после установления стационарного режима ХМП анализируют на газовом хроматографе.
Определение ЛОВ с КGL > 1 наиболее рационально проводить в режиме полного извлечения, поскольку в этом случае для реализации режима равновесного насыщения необходимо отбирать очень большие (несколько литров) объемы проб и поддерживать очень высокие расходы пробы через ХМЯ [55, 56]. При этом наиболее рациональными схемами ХГЭ являются противоточная и двухмерная, обеспечивающие максимально возможные для данного состава пробы концентрации выделяемых веществ в потоке газа-экстрагента.
Определение ЛОВ с КGL < 1 для обеспечения максимальной чувствительности предпочтительнее проводить в режиме равновесного насыщения [57–59]. При лабораторном анализе, когда быстродействие экстракционной системы не имеет принципиального значения, оптимальной является двухмерная схема ХГЭ. Эта схема, в отличие от противоточной, позволяет варьировать соотношения расходов фаз в гораздо более широком диапазоне и, в отличие от прямоточной схемы, – достигать режима равновесного насыщения при гораздо меньшем соотношении W/WE. При анализе в режиме on-line более рациональна прямоточная схема, обеспечивающая максимальное быстродействие [32]. При лабораторном определении ЛОВ с КGL < 0.05 режим равновесного насыщения легко достигается для всех схем, и выбор схемы осуществления ХМП не имеет принципиального значения.
Хроматомембранная газовая экстракция из фиксированного объема пробы. Для осуществления процесса непрерывной ХГЭ с одновременным пропусканием потоков водной пробы и газа-экстрагента через ХМЯ требуются относительно большие объемы пробы и существенная модернизация газовой схемы хроматографа. Значительно проще схема осуществления ХГЭ из фиксированного объема пробы (ХГЭФП) (рис. 7) [60]. При осуществлении ХГЭФП небольшой (1–2 мл) объем водной пробы вводят в бипористую матрицу ХМЯ с помощью шприца через тройник, снабженный самоуплотняющейся резиной, подобно дозированию жидкой пробы в испаритель газового хроматографа. Хроматомембранную ячейку предварительно продувают газом-экстаргентом. Объем пробы составляет до 90% от суммарного объема макропор в используемой ХМЯ. Проба занимает макропоры матрицы, в то время как поток газа-экстаргента проходит через ее микропористую структуру. Выходящий из ХМЯ поток газа-экстрагента направляется в дозирующую петлю объемом 0.5–1 см3 стандартного крана-дозатора. По окончанию анализа водная проба сбрасывается из ХМЯ за счет избыточного давления газа-экстрагента через кран, который при проведении экстракции закрыт.
Рис. 7.
Схема процесса хроматомембранной газовой экстракции с фиксированным объемом пробы: 1, 2 – вход и выход потока газа-экстрагента; 3, 4 – вход и выход полярной водной пробы; 5 – бипористая матрица; 6 – микропористая мембрана; 7 – тройник; 8 – шприц с водной пробой; 9 – кран для слива пробы.

Преимуществом ХГЭФП по сравнению с традиционной газовой экстракцией является сокращение продолжительности газоэкстракционного выделения. В этом случае суммарная продолжительность газовой экстракции и доставки газовой фазы в дозирующую петлю хроматографа не превышает 2–3 мин. Основным ограничением ХГ-ЭФП является адсорбция аналитов на поверхности бипористой матрицы из ПТФЭ, которая проявляется для относительно неполярных, плохо растворимых в воде аналитов с температурой кипения выше 80°С. Определение в воде углеводородов с помощью ХГЭФП неэффективно из-за сильного эффекта памяти.
Независимо от выбранной схемы предел обнаружения аналита (сmin) при использовании ХГЭ в режиме равновесного насыщения можно оценить по формуле:
Хроматомембранная газовая экстракция в сочетании с газоадсорбционным концентрированием. Общим методическим решением, обеспечивающим снижение пределов обнаружения на 2–3 порядка практически любых аналитов, является сочетание газовой экстракции с дополнительным газоадсорбционным концентрированием аналитов из потока газа-экстаргента. В англоязычной литературе подобное сочетание получило название purge and trap pre-concentration (PATP) [62]. При осуществлении хроматомембранного варианта PATP поток газа-экстрагента, выходящий из ХМЯ, направляют в микроколонку с адсорбентом, которую устанавливают на место дозирующей петли крана-дозатора. После пропускания определенного объема газа-экстрагента колонку нагревают и потоком газа-носителя переносят десорбированные аналиты в газовый хроматограф. Преимущество ХГЭ при реализации подобной комбинированной схемы пробоподготовки по сравнению с традиционными вариантами газовой экстракции состоит в возможности при прочих равных условиях в 3–5 раз увеличить скорость потока газа-экстрагента и соответственно уменьшить продолжительность стадии газоэкстракционного извлечения аналитов. Оптимальными сорбентами для газоадсорбционного концентрирования ЛОВ из потока влажного газа являются неполярные полимерные гидрофобные сорбенты [63, 64]. При определении низкокипящих (до 100°С) органических веществ предпочтение отдают углеродным адсорбентам, в частности, активным углям [65, 66].
Реализация хроматомембранного варианта PATP возможна не только в лабораторных, но и в полевых условиях (рис. 8) [63]. Отобранная водная проба 1 под действием собственной силы тяжести вытекает из сосуда 2, проходит через ХМЯ 3 и поступает на сброс 4. За счет создающегося в сосуде разрежения в него поступает поток атмосферного воздуха 5, который проходит через ХМЯ 3, экстрагируя летучие аналиты. Последние удерживаются в сорбционной колонке 6. При этом самопроизвольно поддерживается соотношение расходов водный пробы и экстрагента – воздуха через ячейку, равное 1 : 1, при котором обеспечивается режим равновесного насыщения для большинства классов органических соединений. По окончании процесса извлечения аналитов сорбционную колонку герметизируют и доставляют в лабораторию для термодесорбции и газохроматографического определения сконцентрированных аналитов. Таким образом, можно отказаться от транспортировки в лабораторию больших объемов проб воды, что помимо технических и экономических проблем часто приводит к неконтролируемым потерям летучих аналитов.
Рис. 8.
Схема хроматомембранного варианта PATP (purge and trap pre-concentration), реализуемого в полевых условиях: 1 – водная проба, 2 – сосуд, 3 – хроматомембранная ячейка, 4 – сброс водной пробы, 5 – вход атмосферного воздуха, 6 – сорбционная колонка.

Характеристики разработанных газохроматографических методик, основанных на ХГЭ, приведены в табл. 8 [13]. В случае газоадсорбционного концентрирования применяли микроколонку (6 × 0.3 см) с Porapack QS. Как следует из сопоставления данных табл. 8 и значений ПДК в питьевой воде [67], ХГЭ в сочетании с газоадсорбционным концентрированием аналитов позволяет определять их в водных объектах окружающей среды на уровне самых жестких экологических и санитарно-гигиенических норм.
Таблица 8.
Основные характеристики газохроматографических методик определения летучих веществ в водных растворах с хроматомембранной газовой экстракцией
| Аналит | Вариант ХГЭ | Детектор | сн, мкг/л | Δr, % | t, мин |
|---|---|---|---|---|---|
| Кислород, азот | Противоточный, полное извлечение | ДТП | 20 | 7 | 3 |
| Диоксид углерода | Двухмерный, равновесное насыщение | ДТП | 200 | 6 | 4 |
| Тот же с конверсией в метан | ДИП | 1 | 8 | 4 | |
| Алканы С1–С4 | Противоточный, частичное извлечение | ДИП | 0.1–0.2 | 10 | 5 |
| Этилен | Тот же | ДИП | 0.2 | 10 | 5 |
| Ацетилен | » | ДИП | 0.5 | 10 | 5 |
| 1,2-Дихлорэтан | Прямоточный, равновесное насыщение | ДИП | 60 | 7 | 6 |
| Динамический, равновесное насыщение | ДИП | 100 | 5 | 4 | |
| Бензол | Двухмерный, равновесное насыщение | ДИП | 6 | 6 | 6 |
| Тетрахлорид углерода | Двухмерный, равновесное насыщение | ДИП | 200 | 8 | 6 |
| Тот же | ЭЗД | 0.01 | 9 | 6 | |
| Тот же + ГК | ДИП | 1 | 14 | 10 | |
| Хлороформ | Двухмерный, равновесное насыщение | ДИП | 100 | 6 | 5 |
| Тот же | ЭЗД | 0.1 | 7 | 6 | |
| Тот же + ГК | ДИП | 0.5 | 12 | 10 | |
| ХГЭФП | ДИП | 200 | 6 | 4 | |
| ХГЭФП + ГК | ДИП | 3 | 6 | 6 | |
| Метилацетат | Прямоточный, равновесное насыщение | ДИП | 400 | 5 | 5 |
| Тот же + ГК | ДИП | 2 | 11 | 10 | |
| ХГЭФП | ДИП | 1000 | 4 | 5 | |
| ХГЭФП + ГК | ДИП | 2 | 4 | 7 | |
| Метанол | ХГЭФП | ДИП | 20 000 | 7 | 4 |
| ХГЭФП + ГК | ДИП | 80 | 9 | 5 |
Примечание: сн – нижняя граница определяемых концентраций, Δr – относительная погрешность (P = 0.95, n = 3), t – продолжительность параллельного определения, ДТП –детектор по теплопроводности, ДИП – детектор ионизационно-пламенный, ЭЗД – электронозахватный детектор, ГК – газоадсорбционное концентрирование.
Хроматомембранная газовая экстракция в процессах генерирования стандартных газовых смесей. Одой из актуальных задач аналитической химии является разработка стандартных образцов состава веществ. Известные способы получения стандартных газовых смесей (СГС) в силу специфических особенностей газообразного состояния вещества либо длительны и трудоемки, либо позволяют получать относительно небольшие объемы образцов [68]. Новые возможности для генерирования СГС с постоянной заданной концентрацией целевых компонентов открыла ХГЭ [69]. Если потоки газа-экстрагента и генерирующего водного раствора с постоянной концентрацией целевых компонентов пропускают через ХМЯ одновременно, то объем генерируемой СГС практически неограничен. Однако реализация подобного варианта ХГЭ требует довольно сложного аппаратурного оформления и поддержания расходов фаз в определенных диапазонах, обеспечивающих режим равновесного насыщения.
Более простым является дискретный вариант ХГЭ, когда поток газа-экстрагента проходит через неподвижный генерирующий раствор. В качестве методического решения, позволяющего многократно увеличить объемы генерируемых СГС в этом случае, предложены хроматомембранные матрицы, модифицированные сорбционно-активным по отношению к целевым компонентам материалом (активным углем) [70]. Введение в матрицу сорбента позволяет на несколько порядков увеличить объем генерируемой СГС с квазипостоянной концентрацией целевых компонентов (рис. 9).
Рис. 9.
Изменение относительных концентраций хлороформа (1, 3) и этилацетата (2, 4) в потоке генерируемой газовой смеси от объема газа-экстрагента, пропущенного через хроматомембранную ячейку с немодифицированной (1, 2) и композиционной матрицей, содержащей 8 мас. % активного угля БАУ (3, 4).
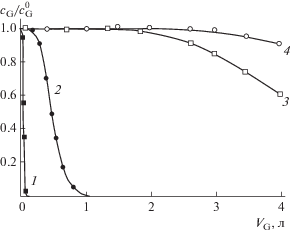
Хроматомембранная газовая экстракция позволяет генерировать СГС с заранее заданной концентрацией целевых компонентов, исходя из их коэффициентов распределения в системе жидкость–газ. При этом наличие сорбента в матрице не влияет на начальную концентрацию целевых компонентов в генерируемой газовой смеси, а обусловливает только величину буферного эффекта, проявляющегося в увеличении объема генерируемой СГС с постоянной концентрацией этих компонентов [70, 71]. Подобный эффект может быть усилен, если для генерирования использовать не традиционные монолитные сорбенты-матрицы, а гранулированные композиционные угольно-фторопластовые сорбенты, в которых содержание активного угля может достигать 35% от массы носителя [72], вместо 5–10% в монолитных сорбентах.
Аналитическое применение хроматомембранной жидкостной абсорбции. Жидкостная абсорбция позволяет одновременно концентрировать определяемые вещества и переводить их из газовой в более удобную для выполнения химического анализа жидкую фазу. Определение большинства веществ даже на уровне ПДК воздуха рабочей зоны становится возможным, если при абсорбционном выделении эти вещества концентрируются не менее, чем на 2–3 порядка. Очевидно, что для этого значение K = сL/сG (K = 1/KGL) должно быть не менее 103. При абсорбции в воду это условие выполняется для всех наиболее полярных органических соединений (см. табл. 7).
Высокие значения K делают рациональным применение полного извлечения. Из-за экспериментальных осложнений реализация непрерывного ХМП ограничена соотношением расходов обменивающихся фаз приблизительно 100 : 1, поэтому для достижения более высокой степени концентрирования необходимо использовать дискретный вариант ХЖА. Среди возможных схем реализации ХЖА предпочтительнее двухмерная, которая при одном и том же объеме абсорбента VL за счет выбора оптимальной конфигурации массообменного слоя (максимального увеличения площади поперечного сечения, перпендикулярного потоку газовой фазы) позволяет пропускать анализируемый воздух через ХМЯ с максимальным расходом.
Подобно другим хроматомембранным методам адекватное описание процесса ХЖА возможно в рамках тарелочной модели [73–75]. Хроматографический принцип межфазного обмена обусловливает превосходство ХЖА над традиционным барботированием, при осуществлении которого эффективность массообмена в лучшем случае характеризуется одной эквивалентной теоретической тарелкой. При физической абсорбции ХЖА позволяет в несколько раз увеличить объемы до проскока выделяемых аналитов и тем самым увеличить коэффициенты их концентрирования. В качестве иллюстрации на рис. 10 приведены выходные кривые удерживания метанола из потока воздуха одним и тем же объемом воды, находящейся в барботере и хроматомембранной ячейке. При хемосорбции, учитывая ничтожно малые концентрации аналитов, поглотительные растворы можно рассматривать как абсорбенты с бесконечно большой емкостью. В этом случае за счет более высокой эффективности массообмена хроматомембранная абсорбция позволяет приблизительно на порядок увеличить допустимую скорость пропускания анализируемого воздуха через абсорбент по сравнению с барботированием и тем самым сократить продолжительность стадии концентрирования для достижения одних и тех же пределов обнаружения. В качестве иллюстрации на рис. 11 сопоставлены выходные кривые удерживания формальдегида водным раствором бисульфита натрия, при взаимодействии которых образуется нелетучий продукт.
Рис. 10.
Кривые абсорбционного удерживания паров метанола из воздуха водой (VL = 1.3 мл) в зависимости от объема пропущенной пробы воздуха: 1 – барботер, 2 – хроматомембранная ячейка.

Рис. 11.
Выходные кривые динамической хемосорбции формальдегида водным 0.1 М раствором бисульфита натрия: 1, 3 – хроматомембранная абсорбция при WG 100 и 200 мл/мин соответственно; 2, 4 – барботирование при WG 50 и 100 мл/мин соответственно.

Пределы обнаружения при использовании ХЖА в режиме полного извлечения можно оценить по формуле:
(11)
${{с }_{{{\text{min}}}}} = {{{{с }_{{{\text{lim}}}}}{{V}_{{\text{L}}}}\gamma } \mathord{\left/ {\vphantom {{{{с }_{{{\text{lim}}}}}{{V}_{{\text{L}}}}\gamma } {{{W}_{{\text{G}}}}t}}} \right. \kern-0em} {{{W}_{{\text{G}}}}t}},$Хроматомембранная жидкостная абсорбция легко сочетается практически с любыми методами определения аналитов в жидких средах [39]. От метода детектирования зависят оптимальные размеры используемых ХМЯ. В случае газохроматографического анализа жидкого концентрата, когда объем вводимой в хроматограф пробы составляет несколько микролитров, рационально применение хроматомембранных микроячеек с объемом массообменного слоя в несколько десятков микролитров. При традиционном фотометрическом анализе этот объем определяется объемом кюветы фотометрического детектора и обычно составляет несколько миллиитров.
Хроматомембранная жидкостная абсорбция позволяет определять многие органические и неорганические загрязнители в воздухе на уровне ПДК. Достигаемые при этом пределы обнаружения в случае хемосорбции лимитируются только продолжительностью стадии хроматомембранного выделения в соответствии с выражением (11). Характеристики разработанных методик с использованием ХЖА приведены в табл. 9. В случае фотометрического определения в скобках указан реагент. При определении метанола и этанола в атмосферном воздухе на уровне фоновых концентраций (1–3 мкг/м3) после их абсорбционного выделения в дистиллированную воду и конверсии в соответствующие алкилнитриты проводили парофазное газохроматографическое определение. Подобный известный методический прием позволил приблизительно в 100 раз снизить пределы обнаружения спиртов по сравнению с непосредственным газохроматографическим анализом водной фазы.
Таблица 9.
Характеристики методик анализа воздуха с хроматомембранной жидкоcтной абсорбцией аналитов
| Определяемое вещество | Абсорбент | t, мин | Метод определения | сн, мкг/м3 | Δr, % | Литература |
|---|---|---|---|---|---|---|
| Диоксид серы | Раствор реагента | 8 | Фотометрия (иод-крахмальный ассоциат) | 20 | 10 | [76] |
| Диоксид азота | Раствор KI | 10 | Фотометрия (4-иод-N,N-диметил-анилин) | 50 | 9 | [77] |
| Гидразин | 0.1 М HCl | 5 | Фотометрия (пара-диметиламино-бензальдегид). | 10 | 12 | [36] |
| Диметил-гидразин | 0.1 М HCl | 15 | Фотометрия (пара-диметиламино-бензальдегид + СuSO4) | 30 | 13 | [33] |
| HF | Вода | 5 | Ионная хроматография | 20 | 15 | [78] |
| HCl | Вода | 5 | Ионная хроматография | 40 | 15 | [78] |
| Формальдегид | 0.2 M NH3 | 10 | Фотометрия (ацетилацетон) | 30 | 11 | [79] |
| 0.2 M NH3 | 10 | Фотометрия (фенилгидразин) | 6 | 10 | [79] | |
| Фенол | Вода | 10 | Флюориметрия | 5 | 8 | [39] |
| Метанол | Вода | 10 | Газовая хроматография | 30 | 17 | [73] |
| Вода | 20 | Тот же (парофазный анализ образовавшегося алкилнитрита) | 0.3 | 20 | [80] | |
| Этанол | Вода | 10 | Газовая хроматография | 40 | 17 | [73] |
| Вода | 20 | Тот же (парофазный анализ образовавшегося метилнитрита) | 0.4 | 20 | [80] | |
| Бутанол | Вода | 10 | Газовая хроматография | 60 | 15 | [73] |
Хроматомембранная жидкостная абсорбция позволяет автоматизировать определение загрязнителей воздуха в сочетании с проточно-инжекционными схемами анализа водного концентрата. Наиболее удобными методами детектирования в этом случае являются фотометрический, флуориметрический и амперометрический. Разработаны методики проточно-инжекционного определения наиболее распространенных загрязнителей атмосферного воздуха, таких как оксиды азота [81, 82], диоксид серы [76, 83], озон и аммиак [84], формальдегид [79, 85]. Предложена методика инверсионно-вольтамперометрического определения паров ртути в воздухе с ХЖА в водный раствор [86]. Общие возможности хроматомембранных методов для автоматизации процессов пробоподготовки и концентрирования в химическом анализе рассмотрены в работах [87–89].
Дальнейшим этапом развития хроматомембранных методов в области аналитической химии явилась двухступенчатая схема выделения аналитов, способных образовывать летучие соединения. Эта схема включает реакционную газовую экстракцию в качестве первой ступени и жидкостно-абсорбционное поглощение целевых компонентов из потока газа-экстрагента – в качестве второй [90, 91]. Подобную комбинированную схему применяли для кондуктометрического определения аммиака [90], ионометрического определения сульфид-ионов [91] и фотометрического определения мышьяка [92] и сурьмы [93] в природных и сточных водах с пределами обнаружения аналитов 50, 0.5, 1 и 1 мкг/л соответственно.
Выполненные исследования по оптимизации пористой структуры бипористых матриц привели к идее создания поликапиллярных матриц, содержащих систему открытых макропор в виде одинаковых, параллельных и равноудаленных друг от друга капилляров [94, 95]. Эти капилляры имеют постоянную конфигурацию, площадь поперечного сечения по всей длине и направлены от системы подачи к системе вывода жидкой фазы. Идентичность, параллельность и равноудаленность друг от друга капилляров, по которым движется жидкая фаза, обеспечивает равномерность потоков жидкой и газовой фаз через бипористую матрицу, и, как следствие, увеличивает эффективность массообмена между жидкой и газовой фазами по сравнению с традиционной матрицей без пространственно ориентированных макропор. К настоящему времени ХМП в поликапиллярных матрицах использован для разработки методик непрерывного определения летучих галогенуглеводородов в водопроводной воде [96] и паров фенола [97] и аммиака [98] в воздухе рабочей зоны.
* * *
С момента открытия ХМП прошло около четверти века. Основанные на его принципах методы разделения пока не вошли широко ни в аналитическую практику, ни в химические технологии, несмотря на большие возможности. При анализе причин подобной ситуации возникает невольная аналогия с хроматографическими методами. С момента открытия хроматографии М.С. Цветом до того времени, когда она нашла широкое применение, прошло полвека. Хроматографические методы стали всеобщим достоянием практически только после начала серийного выпуска соответствующих аналитических приборов: сначала газовых, а потом и жидкостных хроматографов с полным набором аксессуаров в виде всевозможных насадок, сорбентов, носителей и жидких стационарных фаз, а немного позднее и в виде уже готовых хроматографических колонок. Хроматомембранные методы, так же как хроматография, в самом общем их понимании являются не конкретными методами разделения, а общими методологиями разделения веществ, приложимыми к различным по агрегатному состоянию системам фаз. Проведенная параллель, особенно с учетом того, что ХМЯ по своей конструкции существенно сложнее хроматографических колонок, позволяет полагать, что аналитические хроматомембранные методы только тогда привлекут широкое внимание исследователей, когда ХМЯ различной конфигурации и размеров можно будет приобрести в виде готовых изделий.
Авторы выражают благодарность Российскому научному фонду, грант № 16-13-10117-П, за поддержку проведенных исследований.
Список литературы
Москвин Л.Н., Горшков А.И., Гумеров М.Ф. Жидкостно-газовая распределительная хроматография // Докл. АН СССР. 1982. Т. 265. № 2. С. 378.
Москвин Л.Н., Горшков А.И., Гумеров М.Ф. Жидкостно-газовая распределительная хроматография // Журн. физ. химии. 1983. Т. 57. № 8. С. 1979.
Svec F., Tennikova T.B., Deyl Z. Monolithic Materials: Preparation, Properties and Applications. Amsterdam: Elsevier, 2003. V. 67. P. 457.
Химич Г.Н., Рахматуллина Е.Н., Слабоспитская М.Ю., Тенникова Т.Б. Синтез и пористая структура монолитных полимерных сорбентов // Журн. прикл. химии. 2005. Т. 78. № 4. С. 623. (Khimich E. N., Rakhmatyllina E.N., Slabospitskaya M.Yu., Tennikova T.B. Synthesis and pore structure of monolithic polymeric sorbent // Russ. J. Appl. Chem. 2005. V. 78. № 4. P. 617.)
Преображенский Б.К., Москвин Л.Н., Калямин А.В., Лилова О.М., Усиков Б.С. Использование компактного пористого фторопласта в распределительной хроматографии // Радиохимия. 1968. Т. 10. № 3. С. 377.
Москвин Л.Н., Гумеров М.Ф., Горшков А.И., Ефимов А.А. Исследование блочных сорбентов для газо-адсорбционной хроматографии // Журн. прикл. химии. 1974. Т. 47. № 7. С. 1973.
Москвин Л.Н., Мирошников В.С., Мельников В.А. Экспрессный хроматографический радиохимический анализ // Радиохимия. 1979. Т.21. № 2. С. 311.
Giddings J.C., Myers M.N. Liquid gas partition chromatography. Liquid chromatography system with gaseous phase for determination gases // J. High Resol. Chromatogr. 1983. V. 6. № 7. P. 831.
Фролов Ю.Г. Курс коллоидной химии. Поверхностные явления и дисперсные системы. М.: Химия, 1988. 464 с.
Гумеров М.Ф., Родинков О.В., Москвин Л.Н., Горшков А.И. Влияние носителя неподвижной газовой фазы на процесс жидкостно-газовой распределительной хроматографии // Журн. физ. химии. 1988. Т. 62. № 9. С. 2249.
Гумеров М.Ф., Родинков О.В., Москвин Л.Н. Жидкостно-газовая распределительная хроматография на тонкопористых носителях // Журн. физ. химии. 1990. Т. 64. № 2. С. 501.
Сумм Б.Д., Горюнов Ю.В. Физико-химические основы смачивания и растекания. М.: Химия, 1976. 232 с.
Москвин Л.Н., Родинков О.В. Хроматомембранные методы разделения веществ. СПб: Изд-во С.-Петерб. ун-та, 2014. С. 27.
Горшков А.И., Гумеров М.Ф., Москвин Л.Н. Хроматографический анализ газов и перекиси водорода в водном теплоносителе // Теплоэнергетика. 1980. № 10. С. 25.
Горшков А.И., Гумеров М.Ф., Леонтьева Е.И., Москвин Л.Н. Хроматографическое определение газов, растворенных в воде // Журн. аналит. химии. 1986. Т. 41. № 1. С. 146.
Родинков О.В., Москвин Л.Н. Жидкостно-газовая хроматография и ее аналитические возможности // Журн. аналит. химии. 1996. Т. 51. № 1. С.102. (Ro-dinkov O.V., Moskvin L.N. Liquid-gas chromatography and its analytical capabilities // J. Analyt. Chem. 1996. V. 51. № 1. P. 92).
Родинков О.В., Гумеров М.Ф. Способ хроматографического анализа газов, растворенных в жидкости. А. с. № 1545767 СССР // Б. и. 1991. № 16.
Родинков О.В., Гумеров М.Ф., Москвин Л.Н. Удерживание хроматографируемых компонентов в системе жидкость–газ–твердый сорбент // Журн. физ. химии. 1989. Т. 63. № 11. С. 3128.
Березкин В.Г. Газо-жидко-твердофазная хроматография. М.: Химия, 1986. 112 с.
Родинков О.В. Автореф. дис. … докт. хим. наук. Санкт-Петербург, 2004. 32 с.
Moskvin L.N., Rodinkov O.V. Anaytical application of liquid-gas and liquid-gas-solid chromatography // Crit. Rev. Anal. Chem. 1994. V. 24. № 5–6. P. 317.
Родинков О.В., Москвин Л.Н. Улучшение процесса адсорбционного концентрирования органических примесей из водных растворов // Журн. аналит. химии. 1995. Т. 50. № 2. С. 147.
Родинков О.В., Москвин Л.Н. Жидкостно-газоадсорбционная хроматография в процессах концентрирования летучих органических веществ из водных растворов // Журн. аналит. химии. 1996. Т. 51. № 11. С. 1133. (Rodinkov O.V., Moskvin L.N. Liquid-gas adsorption chromatography in the processes of preconcentration of volatile organic substances from aqueous solutions // J. Analyt. Chem. 1996. V. 51. № 11. P. 1034.)
Родинков О.В., Сбитнев С.В., Москвин Л.Н. Влияние температуры на удерживание летучих органических веществ из водных растворов на неполярных полимерных сорбентах // Журн. физ. химии. 1998. Т. 72. № 2. С. 337. (Rodinkov O.V., Sbitnev S.V., Moskvin L.N. Effect of temperature on retention of volatile organic substances from aqueous solutions by nonpolar polymeric sorbents // Russ. J. Phys. Chem. 1998. V. 72. № 2. P. 273.)
Родинков О.В., Москвин Л.Н., Сбитнев С.В. Температурная оптимизация сорбционного концентрирования полярных летучих органических веществ из водных растворов на неполярных полимерных сорбентах // Журн. аналит. химии. 1998. Т. 53. № 12. С. 12. (Rodinkov O.V., Moskvin L.N., Sbitnev S.V. Temperature optimization of the adsorption preconcentration of polar volatile organic compounds from aqueous solutions on nonpolar polymeric sorbents // J. Analyt. Chem. 1996. V. 53. № 12. P. 1104.)
Москвин Л.Н., Родинков О.В. Жидкостно-газоадсорбционная хроматография // Журн. аналит. химии. 2004. V. 59. № 12. С. 1283. (Moskvin L.N., Ro-dinkov O.V. Liquid – gas adsorption chromatography // J. Analyt. Chem. 2004. V. 59. № 12. P. 1165.)
Москвин Л.Н. Хроматомембранный метод разделения веществ // Доклады РАН. 1994. Т. 334. № 5. С. 599.
Moskvin L.N. Chromatomembrane method for the continuous separation of substances // J. Chromatogr. A. 1994. V. 669. P. 81.
Москвин Л.Н., Родинков О.В., Катрузов А.Н. Хроматомембранный метод разделения веществ и его аналитические возможности // Журн. аналит. химии. 1996. Т. 51. № 8. С. 835. (Moskvin L.N., Ro-dinkov O.V., Katruzov A.N. Chromatomembrane method for substance separation and its analytical potentialities // J. Analyt. Chem. 1996. V. 51. № 8. P. 770.)
Родинков О.В., Москвин Л.Н., Папсуева А.Г., Григорьев Г.Л. Условия осуществления хроматомембранного массообменного процесса в системах жидкость–жидкость и жидкость–газ // Журн. физ. химии. 1999. Т. 73. № 9. С. 1638. (Rodinkov O.V., Moskvin L.N., Papsueva A.G., Grigor’ev G.L. Chromatomembrane mass transfer in liquid-liquid and li-quid-gas systems // Russ. J. Phys. Chem. A. 1999. V. 73. № 9. P. 1470.)
Родинков О.В. Закономерности непрерывной хроматомембранной газовой экстракции при движении фаз в одном направлении // Вестн. СПбГУ. Сер. 4. 2001. Вып. 3 (№ 20). С. 68.
Родинков О.В., Москвин Л.Н., Майорова Н.А. Быстродействие различных схем непрерывной хроматомембранной газовой экстракции // Журн. аналит. химии. 2005. Т. 60. № 8. С. 727. (Rodinkov O.V., Moskvin L.N, Maiorova N.A. Operation rates of different schemes of continuous chromatomembrane gas extraction // J. Analyt. Chem. 2005. V. 60. № 8. P. 820.)
Москвин Л.Н., Родинков О.В. Хроматомембранные методы. Физико-химические принципы, аналитические и технологические возможности // Изв. АН. Сер. хим. 2012. Т. 61. № 4. С. 719. (Moskvin L.N., Rodinkov O.V. Chromatomembrane methods: Physicochemical principles, analytical and technological possibilities // Russ. Chem. Bull. 2012. V. 61. P. 723.)
Родинков О.В., Москвин Л.Н., Васькова Е.А. Оптимизация пористой структуры гидрофобной матрицы для осуществления хроматомембранных массообменных процессов // Журн. физ. химии. 2005. Т. 79. № 3. С. 539. (Rodinkov O.V., Moskvin L.N., Vaskova E.A. Optimization of the porous structure of a hydrophobic matrix for chromatomembrane mass-exchange processes // Russ. J. Phys. Chem. 2005. V. 79. № 3. P. 453.)
Баскин З.Л. Промышленный аналитический контроль. Хроматографические методы анализа фтора и его соединений. М.: Энергоатомиздат, 2008. С. 193.
Москвин Л.Н., Родинков О.В., Синицына Т.В. Фотометрическое определение гидразина в воздухе с хроматомембранным концентрированием // Журн. аналит. химии. 1999. Т. 54. № 1. С. 61. (Moskvin L.N., Rodinkov O.V., Sinitzyna T.V. Determination of hydrazine in air by photometry with chromatomembrane preconcentration // J. Analyt. Chem. 1999. V. 54. № 1. P. 53.)
Родинков О.В., Москвин Л.Н. Закономерности противоточной хроматомембранной газовой экстракции // Журн. аналит. химии. 2003. Т. 58. № 6. С. 611. (Rodinkov O.V., Moskvin L.N. Regularities in counterflow chromatomembrane gas extraction // J. Analyt. Chem. 1999. V. 54. № 1. P. 53.)
Родинков О.В., Москвин Л.Н. Непрерывная двухмерная хроматомембранная газовая экстракция. Тарелочная модель и практические следствия // Журн. аналит. химии. 2000. Т. 55. № 9. С. 950. (Ro-dinkov, O.V., Moskvin, L.N. Continuous two-dimensional chromatomembrane gas extraction: A plate model and its practical consequences // J. Analyt. Chem. 2000. V. 55. № 9. P. 854.)
Москвин Л.Н., Родинков О.В. Хроматомембранное концентрирование микропримесей органических загрязнителей природных вод и атмосферного воздуха // Журн. аналит. химии. 2002. Т. 57. № 10. С. 1057. (Moskvin L.N., Rodinkov O.V. Chromatomembrane preconcentration of trace impurities of organic pollutants from natural waters and atmospheric air // J. Analyt. Chem. 2002. V. 57. № 10. P. 894.)
Moskvin L.N., Simon J. Gas/liquid and liquid/liquid solvent extraction in flow analysis with the chromatomembrane cell // Sensors. 2006. V. 6. P. 1321.
Москвин А.Л., Москвин Л.Н., Родинков О.В. Хроматомембранные методы – новый принцип функционирования устройств для пробоподготовки в аналитических приборах // Научное приборостроение. 1999. Т. 9. № 4. С. 62.
Золотов Ю.А., Цизин Г.И., Дмитриенко С.Г., Моросанова Е.И. Сорбционное концентрирование микрокомпонентов из растворов. Применение в неорганическом анализе. М.: Наука, 2007. 320 с.
Valcarcel M., M.D. Lique de Castro M.D. Non-chromatographic Continuous Separation Techniques. Cambridge: RSC publ., 1991. 290 p.
Moskvin L.N., Simon J. Flow injection analysis with the chromatomembrane – a new device for gaseous/liquid and liquid/liquid extraction // Talanta. 1994. V. 41. № 10. P. 1765.
Никоноров В.В., Москвин Л.Н. Проточно-инжекционное определение нитрит-ионов с хроматомембранным экстракционным предконцентрированием // Журн. аналит. химии. 1998. V. 53. № 10. С. 925. (Nikonorov V.V., Moskvin N.L. Flow injection determination of nitrite with chromatomembrane extraction preconcentration. // J. Analyt. Chem. 1998. V. 53. № 10. P. 948.)
Москвин Л.Н., Николаева Д.Н., Михайлова Н.В. Экстракционно-фотометрическое определение анионных поверхностно-активных веществ с хроматомембранным концентрированием // Журн. аналит. химии. 1996. Т. 51. № 8. С. 845. (Moskvin L.N., Nikolaeva D.N., Michailova N.V. Solvent extraction-photometric determination of anionic surfactants using chromatomembrane preconcentration // J. Analyt. Chem. 1996. V. 51. № 8. P. 779.)
Moskvin A.L., Moskvin L.N., Moszhuchin A.V., Fomin V.V. Extraction-chromatographic preconcentration with chromatomembrane separation of extract from aqueous phase for luminescence determination of oil products and phenols in natural water by flow injection analysis // Talanta. 1999. V. 50. P. 113.
Москвин А.Л., Мухина Е.А. Экстракционное концентрирование в потоке: экстракционно-хроматографическое выделение с хроматомембранным отделением экстракта от водной фазы // Журн. аналит. химии. 2003. Т. 58. № 7. С. 710.
Москвин А.Л., Москвин Л.Н., Мозжухин А.В., Фомин В.В. Проточно-инжекционное люминесцентное определение фенолов с экстракционно-хроматографическим концентрированием и хроматомембранным отделением экстракта от водной фазы // Журн. аналит. химии. 2000. Т. 55. № 4. С. 369. (Moskvin A.L., Moskvin L.N., Mozzhukhin A.V., Fomin V.V. Flow-injection luminescence determination of phenols with extraction-chromatographic preconcentration and chromatomembrane separation of the extract from the aqueous phase // J. Analyt. Chem. 2000. V. 55. № 4. P. 354.)
Москвин Л.Н., Булатов А.В., Николаева Д.Н., Григорьев Г.Л. Проточно-инжекционное экстракционно-фотометрическое определение микроколичеств фосфат- и силикат-ионов // Журн. аналит. химии. 2002. Т. 57. № 7. С. 709. (Moskvin L.N., Bulatov A.V., Nikolaeva D.N., Grigor’ev G.L. Flow-injection extraction-photometric trace determination of phosphate and silicate ions // J. Analyt. Chem. 2002. V. 57. № 7. P. 590.)
Supriyanto G., Simon J. The chromatomembrane method used for sample preparations in the spectrophotometric determination of zinc and copper in pharmaceuticals // Talanta. 2005. V. 68. P. 318.
Simon J., Kirchhoff A., Gültzow O. Advanced sample pretreatment for the monitoring of polycyclic aromatic hydrocarbons and extractable organic halogens in waste water. Flow based procedures with chromatomembrane cells // Talanta. 2002. V. 58. P. 1335.
Витенберг А.Г., Иоффе Б.В. Газовая экстракция в хроматографическом анализе. Л.: Химия, 1982. 279 с. (Ioffe B.V., Vitenberg A.G. Head-space Analysis and Related Methods in Gas Chromatography. N.Y.: Wiley Inters, 1984. 276 p.)
Moskvin L.N., Rodinkov O.V. Continuous chromatomembrane headspace analysis // J. Cromatogr. A. 1996. V. 725. P. 351.
Родинков О.В., Москвин Л.Н., Зыкин И.А. Газохроматографическое определение газообразных углеводородов в водных растворах с хроматомембранной газовой экстракцией // Журн. аналит. химии. 2003. Т. 58. № 1. С. 82. (Rodinkov O.V., Moskvin L.N., Zykin I.A. Gas-chromatographic determination of gaseous hydrocarbons in aqueous solutions using chromatomembrane gas extraction // J. Analyt. Chem. 2003. V. 58. № 1. P. 71.)
Москвин Л.Н., Родинков О.В., Катрузов А.Н. Непрерывное выделение газообразных и леколетучих примесей из водных растворов с использованием хроматомембранного метода // Журн. аналит. химии. 1996. Т. 51. № 2. С. 215.
Bloch K., Simon J., Moskvin L.N., Rodinkov O.V. The properties of chromatomembrane cells in flow systems coupled to gas chromatography – analysis of volatile organic compounds // Talanta. 2000. V. 52. P. 123.
Родинков О.В., Москвин Л.Н., Майорова Н.А., Зеймаль А.Е. Газохроматографическое определение алкилацетатов в водных растворах с хроматомембранной газовой экстракцией // Журн. аналит. химии. Т. 58. 2003. № 6. С. 61. (Rodinkov O.V., Moskvin L.N., Maiorova N.A., Zeimal’ A.E. Gas-chromatographic determination of alkyl acetates in aqueous solutions with chromatomembrane gas extraction // J. Analyt. Chem. 2003. V. 58. 6. P. 554.)
Майорова Н.А., Родинков О.В., Москвин Л.Н. Газохроматографическое определение метилакрилата и метилметакрилата в водных растворах с хроматомембранной газовой экстракцией и газоадсорбционным концентрированием // Вестн. СПбГУ. Сер. 4. Вып. 4. С. 84.
Родинков О.В., Москвин Л.Н., Ратхоур Д.К. Закономерности динамической хроматомембранной газовой экстракции из неподвижной жидкой фазы // Журн. аналит. химии. 2004. Т. 59. № 11. С. 1188. (Rodinkov O.V., Moskvin L.N., Rathour J.K. Features of dynamic chromatomembrane gas extraction from a stationary liquid phase // J. Analyt. Chem. 2004. V. 59. № 11. P. 1073.)
Гольберт К.П., Вигдергауз М.С. Введение в газовую хроматографию. М.: Химия, 1990. 385 с.
Saridara C., Brukh R., Mitra S. Development of continuous on-lline purge and trap analysis // J. Sep. Sci. 2006. V. 29. № 3. P. 446.
Москвин Л.Н., Родинков О.В., Ли Бухай. Определение бензола, толуола, хлороформа и тетрахлорида углерода в водных растворах методом хроматомембранного парофазного анализа с газоадсорбционным концентрированием // Журн. аналит. химии. 1998. Т. 53. № 11. С. 1174. (Moskvin L.N., Rodinkov O.V., Bukhai L. Determination of benzene, toluene, chloroform, and carbon tetrachloride in aqueous solutions by chromatomembrane headspace analysis with gas-adsorption preconcentration // J. Analyt. Chem. 1998. V. 53. № 11. P. 1018.)
Rodinkov O.V., Moskvin L.N., Viktorova M.I., Dyakin A.A., Yakimova N.M. Chromatomembrane headspace analysis of aqueous solutions at elevated temperatures // Chromatographia. 2015. V. 78. P. 1211.
Родинков О.В., Бугайченко, А.С., Москвин Л.Н., Горбачева А.Р., Вагнер Е.А. Выбор условий хроматомембранной газовой экстракции при ее сочетании с газоадсорбционным концентрированием аналитов // Журн. аналит. химии. 2018. Т. 73. № 5. С. 358. (Rodinkov O.V., Bugaichenko A.S., Moskvin L.N., Gorbacheva A.R., Vagner E.A. Selection of conditions for chromatomembrane gas extraction at its combination with the gas-adsorption preconcentration of analytes // J. Analyt. Chem. 2018. V. 73. № 5. P. 432.)
Родинков О.В., Вагнер Е.А., Бугайченко А.С., Москвин Л.Н. Сравнение эффективности углеродных сорбентов для концентрирования легколетучих органических веществ из влажных газовых сред для последующего газохроматографического определения // Журн. аналит. химии. 2019. Т. 74. № 9.
Гигиенические нормативы ГН 2.1.5.1315-03. Предельно допустимые концентрации (ПДК) химических веществ в воде водных объектов хозяйственно-питьевого и культурно-бытового водопользования // Бюллетень нормативных актов федеральных органов исполнительной власти. № 50. 10.12.2007.
Платонов И.А. Родинков О.В., Горбачева А.Р., Москвин Л.Н., Колесниченко И.Н. Методы и средства приготовления стандартных газовых смесей // Журн. аналит. химии. 2018. Т. 73. № 2. С. 83. (Platonov I.A., Rodinkov O.V., Gorbacheva A.R., Moskvin L.N., Kolesnichenko I.N. Methods and devices for the preparation of standard gas mixtures // J. Analyt. Chem. 2018. V. 73. № 2. P. 109).
Москвин Л.Н., Никитина Т.Г. Хроматомембранное генерирование стандартных газовых смесей загрязнителей атмосферного воздуха на уровне микроконцентраций // Заводск. лаборатория. 2005. Т. 71. № 3. С. 3.
Родинков О.В., Рачковский И.Н., Москвин Л.Н. Хроматомембранное газоэкстракционное генерирование стандартных газовых смесей с применением композиционных угольно-фторопластовых матриц // Журн. аналит. химии. 2008. Т. 63. № 9. С. 941. (Rodinkov O.V., Rachkovskii I.N., Moskvin L.N. Chromatomembrane gas extraction generation of standard gas mixtures using composite carbon–fluoroplastic matrices // J. Analyt. Chem. 2008. V. 63. № 9. P. 857.)
Горбачева А.Р., Родинков О.В. Хроматомембранное генерирование стандартных газовых смесей летучих органических соединений на уровне ppm // Аналитика и контроль. 2018. Т. 22. № 1. С. 75. (Gorbacheva A.R., Rodinkov O.V. Chromatomembrane generation of volatile organic compounds standard gas mixtures at the ppm level // Analitika i Kontrol. 2018. V. 22. № 1. P. 75.)
Родинков О.В., Москвин Л.Н., Горбачева А.Р., Бугайченко А.С. Оценка возможности осуществления хроматомембранного массообменного процесса на гранулированных композиционных угольно-фторопластовых сорбентах для генерирования стандартных газовых смесей // Журн. аналит. химии. 2017. Т. 72. № 3. С. 237. (Rodinkov O.V., Moskvin L.N., Gorbacheva A.R., Bugaichenko A.S. Assessment of the possibility of the performance of a chromatomembrane mass-exchange process on granular composite carbon–fluoroplastic sorbents for the generation of standard gas mixtures // J. Analyt. Chem. 2017. V. 72. № 3. P. 289.)
Родинков О.В., Москвин Л.Н., Синицына Т.В. Григорьев Г.Л. Хроматомембранная абсорбция микропримесей полярных органических веществ из воздуха водными растворами // Журн. аналит. химии. 1998. Т. 53. № 4. С. 373. (Rodinkov O.V., Moskvin L.N., Sinitsina T.V., Grigor’ev G.L. Chromatomembrane absorption of trace impurities of polar organic substance from air to aqueous solutions // J. Analyt. Chem. 1998. V. 53. № 4. P. 326.)
Родинков О.В., Москвин Л.Н. Физико-химическая модель хроматомембранной жидкостной хемосорбции микропримесей из газовой фазы // Журн. физ. химии. 2001. Т. 75. № 2. С. 329. (Rodinkov O.V., Moskvin L.N. A physicochemical model of chromatomembrane liquid chemisorption of trace amounts of admixtures from the gas phase // Russ. J. Phys. Chem. A. 2001. V. 75. № 2. P. 272.)
Родинков О.В., Бугайченко А.С., Москвин Л.Н. Динамическая хроматомембранная жидкостная хемосорбция микропримесей из газовой фазы в политетрафторэтиленовых матрицах, модифицированных сорбционно-активным материалом // Журн. физ. химии. 2010. Т. 84. № 8. С. 1568. (Ro-dinkov O.V., Bugaichenko A.S., Moskvin L.N. Dynamic chromatomembrane liquid chemisorption of trace impurities from a gas phase in polytetrafluoroethylene matrices modified with sorptionactive material // Russ. J. Phys. Chem. A. 2010. V. 84. № 8. P. 1428.)
Москвин Л.Н., Никоноров В.В. Проточно-инжекционное определение диоксида серы в воздухе с предварительным хроматомембранным концентрированием // Журн. аналит. химии. 1996. Т. 51. № 8. С. 891.
Никоноров В.В., Москвин Л.Н. Новый реагент для определения нитритов // Журн. аналит. химии. 1996. Т. 51. № 7. С. 737–740. (Nikonorov V.V., Moskvin L.N. New reagent for determining nitrites // J. Analyt. Chem. 1996. V. 51. № 7. P. 679.)
Москвин Л.Н., Родинков О.В., Катрузов А.Н., Томилова Е. С. Ионохроматографическое определение полярных неорганических примесей в воздухе с хроматомембранным предконцентрированием // Журн. аналит. химии. 1996. Т. 51. № 11. С. 1214. (Moskvin L.N., Rodinkov O.V., Katruzov A.N., Tomilova E.S. Ion-chromatographic determination of polar inorganic impurities in air with chromatomembrane preconcentration // J. Analyt. Chem. 1996. V. 51. № 11. P. 1109.)
Rodinkov O.V., Moskvin L.N., Vaskova E.A. Photometric flow injection determination of formaldehyde in atmospheric air using chromatomembrane absorption // J. Flow Injection Analysis. 2005. V. 22. № 1. P. 11.
Москвин Л.Н., Родинков О.В., Синицына Т.В. Газохроматографическое определение низших спиртов в воздухе с хроматомембранным предконцентрированием // Заводск. лаборатория. 1998. Т. 64. № 5. С. 3.
Wei Yanlin, Oshima Mitsuko, Simon J. Absorption concentration and determination of trace amounts of air pollutants by flow injection method coupled witch a chromatomembrane cell system: application to nitrogen dioxide determination // Talanta. 2002. V. 58. № 6. P. 1343.
Wei Y., Oshima M., Simon J., Motomizu S. The application of the chromatomembrane cell for the absorptive sampling of nitrogen dioxide followed by continuous determination of nitrite using a micro-flow injection system // Talanta. 2002. V. 57. № 2. P. 355.
Sritharathikhun P., Oshima M., Wei Y., Simon J., Motomizu S. On-line collection/concentration and detection of sulfur dioxide in air by flow-injection spectrophotometry coupled with a chromatomembrane cell // Anal. Sci. 2004. V. 20. № 1. P. 113.
Erxleben H., Simon J. Moskvin L.N., Lozhkina O.V., Nikitina T.G. Automized procedures for the determination of ozone and ammonia contens in air by using the chromatomembrane method for gas-liquid extraction // Fresenius J. Anal. Chem. 2000. V. 366. № 4. P. 332.
Sritharathikhun P., Oshima M., Motomizu S. On-line collection/concentration of trace amounts of formaldehyde in air with chromatomembrane cell and its sensitive determination by flow injection technique coupled with spectrophotometric and fluorometric detection // Talanta. 2005. V. 67. № 5. P. 1014.
Аверяскаина Е.О., Ермаков С.С., Москвин Л.Н. Инверсионно- вольтамперометрическое определение ртути в воздухе с предварительным хроматомембранным концентрированием // Журн. аналит. химии. 2006. Т. 61. № 11. С. 1192. (Averyaskina Е.O., Ermakov S.S, Moskvin L.N. Determination of mercury in air by stripping voltammetry with chromatomembrane pre-concentration // J. Analyt. Chem. 2006. V. 61. № 11. P. 1100.)
Moskvin L.N., Moskvin A.L. Chromatomembrane methods – Novel automatization possi-bilities of substances separation processes // Lab. Rob. Automat. 1998. V. 10. P. 3.
Simon J., Moskvin L.N. Does the chromatomembrane cell improve the quality of enviromental analysis? // Talanta. 1999. V. 49. P. 985.
Москвин А.Л., Москвин Л.Н., Родинков О.В. Хроматомембранный метод – новый принцип функционирования устройства для пробоподготовки в аналитических приборах // Научное приборостроение. 1999. Т. 9. № 4. С. 62.
Москвин Л.Н., Григорьев Г.Л., Папсуева А.Г. Проточное кондуктометрическое определение микропримесей аммиака в природных водах с хроматомембранным выделением в деионизованную воду // Журн. прикл. химии. 2001. Т. 74. № 6. С. 949. (Moskvin L.N., Grigor’ev G.L., Papsueva A.G. Flow-through conductometric analysis for microconcentrations of ammonia in natural water with chromatomembrane separation into deionized water // Russ. J. Appl. Chem. 2001. V. 74. № 6. P. 976.)
Москвин Л.Н., Папсуева А.Г., Григорьев Г.Л. Проточное ионометрическое определение сульфид-ионов с хроматомембранным выделением в природных и сбросных водах // Вестн. СПбГУ. Сер. 4. 2001. № 3. С. 62.
Москвин Л.Н., Булатов А.В., Коломиец Н.А., Москвин А.Л. Циклическое инжекционное фотометрическое определение мышьяка в водных средах // Журн. аналит.химии. 2007. Т. 62. № 12. С. 1267. (Moskvin L.N., Bulatov A.V., Kolomiets N.A., Moskvin A.L. Photometric cyclic-injection determination of arsenic in aqueous solutions // J. Analyt. Chem. 2007. V. 62. № 12. P. 1143.)
Москвин Л.Н., Булатов А.В., Назарова Н.В. Проточно-инжекционное определение микропримесей сурьмы в природных водах с экстракционно-хроматографическим концентрированиеи // Журн. аналит. химии. 2004. Т. 59. № 10. С. 1097. (Moskvin L.N., Bulatov A.V., Nazarova N.V. Flow-injection determination of trace antimony in natural waters with extraction–chromatographic preconcentration // J. Analyt. Chem. 2004. V. 59. № 10. P. 991.)
Москвин Л.Н., Родинков О.В., Москвин А.Л., Григорьев Г.Л., Мельниченко А.Н. Устройство для осуществления массообмена между жидкой и газовой фазами. Патент № 2392038 РФ. Опубл. 20.06.2010.
Москвин А.Л., Мельниченко А.Н., Диченко О.Ю. Влияние структуры массообменных матриц и фазоразделительных мембран на “эффект памяти” хроматомембранных ячеек в парофазном анализе // Вестн. СПбГУ. Сер. 4. 2011. № 1. С. 94.
Мельниченко А.Н., Москвин А.Л., Поваров В.Г. Определение содержания легколетучих галогенсодержащих органических веществ в водопроводной воде on-line с непрерывной хроматомембранной газовой экстракцией // Аналитика и контроль. 2012. Т. 16. № 4. С. 383.
Москвин А.Л., Мельниченко А.Н. Флуориметрическое определение фенола в воздухе рабочей зоны с хроматомембранной жидкостной абсорбцией // Аналитика и контроль. 2013. Т. 17. № 4. С. 485.
Москвин А.Л., Мельниченко А.Н., Диченко О.Ю. Фотометрическое определение аммиака в воздухе рабочей зоны с хроматомембранным концентрированием // Вестн. СПбГУ. Серия 4. 2011. № 4. С. 55.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии