

Журнал аналитической химии, 2019, T. 74, № 5, стр. 350-355
Прямое определение свинца в морской воде методом атомно-абсорбционной спектроскопии высокого разрешения с использованием смешанного модификатора нитрат бария–фтороводородная кислота
Н. А. Соболев 1, Н. Л. Иванченко 1, А. Ю. Кожевников 1, *
1 Северный (Арктический) федеральный университет имени М.В. Ломоносова,
Центр коллективного пользования научным оборудованием “Арктика”
163002 Архангельск, наб. Северной Двины, 14, Россия
* E-mail: a.kozhevnikov@narfu.ru
Поступила в редакцию 04.10.2017
После доработки 22.06.2018
Принята к публикации 22.06.2018
Аннотация
Предложена методика прямого определения свинца в морской воде методом атомно-абсорбционной спектрометрии высокого разрешения с электротермической атомизацией. Для устранения влияния фоновых помех, вызванных матрицей морской воды, предложено использование смешанного модификатора нитрат бария–фтороводородная кислота. Оптимальное соотношение компонентов модификатора составляет 60 мкг Ba(NO3)2 + 1.52 мкг HF. Условия проведения анализа: температура пиролиза 600°C, температура атомизации 1200°C. Предел обнаружения для разработанной методики составляет 0.0003 мг/л.
Известно, что свинец является экотоксикантом, который негативно влияет на протекание различных биологических процессов [1]. Определение его содержания в объектах окружающей среды относится к приоритетным задачам. Содержание свинца в природных незагрязненных водах в среднем по мировому океану составляет 0.01–0.02 мкг/л [2], однако предельно допустимая концентрация свинца в Российской Федерации для морских водоемов рыбохозяйственного назначения установлена в размере 0.01 мг/л [3].
Морская вода является сложным объектом для проведения прямого анализа в силу относительно высокого содержания в ней растворенных солей. Основная часть неорганической матрицы морской воды – это соли щелочных и щелочноземельных металлов [4]. Показано [5], что солевая матрица отрицательно влияет на воспроизводимость и точность результатов определения металлов в морской воде. Это обусловлено значительными химическими и спектральными помехами, вызванными в том числе совместной атомизацией матрицы и аналита.
Большинство современных методик определения тяжелых металлов в морской воде основано на использовании метода атомно-абсорбционной спектрометрии с электротермической атомизацией. При этом зачастую необходима стадия пробоподготовки, которая заключается в экстракции металлов органическими комплексообразователями [6–10]. Эта стадия является достаточно трудоемкой и занимает много времени. Необходимость пробоподготовки обусловлена тем, что неорганические соли, составляющие основу матрицы морской воды, имеют высокие температуры кипения и их невозможно удалить на стадии пиролиза. Температуры атомизации большинства солей сопоставимы с температурами атомизации свинца [11], что вызывает образование молекулярного пара на стадии атомизации, который является источником фоновых помех.
Существует вариант прямого определения свинца в морской воде без использования стадии пробоподготовки. В этом случае основными компонентами морской воды, мешающими определению свинца, являются хлорид и сульфат натрия [5, 12–15]. Для снижения влияния матрицы широкое распространение получили химические модификаторы, которые добавляют совместно с морской водой в атомизатор. Модификаторы применяют для температурной стабилизации элементов с относительно низкой температурой атомизации и предотвращения их предварительной атомизации во время стадии пиролиза (нитрат магния, соли благородных металлов – нитраты палладия, платины, иридия) [12–18]. Используют также модификаторы, способные вступать в химические реакции с матрицей: нитрат и оксалат аммония, щавелевую кислоту. Они образуют легколетучие компоненты при взаимодействии с матрицей, которые удаляются на стадии пиролиза [19]. Например, температура возгонки NH4Cl составляет 210°С; температуры разложения нитратов аммония и натрия лежат в пределах от 380 до 600°С [20]. В работе [5] в качестве химического модификатора использовали фтороводородную кислоту для уменьшения фонового сигнала хлоридов. Модификатор взаимодействует с хлоридами с образованием летучей хлороводородной кислоты, которая удаляется во время стадии сушки. Образующийся фторид натрия атомизируется при 1700°С [20], т.е. при температуре более высокой, чем температура атомизации свинца, и не влияет на его определение. Подбор новых модификаторов для определения свинца в морской воде без стадии пробоподготовки с целью повышения надежности и воспроизводимости результатов анализа – актуальная задача.
Цель данной работы – подбор модификаторов для снижения влияния солевой матрицы и разработка методики определения свинца в морской воде методом атомно-абсорбционной спектроскопии.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Объект исследования – морская вода, отобранная с глубины 200 м в Гренландском море. Пробу отбирали в пластиковую тару, консервировали азотной кислотой до рН 1–2. Соленость воды составляла 33 г/л.
Оборудование. Использовали атомно-абсорбционный спектрофотометр высокого разрешения ContrAA 700 (Analytic Jena, Германия) с пиксельной коррекцией фона, источником излучения сплошного спектра (дуговая ксеноновая лампа высокого давления) и графитовой печью поперечного нагрева с платформой. Длина волны для определения свинца – 283.3060 нм. Измеренный сигнал оценивали по площади пика, записанного на трех пикселях: основном (283.3060 нм) и боковых (±12 пм). Инертный газ – аргон высокой чистоты (99.998%) – использовали в качестве защитного газа. Исследуемый раствор помещали в графитовую кювету с помощью автодозатора AS-52s. Объем вводимой пробы – 10 мкл. Температурная программа определения свинца представлена в табл. 1.
Таблица 1.
Температурная программа для определения свинца в морской воде
| Параметр | Сушка | Пиролиз | Стадия без подачи газа | Атомизация | Чистка | ||
|---|---|---|---|---|---|---|---|
| Температура, °С | 80 | 90 | 350 | Варьировалась | Варьировалась | Варьировалась | 2450 |
| Нагрев, °С/с | 6 | 1 | 50 | 300 | 0 | 1500 | 500 |
| Удерживание температуры, с | 30 | 30 | 20 | 10 | 5 | 4 | 4 |
| Поток газа | Макс. | Макс. | Макс. | Макс. | 0 | 0 | Макс. |
Реактивы и материалы. Для построения градуировочной зависимости применяли мультиэлементный стандарт фирмы “Merck” в 0.1 М HNO3. В качестве модификатора использовали фтороводородную кислоту х. ч. (45%) (Сигматек), нитраты палладия и магния (98%) (Merck), нитрат бария х. ч. (Нева Реактив).
Приготовление растворов. Модельный раствор морской воды [4] готовили с использованием реактивов классификации х. ч. и выше. В мерную колбу емк. 1 л вносили предварительно взвешенные на аналитических весах навески: 23.476 г хлорида натрия, 4.981 г хлорида магния, 3.917 г сульфата натрия, 1.102 г хлорида кальция, 0.664 г хлорида калия, разбавляли до метки деионированной водой. Индивидуальные растворы солей готовили аналогично, добавляя в колбу емк. 1 л навеску соли.
Модификаторы готовили следующим образом: в мерную колбу емк. 100 мл вносили навеску 0.5 г нитрата бария, взвешенную на аналитических весах, и разбавляли до метки деионизи-рованной водой. Модификатор HF (45%) вводили непосредственно в графитовую печь.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Выявление фоновых помех. Известно [16–19], что хлориды и сульфаты существенно влияют на воспроизводимость результатов анализа, особенно при низких пределах обнаружения. Проведенные исследования показали, что компоненты матрицы существенно влияют на результаты определения свинца. Полезный сигнал, наблюдаемый при длине волны 273.8 нм, перекрывается помехами, вызванными солевой матрицей (рис. 1).
Рис. 1.
Спектр сигнала абсорбции при определении свинца в морской воде (а) и в модельном растворе, имитирующем морскую воду (б).
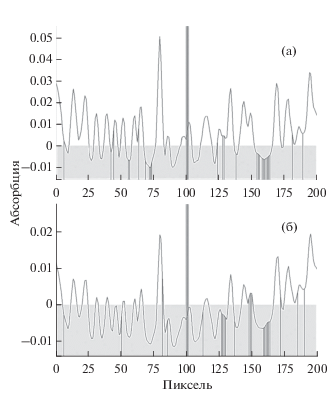
Для исключения возможного влияния различных компонентов морской воды, помимо сульфатов и хлоридов, на определение свинца мы получили спектры модельной морской воды. Сопоставление спектров реальной и модельной морской воды показывает их идентичность (рис. 1).
Регистрировали спектры растворов индивидуальных солей при одинаковых условиях. Из рис. 2а видно, что спектры хлоридов магния, кальция и калия свободны от сильных флуктуационных помех, характерных для спектров морской воды. Спектр сульфата натрия (рис. 2б) оказался практически идентичным спектру морской воды. Сульфат-ионы сильно влияют на результаты определения свинца за счет спектрального наложения.
Рис. 2.
Спектр сигнала абсорбции при определении свинца в присутствии хлорида натрия (а), сульфата натрия (б).
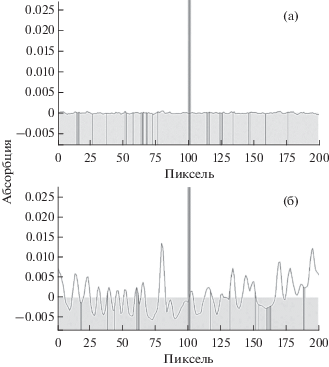
Таким образом, установлено, что мешающее влияние матрицы морской воды обусловлено только наличием в ней сульфатов и хлоридов. Дальнейшие исследования были направлены на устранение их влияния.
Подбор модификаторов для прямого определения свинца. Для прямого определения свинца в морской воде необходимо удалить мешающую матрицу. Для удаления сульфатов в качестве химического модификатора выбрали раствор нитрата бария с концентрацией 5 г/л, который добавляли в избытке для обеспечения полного взаимодействия с сульфат-ионами морской воды с образованием сульфата бария, температура атомизации которого выше температуры атомизации свинца [11].
Определение содержания ионов свинца в морской воде с добавкой ионов свинца (общая концентрация свинца 0.05 мг/л) и с применением модификатора нитрата бария (рис. 3а) позволило существенно снизить спектральные помехи. Однако при этом в спектре наблюдется достаточно интенсивный фоновый сигнал, обусловленный присутствием хлорид-ионов, который накладывается на пик аналита (рис. 3б). С целью исключения совместного влияния сульфатов и хлоридов при прямом определении содержания свинца в морской воде нами предложено добавлять к пробе смешанный модификатор нитрат бария–фтороводородная кислота. Нитрат бария препятствует совместной атомизации сульфатов и свинца, а фтороводородная кислота удаляет хлориды из пробы на стадии пиролиза. Спектр не имеет флуктуаций, а фоновый сигнал незначителен и не перекрывает максимум аналитического сигнала (рис. 4).
Рис. 3.
Спектр сигнала абсорбции морской воды с добавлением модификатора – нитрата бария (температура пиролиза 600°C, температура атомизации 1200°C, концентрация свинца 0.05 мг/л) (а); изменение сигналов абсорбции во времени (1 – фоновый сигнал, 2 – полезный сигнал) (б).

Рис. 4.
Спектр сигнала абсорбции морской воды с добавлением модификатора нитрат бария–фтороводородная кислота (температура пиролиза 600°C, температура атомизации 1200°C, концентрация свинца 0.05 мг/л) (а), изменение сигнала абсорбции во времени (1 – фоновый сигнал, 2 – полезный сигнал) (б).
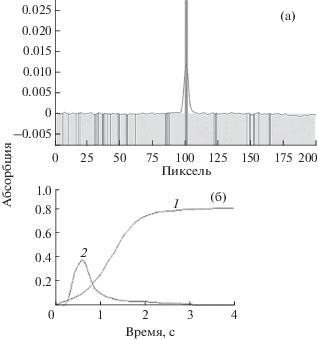
Следующий этап исследования – подбор оптимальных температур атомизации и пиролиза, а также соотношения масс выбранных модификаторов – нитрата бария и фтороводородной кислоты – с целью достижения наилучшей чувствительности при наименьшем влиянии матрицы на результаты анализа и снижения воздействия агрессивных сред на графитовую печь. Установлено (табл. 2), что наименьшие объемы растворов модификаторов, полностью устраняющие влияние матрицы на определение свинца в морской воде, составляют 12 мкл раствора нитрата бария с концентрацией 5 г/л и 3 мкл 45%-ной фтороводородной кислоты, которые добавляют к 10 мкл пробы морской воды. Таким образом, оптимальная масса модификаторов составляет: 60 мкг Ba(NO3)2 и 1.52 мкг HF.
Таблица 2.
Сигналы абсорбции при изменении концентраций модификаторов
| Состав смеси модификаторов* | Полезный сигнал в максимуме (абс.) | Фоновый сигнал в максимуме полезного сигнала (абс.) |
|---|---|---|
| 12 мкл Ba(NO3)2 + 2 мкл HF | 0.28 | 0.10 |
| 12 мкл Ba(NO3)2 + 4 мкл HF | 0.36 | 0.078 |
| 12 мкл Ba(NO3)2 + 5 мкл HF | 0.37 | 0.070 |
| 4 мкл Ba(NO3)2 + 3 мкл HF | 0.25 | 0.099 |
| 6 мкл Ba(NO3)2 + 3 мкл HF | 0.28 | 0.078 |
| 10 мкл Ba(NO3)2 + 3 мкл HF | 0.38 | 0.11 |
| 12 мкл Ba(NO3)2 + 3 мкл HF | 0.37 | 0.079 |
| 14 мкл Ba(NO3)2 + 3 мкл HF | 0.36 | 0.11 |
Для определения оптимальной температуры атомизации сравнивали сигналы абсорбции стандартных растворов свинца с концентрацией 0.05 мг/л в 0.1 М азотной кислоте и в морской воде при варьировании данного параметра. Из рис. 5 видно, что оптимальная температура атомизации составляет 1200°C. Аналогичным образом подбирали оптимальную температуру пиролиза (рис. 6), которая составила 600°C. В выбранных условиях наблюдаются минимальные различия аналитических сигналов стандартного раствора и раствора свинца в морской воде.
Рис. 5.
Зависимость аналитического сигнала от температуры атомизации. Температура пиролиза 600°C, концентрация свинца 0.05 мг/л; 1 – реальная проба, 2 – стандарт.

Рис. 6.
Зависимость аналитического сигнала от температуры стадии пиролиза. Температура атомизации 1200°C, концентрация свинца 0.05 мг/л; 1 – реальная проба, 2 – стандарт.

Апробация методики определения свинца. В выбранных условиях провели анализ реальных проб морской воды. Концентрацию свинца рассчитывали по градуировочной зависимости (коэффициент корреляции 0.9996), определили характеристическую концентрацию и предел обнаружения свинца.
Для подтверждения надежности определения свинца в морской воде в подобранных условиях использовали метод введено–найдено. Для определения свинца применяли смешанный модификатор 12 мкл раствора Ba(NO3)2 (5 г/л) + 3 мкл 45%-ной HF и выбранный температурный режим.
Показано, что предложенный подход позволяет устранить матричные влияния морской воды при определении свинца без предварительной пробоподготовки. При введении в морскую воду ионов свинца в концентрации 0.050 мг/л найдено 0.047 мг/л. Погрешность определения по методу введено–найдено составила 6.0%, стандартное отклонение – 0.0041 мг/л.
По результатам 10 параллельных определений ионов свинца в морской воде в выбранных ранее условиях (температура пиролиза и атомизации, соотношение модификаторов) характеристическая концентрация свинца составила 0.00035 мг/л, предел обнаружения свинца – 0.0003 мг/л, что примерно в 30 раз ниже уровня ПДК для рыбохозяйственных водоемов. Относительная погрешность при P = 0.95 составила 2.9% при концентрации аналита 0.005 мг/л [21, 22].
Работа выполнена с использованием оборудования Центра коллективного пользования научным оборудованием “Арктика” Северного (Арктического) федерального университета имени М.В. Ломоносова (проект RFMEFI59417X0013).
Список литературы
Juberg D.R. Lead and Human Health: An Update. New York: American Council on Science and Health, 2000. 64 p.
Chester R. Marine Geochemistry. London.: Unwin Hyman, 1990. 698 p.
Перечень рыбохозяйственных нормативов: предельно-допустимых концентраций и ориентировочно безопасных уровней веществ для воды, водных объектов, имеющих рыбохозяйственное значение. М.: ВНИРО, 1999. 188 с.
Хорн Р. Морская химия. Структура воды и химия гидросферы. М.: Мир, 1972. 400 с.
Cabon J.Y. Determination of Cd and Pb in seawater by graphite furnace atomic absorption spectrometry with the use of hydrofluoric acid as a chemical modifier // Spectrochim. Acta. 2002. V. 57. P. 513.
Creed J.T., Martin T.D. Determination of Trace Elements in Marine Waters by Off-Line Chelation Preconcentration with Graphite Furnace Atomic Absorption. Method 200.13. Cincinnati: United States Environmental Protection Agency, 1992. 21 p.
Wells M.L., Bruland K.W. An improved metal extraction procedure for the determination of trace metals in sea water by atomic absorption spectrometry // Marine Chemistry. 1998. V. 63. P. 145.
Kojuncu Y., Bundalevska J.M., Ay U., Čundeva K., Stafilov T., Akçin G. Atomic Absorption Spectrometry Determination of Cd, Cu, Fe, Ni, Pb, Zn and Tl Traces in Seawater Following Flotation Separation // Sep. Sci. Technol. 2004. V. 39. P. 2751.
Gladis J.M., Biju V.M., Prasada Rao T. On-line solid phase extraction of the 5,7-dichloroquinoline-8-ol complex onto C18 bonded silica gel and flame AAS determination of Cu in seawater samples // At. Spectrosc. 2002. V. 23. P. 143.
Shiowatana J., Benyatianb K., Siripinyanond A. Determination of Cd, Co, Hg, and Ni in seawater after enrichment on activated carbon by slurry sampling electrothermal AAS // At. Spectrosc. 2000. V. 21. P. 179.
Хавезов И. Атомно-абсорбционный анализ. Л.: Химия, 1983. 144 с.
Cabon J.Y., Le Bihan A. Determination of lead in seawater by electrothermal atomic absorption spectrometry with transversely heated furnace by using oxalic acid or Pd/Mg as modifiers // Spectrochim. Acta. 1996. P. 1245.
Ortner H.M., Bulska E., Rohr U., Schlemmer G., Weinbruch S., Welz B. Modifiers and coatings in graphite furnace atomic absorption spectrometry – mechanisms of action (A tutorial review) // Spectrochim. Acta. 2002. V. 57. P. 1835.
Алемасова А.С. Высокотемпературные процессы превращения комплексообразователей и комплексов металлов в атомно-абсорбционном анализе. Донецк: ДонГУ, 1997. 297 с.
Tsalev D.L., Slaveikova V.I., Mandjukov P.B. Chemical modification in graphite-furnace atomic absorption spectrometry // Spectrochim. Acta. Rev. 1990. V. 13. № 3. P. 225.
Bin H., Zhe-Min N. Minimization of sulfate interference on lead atomization with palladium-strontium nitrate as chemical modifier in electrothermal atomic absorption spectrometry // J. Anal. At. Spectrosc. 1996. V. 11. P. 165.
Volynsky A.B., Akmana S., Dogana C.E., Koklua U. Application of colloidal palladium modifier for the determination of As, Sb and Pb in a spiked sea water sample by electrothermal atomic absorption spectrometry // Spectrochim. Acta B. 2001. V. 56. P. 2361.
Acar O. Determination of cadmium, copper and lead in soils, sediments and sea water samples by ETAAS using a Sc + Pd + NH4NO3 chemical modifier // Talanta. 2005. V. 65. P. 672.
Пупышев А.А. Атомно-абсорбционный спектральный анализ. М.: Техносфера, 2009. 784 с.
Dean J.A. Lange’s Handbook of Chemistry. 15th ed. New York: MGH, 1999. 1291 p.
Смагунова А.Н. Методы математической статистики в аналитической химии. Ростов-на-Дону: Феникс, 2012. 346 с.
Закс Л. Статистическое оценивание. М.: Статистика, 1976. 598 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии