

Журнал аналитической химии, 2019, T. 74, № 6, стр. 403-409
Применение микроэмульсий для определения тетрафенилолова методом высокоэффективной жидкостной хроматографии со спектрофотометрическим детектированием
А. В. Пирогов 1, *, В. В. Левкина 1, Е. А. Овинова 1, В. Ю. Савицкая 1, И. А. Дьячков 1, А. Д. Смоленков 1, И. А. Ананьева 1, М. В. Попик 1, О. А. Шпигун 1
1 Московский государственный университет имени М.В. Ломоносова, химический факультет
119991 Москва, ГСП-1, Ленинские горы, 1, стр. 3, Россия
* E-mail: pirogov@analyt.chem.msu.ru
Поступила в редакцию 19.01.2018
После доработки 27.03.2018
Принята к публикации 30.11.2018
Аннотация
На примере тетрафенилолова (ТеФО) разработан и апробирован новый способ выделения и концентрирования токсичных металлорганических соединений из почв. Он заключается в концентрировании целевого компонента в органическом слое, образующемся после расслаивания микроэмульсии (МЭ), с последующим определением ТеФО методом обращенно-фазовой ВЭЖХ со спектрофотометрическим детектированием. Изучена зависимость коэффициента концентрирования ТеФО в органическом слое, образующемся после расслаивания МЭ, от состава МЭ. Установлено, что при использовании МЭ состава додецилсульфат натрия–н-гексан–н-бутанол–вода (3 : 1.5 : 6 : 89.5, по объему), коэффициент концентрирования максимален и равен 10. Предел обнаружения ТеФО составляет 0.1 нг/мл.
В связи с высокой токсичностью в Российской Федерации нормируется содержание 16 оловоорганических соединений в питьевой воде, причем некоторые из них относятся к 1 классу опасности, их ПДК составляют 0.2 мкг/л [1]. Наиболее токсичными являются соединения триалкиолова, токсичность соединений диалкилолова несколько ниже. Тетраалкилолово еще менее токсично, так как с увеличением длины углеводородной цепи токсичность оловоорганических соединений для живых организмов снижается. Однако четырехзамещенные оловоорганические соединения могут проявлять отсроченный токсичный эффект, связанный с их разложением до соединений с меньшим числом заместителей [2].
Присутствие оловоорганических соединений в окружающей среде и их неблагоприятное воздействие на здоровье человека стимулировали разработку методов прямого определения оловоорганических соединений в объектах окружающей среды на уровне ПДК (табл. 1). Однако анализ таких объектов затруднен из-за очень низкого содержания оловоорганических соединений и матричных эффектов [3–12]. Следовательно, проблема количественного извлечения и предварительного концентрирования таких соединений актуальна. В табл. 2 представлены примеры использования метода ВЭЖХ для определения оловоорганических соединений.
Таблица 1.
Определение оловоорганических соединений методом газовой хроматографии
| Соединения | Пробоподготовка | Метод [3–8] | сmin | Объект анализа | Литера-тура |
|---|---|---|---|---|---|
| Монобутилолово Дибутилолово, Трибутиолово |
Твердофазнвая микроэкстракция, дериватизация тетрапропилборатом натрия | ГХ–МС/МС | 1.05–1.96 нг/л | Вода | [3] |
| Тетрабутилолово, Tрибутилолово, Дибутилолово, Монобутилолово, Трифенилолово, Дифенилолово, Монофенилолово |
Экстракция гексаном смеси образца с ледяной уксусной кислотой (рН 4.5) и NaCl | ГХ–МС | 7–29 нг/л; 0.35–1.45 мкг/л |
Вода Морские отложения |
[4] |
| Монобутилолово, Дибутилолово,
Tрибутилолово, Tетрабутиолово |
Экстракция метанолом, содержащим трополон, дериватизация, очистка | ГХ–МС | 13–25 нг/г | Морские организмы | [5] |
| Трибутилолово | Экстракция смесью толуол–уксусная кислота в ультразвуковой ванне, дериватизацией | ГХ-ПФД | 66.3 нг/г | Морские отложения | [6] |
| Mетилбутилолво, Дибутилолово, Tрибутилолово |
Экстракция смесью толуол–уксусная кислота с последующей дериватизацией | ГХ-ИПФД | 7–25 нг/мл | Поверхность побережья | [7] |
| Mетилбутилолово, Дибутилолово, Tрибутилолово, Дифенилолово, Tрифенилолово |
Экстракция гексаном, дериватизация тетраэтилбутилоловоратом натрия | ГХ–МС | ≈10 нг/л | Прибрежные воды | [8] |
Таблица 2.
Определение оловоорганических соединений методом ВЭЖХ
| Соединение | Пробоподготовка | Метод [9–12] | сmin | Объект анализа | Литература |
|---|---|---|---|---|---|
| Диметилолово, Дибутилолово |
Растворение в тетрагидрофуране с добавлением конц. HCl, экстракция гексаном | ВЭЖХ-СФД | 1.7–2.1 нг/г | Поливинил-хлорид | [9] |
| Метилбутилолово Дибутилолово, Tрибутилолово, Дифенилолово, Tрифенилолово (в образцах воды) |
Растворение в тетрагидрофуране с уксусной кислотой, содержащей некоторое количество трополона. | ВЭЖХ (ААС) | 0.03 мкг/л | Вода | [10] |
| Tрифенилолово, Тетраэтилолово |
Экстракция смесью гексана и уксусной кислоты под воздействием микроволнового излучения | ВЭЖХ-УФ | 0.14 мкг/мл 0.31 мкг/мл |
Мука | [11] |
| Дибутилолово, Диметилолово, Диэтилолово | Экстракция этилацетатом, концентрирование в гексане | ВЭЖХ (ФЛД) | 0.1–1 нг/г | Морские организмы | [12] |
Цель данного исследования – применение микроэмульсий для извлечения и концентрирования тетрафенилолова как гидрофобного представителя класса оловоорганических соединений с последующим ВЭЖХ-анализом.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты. Использовали следующие реактивы: н-гексан (Panreac, Испания), н-бутанол (CarlRoth, Германия), ацетонитрил (Panсreac, Испания), метанол (Panсreac, Испания), уксусную кислоту (Panсreac, Испания), толуол (Panсreac, Испания), хлорид кальция (Merck, Германия), додецилсульфат натрия (ДДСН) (Panreac, Испания), тетрафенилолово (Aldrich Chemical Company, США), хлорид натрия (Panreac, Испания). В работе использовали дистиллированную воду.
Приборы и материалы. Эксперименты проводили на хроматографической системе Agilent 1200, снабженной бинарным градиентным насосом, дегазатором подвижной фазы в режиме реального времени, автоматическим устройством ввода пробы, термостатом колонок, диодно-матричным детектором (Agilent Technologies, США). Сбор данных и обработку хроматограмм осуществляли с помощью программного обеспечения Chemstation (Agilent Technologies, США). В работе использовали колонки Luna-C18 (2) (250 × 4.6 мм, размер частиц 5 мкм) фирмы “Phenomenex”, ODS-AQ (250 × 4.0 мм, 5 мкм) фирмы “YMC”, Synergi Hydro-RP (250 × 4.6 мм, 4 мкм) фирмы “Phenomenex”, Synergi 4u Polar-RP (250 × × 2.0 мм, 4 мкм), а также спектрофотометр Cary-60 UV-Vis (Agilent Technologies, США).
Аликвоты отбирали автоматическими дозаторами с диапазонами установки отбираемого объема в интервалах 10–100, 20–200, 100–1000, 1000–5000 мкл соответственно (LABMATE, Польша). Предел допустимой относительной погрешности измерения не более ±5%.
Точные навески взвешивали на весах ExplorerPro (OhausCorporation, США), точность которых составляла 0.0001 г.
Для ультразвуковой обработки использовали ультразвуковую термостатируемую ванну САПФИР (Россия).
Приготовление микроэмульсий. Точную навеску ПАВ растворяли в рассчитанном количестве воды и выдерживали в ультразвуковой ванне до полного растворения в течение 5 мин. К полученному раствору добавляли необходимый объем н-гексана, со-ПАВ (н-бутанол) и помещали в ультразвуковую ванну на 10 мин до образования стабильной МЭ. Для расслоения МЭ добавляли навеску сухого хлорида кальция и выдерживали в ультразвуковой ванне в течение 5 мин до образования двух несмешивающихся фаз.
Пробоподготовка почвы. К навеске глинистой почвы добавляли ТеФО (с = 0.3 нг/г) и высушивали до воздушно-сухого состояния. Навеску высушенной почвы помещали в коническую колбу, добавляли 50 мл МЭ и помещали в ультразвуковую ванну на 10 мин. Для расслоения смеси добавляли десятикратный избыток сухого хлорида кальция (по отношению к ДДСН). Полученный экстракт центрифугировали в течение 3 мин при 16 000 об./мин. Отбирали надосадочную жидкость для дальнейшего хроматографического определения.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Термином “микроэмульсии” обозначают системы, принципиально отличные от обычных макроэмульсий. Обычно МЭ содержат четыре компонента: воду, углеводород (“масло”), коллоидное ПАВ и немицеллообразующее ПАВ (со-ПАВ), например спирт [13]. Микроэмульсия – термодинамически стабильная система, представляющая собой дисперсную систему масла и воды, стабилизированную с помощью ПАВ и со-ПАВ. Под маслом в данном случае понимают любое органическое вещество, ограниченно растворимое в воде. Поскольку возникновение термодинамически стабильной фазы основано на принципах самоорганизации ПАВ, микроэмульсионные фазы во многом похожи на другие организованные системы ПАВ, например мицеллярные растворы. Микроэмульсии типа “масло в воде” являются очень хорошими экстрагентами для количественного извлечения гидрофобных соединений из различных объектов. Ранее показано [14], что при добавлении хлорида кальция к МЭ на основе ДДСН система теряет свою термодинамическую стабильность и происходит образование двух несмешивающихся фаз: масла и воды. Гидрофобные вещества переходят в масляную фазу после расслоения и тем самым концентрируются за счет уменьшения объема растворителя (примерно в 10 раз). Этот эффект использован в данной работе при определении ТеФО в объектах окружающей среды для его извлечения с помощью МЭ, концентрирования целевого компонента в органическом слое МЭ, образующемся после ее расслаивания с последующим определением ТеФО методом обращенно-фазовой ВЭЖХ с УФ-детектированием. Такой подход уменьшает общую продолжительность анализа и сводит процесс пробоподготовки к простым шагам: приготовление МЭ, растворение образца в МЭ, расслоение МЭ на две фазы и хроматографический анализ одной из фаз, в которую перешло исследуемое соединение.
Готовили растворы ТеФО в микроэмульсиях различного состава. Коэффициент концентрирования рассчитывали как отношение площади пика ТеФО в органической фазе, полученной после расслаивания МЭ, к площади пика ТеФО, полученного в микроэмульсии без расслаивания. Результаты приведены в табл. 3. На рис. 1 представлены хроматограммы ТеФО в МЭ без расслаивания (рис. 1, хроматограмма 1) и в органическом слое, полученном после расслаивания МЭ (рис. 1, хроматограммы 1−6).
Таблица 3.
Коэффициенты концентрирования для микроэмульсий разного состава
| Микроэмульсия | ДДСН, об. % | н-Гексан, об. % | н-Бутанол, об. % | Коэффициент концентрирования |
|---|---|---|---|---|
| 1 | 3 | 1.5 | 6 | 10.0 |
| 2 | 3 | 1.5 | 8 | 9.7 |
| 3 | 3 | 2 | 6 | 5.3 |
| 4 | 3 | 0.8 | 8 | 6.0 |
| 5 | 3 | 3 | 8 | 6.3 |
Рис. 1.
Хроматограммы тетрафенилолова в МЭ 1 без расслаивания (1) и в органическом слое, полученном после расслаивания микроэмульсий различных составов: МЭ 1 (2), МЭ 2 (3), МЭ 3 (4), МЭ 4 (5) и МЭ 5 (6).
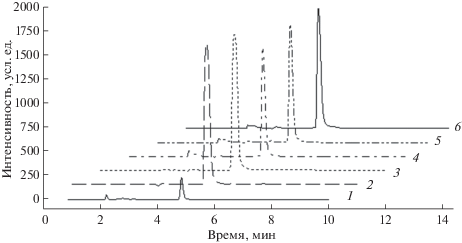
Видно, что при использовании МЭ 1 достигается наибольший коэффициент концентрирования, который полностью соответствует теоретическому (рассчитан как отношение общего объема МЭ до расслаивания к объему органического слоя, образовавшегося после расслаивания МЭ). Таким образом, в этих условиях ТеФО экстрагируется количественно.
Для выбора подвижной фазы использовали растворы: метанол–уксусная кислота–вода (80 : 3 : 17, по объему) и ацетонитрил–уксусная кислота–вода (80 : 3 : 17, по объему). При практически одинаковом времени удерживания ТеФО давление в хроматографической системе во втором случае почти в 2 раза меньше (220 и 140 Бар соответственно). Для дальнейшей работы использовали элюент, содержащий ацетонитрил.
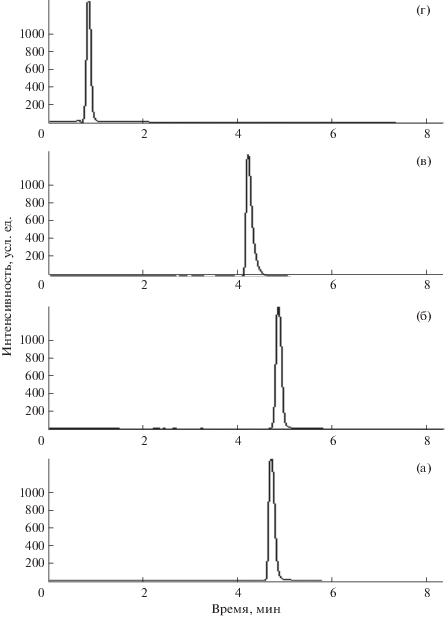
Природа неподвижной фазы является основным параметром, влияющим на селективность и удерживание в жидкостной хроматографии. Тетрафенилолово содержит фенильные радикалы, за счет чего обладает большой гидрофобностью (lgP = 8.34). Для сравнения селективности и эффективности различных неподвижных фаз использовали колонки с обращенно-фазовым силикагелем Luna-C18 (2) и ODS-AQ, колонку Synergi Hydro-RP, которая храктеризуется высоко гидрофобной поверхностью в совокупности блокированием остаточных силанольных групп, что обеспечивает хорошее удерживание неполярных и сильно полярных соединений, и колонку Synergi 4µm Polar-RP, которая предназначена для разделения сильно полярных и ароматических соединений, благодаря уникальной фазе на основе фенильных групп, пришитых к силикагелю. Хроматограммы модельного раствора ТеФО представлены на рис. 2. Видно, что колонка Synergi 4µm Polar-RP имеет минимальную емкость, которая сильно отличается от остальных колонок. Для трех остальных неподвижных фаз времена удерживания ТеФО отличаются незначительно. Однако на сорбенте ODS-AQ пик ТеФО сильно ассиметричен. Эффективности сорбентов Synergi Hydro-RP и Luna-C18 (2) сопоставимы и составляют 95 000 и 102 000 теоретических тарелок на метр соответственно. В дальнейшей работе использовали колонку Luna-C18 (2).
Рис. 2.
Хроматограммы модельного раствора тетрафенилолова в тетрагидрофуране (10 мкг/мл) на колонках: Synergi Hydro-RP (а), Luna-C18 (2) (б), ODS- AQ (в), Synergi 4u Polar-RP (г). Подвижная фаза: ацетонитрил–вода–уксусная кислота (80 : 17 : 3, по объему) с рН 3.0. Термостатирование при 40°С, расход подвижной фазы 1 мл/мин, объем вводимой пробы 20 мкл, спектрофотометрическое детектирование при 270 нм.
Предел обнаружения ТеФО (с учетом концентрирования в 10 раз) составил 0.1 нг/мл. Диапазон линейности градуировочного графика 0.3–500 нг/мл (коэффициент корреляции R = = 0.9996).
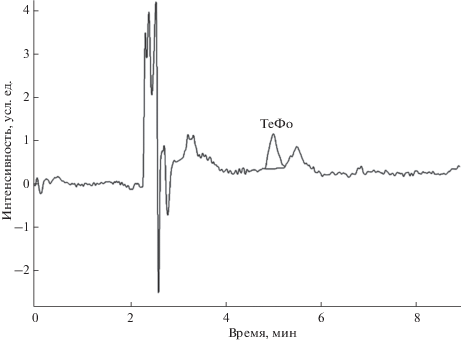
Для проверки правильности разработанной методики проанализировали реальный объект (глинистая почва) с добавкой ТеФО. Полученная хроматограмма представлена на рис. 3. Степень извлечения составила 98 ± 3% (n = 3).
Рис. 3.
Хроматограмма тетрафенилолова из образца почвы (добавлено 0.3 нг/г) после экстракции и расслоения микроэмульсии на колонке Luna-C18 (2) (250 × 4.6 мм, 5 мкм). Остальные условия хроматографирования см. в подписи под рис. 2.
* * *
Таким образом, использование микроэмульсия состава ДДСН–н-гексан–н-бутанол–вода (3 : 1.5 : 6 : 89.5, по объему) позволило количественно экстрагировать ТеФО из глинистой почвы для его последующего определения методом обращенно-фазовой ВЭЖХ со спектрофотометрическим детектированием. Коэффициент концентрирования равен 10. Предел обнаружения ТеФО (с учетом концентрирования в 10 раз) составил 0.1 нг/мл. Диапазон линейности градуировочного графика 0.3–500 нг/мл.
Работа выполнена при финансовой поддержке гранта РФФИ 16-03-00257 (выбор условий хроматографического определения тетрафенилолова, оценка метрологических характеристик метода) и гранта РНФ 16-13-10079 (композиция микроэмульсий, условия их разложения и количественной экстракции оловоорганики).
Список литературы
Alzieu C., Sanjuan J., Michel P., Borel M., Dreno J.P. Monitoring and assessment of butyltins in atlantic coastal waters // Marine Pollut. Bull. 1989. V. 20. № 1. P. 22.
Rabadan J.M., Galban J., Vidal J.C., Aznarez J. Determination of tin in organotin compounds by hydride generation atomic Absorption Spectrometry in organic media // J. Anal. Atom. Spectrom. 1990. V. 5. P. 1.
Moscoso-Perez C., Fernandez-Gonzalez V., Moreda-Pineiro J., Lopez-Mahia P., Muniategui-Lorenzo S., Prada-Rodriguez D. Determination of organotin compounds in waters by headspace solid phase microextraction gas chromatography triple quadrupole tandem mass spectrometry under the European Water Framework Directive // J. Chromatogr. A. 2015. V. 1385. P. 85.
Ikonomou M.G., Fernandez M.P., He T., Cullon D. Gas chromatography–high-resolution mass spectrometry based method for the simultaneous determination of nine organotin compounds in water, sediment and tissue // J. Chromatogr. A. 2002. V. 975. № 12. P. 319.
Liscio C., Di Carro M., Magi E. Comparison of two analytical methods for the determination of organotin compounds in marine organisms // Comptes Rendus Chimie. 2009. V. 12. P. 831.
Godoi A.F.L., Montone R.C., Santiago-Silva M. Determination of butyltin compounds in surface sediments from the Sao Paulo State coast (Brazil) by gas chromatography–pulsed flame-photometric detection // J. Chromatogr. A. 2003. V. 985. № 1–2. P. 205.
Rossi de Oliveira C., Moscardi dos Santos D., Augusto dos Santos Madureira L., Rodrigues de Marchi M. R. Speciation of butyltin derivatives in surface sediments of three southern Brazilian harbors // J. Hazard. Mater. 2010. V. 181. P. 851.
Cole R.F., Mills G.A., Bakir A., Townsend I., Gravell A., Fones G.R. A simple, low cost GC/MS method for the sub-nanogram per litre measurement of organotins in coastal water // MethodsX. 2016. V. 3. P. 490.
Row I., Row Y., Whang C. Determination of dialkyltin compounds in PVC by high – performance liquid chromatography // J. Chin. Chem. Soc. 1990. V. 37. P. 203.
Kadokami K., Uehiro T., Morita M., Fuwa K. Determination of organotin compounds in water by bonded-phase extraction and high-performance liquid chromatography with long-tube atomic absorption spectrometric detection // J. Anal. Atom. Spectrom. 1988. V. 3. P. 187.
Vickrey T.M., Howell H.E., Harrison G.V., Ramelow G.J. Post column digestion methods for liquid chromatography-graphite furnace atomic absorption speciation of organolead and organotin compounds // Anal. Chem. 1980. V. 52. № 11. P. 1743.
Yu T., Arakawa Y. High-performance liquid chromatographic determination of dialkyltin homologues using fluorescence detection // J. Chromatogr. 1983. V. 258. P. 189.
Hoar T.P., Schulman J.H. Transparent water-in-oil dispersions: the oleophatic hydromicelle // Nature. 1943. V. 152. № 3847. P. 102.
Толмачева Н.Г., Пирогов А.В., Шпигун О.А. Метиленовая селективность в режиме микроэмульсионной жидкостной хроматографии // Вестн. Моск. ун-та. Сер. 2. Химия. 2017. Т. 58. № 2. С. 83.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии