

Журнал аналитической химии, 2019, T. 74, № 7-app, стр. 57-62
Моделирование адсорбции ароматических полихлорзамещенных углеводородов на графитированной термической саже для предсказания величин хроматографического удерживания
Д. Д. Матюшин 1, *, А. К. Буряк 1
1 Институт физической химии и электрохимии им. А.Н. Фрумкина Российской академии наук
119071 Москва, Ленинский просп., 31, корп. 4, Россия
* E-mail: dm.matiushin@mail.ru
Поступила в редакцию 08.02.2018
После доработки 13.01.2019
Принята к публикации 13.01.2019
Аннотация
Выполнено моделирование адсорбции полихлорзамещенных ароматических углеводородов на графитированной термической саже молекулярно-статистическим методом. Результаты вычислений сопоставлены с данными по хроматографическому удерживанию в условиях газовой хроматографии. Для полихлордифенилов использовали вариант, учитывающий конформационную нежесткость молекул. Рассмотрены разные методы вычислительной химии (методы классической молекулярной динамики и полуэмпирические квантовые методы) для оценки внутренней энергии молекулы. Показано, что применение молекулярно-статистического метода и полуэмпирического метода AM1 для оценки внутренней энергии молекул позволяет правильно предсказать порядок элюирования изомерных дихлордифенилов. Показано, что такой подход можно использовать для подтверждения отнесения пиков на хроматограмме к тому или иному изомеру.
Теоретическое предсказание адсорбции на хроматографических сорбентах и соответственно предсказание величин удерживания может быть использовано в химическом анализе. При сопоставлении времен удерживания неизвестных аналитов с результатами такого расчета для возможных структур можно подтвердить или опровергнуть предположение о структуре определяемых соединений. В некоторых случаях такая информация позволяет с высокой вероятностью узнать структуру соединения, например, когда отсутствуют нужные стандартные образцы или сведения о фрагментации в масс-спектрометрическом эксперименте недостаточны. Кроме того, такие предсказания могут быть полезными при планировании эксперимента, для предварительной оценки возможности разделения веществ на используемой хроматографической колонке.
Для предсказания хроматографического удерживания применяют эмпирические формулы и аддитивные схемы, формулы, использующие молекулярные деcкрипторы, методы машинного обучения [1–3]. Точность таких подходов недостаточно велика – коэффициент корреляции между экспериментальными и рассчитанными временами удерживания часто находится в диапазоне 0.7–0.9.
Киселев и Пошкус разработали метод молекулярно-статистических расчетов, позволяющий точно вычислять константу Генри адсорбции на графитированной термической саже (ГТС). Этот сорбент имеет однородную плоскую поверхность, что позволяет относительно легко аналитически описать взаимодействие сорбат–сорбент и вычислить конфигурационные интегралы в адсорбированном состоянии [4, 5]. Потенциальная энергия взаимодействия сорбата и сорбента рассчитывается в атом-атомном приближении с использованием эмпирически подобранных потенциалов в форме Леннард-Джонса или Бакингема−Корнера. Данный метод дает очень точные результаты (погрешность не более 1%) для некоторых классов органических соединений и широко применялся в исследованиях. Однако метод имеет и ряд ограничений: он применим только для конформационно-жестких молекул, погрешность при его применении к сильнополярным молекулам высока.
Нами разработан [6] новый вариант молекулярно-статистического метода, пригодный для конформационно-нежестких молекул, с учетом внутреннего вращения, особенность которого состоит в том, что с помощью метода Монте-Карло учитываются все возможные конформации. Внутренняя энергия молекулы оценивается с помощью методов классической молекулярной динамики. Рассмотрено также применение молекулярно-статистического метода к высокоэффективной жидкостной хроматографии [7].
Углеродные сорбенты применяют для разделения смесей полихлорбензолов, полихлордифенилов и других полихлорзамещенных ароматических соединений методами как газовой, так и жидкостной хроматографии [8–11]. С помощью молекулярно-статистического метода предсказывали порядок элюирования таких соединений, однако при использовании потенциала в форме Бакингема−Корнера наблюдалось отличие экспериментального порядка элюирования от теоретически предсказанного. Для объяснения данного эффекта сделано предположение о различном характере взаимодействия с поверхностью сорбента двух атомов хлора, расположенных в орто-положении по отношению друг к другу, по сравнению с другими атомами хлора (так называемый “орто-эффект”) [12]. При использовании потенциала в форме Леннард-Джонса предсказанный порядок удерживания совпадал с наблюдаемым без дополнительных поправок на “орто-эффект” [13].
Для нежестких систем, таких как полихлордифенилы, применение молекулярно-статистического метода для предсказания удерживания напрямую невозможно, так как необходим учет всех возможных углов вращения и соответственно оценка зависимости внутренней энергии молекулы от угла между бензольными кольцами. В работе [14] для решения данной проблемы использовали “эффективные” углы между бензольными кольцами, зависящие от количества заместителей в орто-положении. Однако такой подход не достаточно универсален и требует предварительно подобранных для данного типа систем параметров.
В настоящей работе изучена применимость варианта метода молекулярно-статистических расчетов с использованием параметров взаимодействий из набора GAFF (general AMBER force field) [15] к моделированию адсорбции полихлорзамещенных ароматических соединений на ГТС и проведено сравнение различных методов оценки внутренней энергии конформаций. Рассмотрены все возможные хлорбензолы, изомеры хлордифенила и дихлордифенила. Цель данной работы – выяснить, можно ли использовать расчетные методы для надежного предсказания порядка элюирования изомеров этих и подобных соединений для отнесения пиков на хроматограмме к тому или иному из изомеров с высокой вероятностью на основе только расчетных данных. В отличие от предыдущих работ, в настоящей работе не использовали эмпирически подобранные параметры специально для решения этой задачи (такие как поправки на “орто-эффект” и “эффективные” углы между бензольными кольцами), а ограничивались только универсальными методами и подходами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В данной работе использовали экспериментальные данные по удерживанию на ГТС хлорбензолов [12] и хлордифенилов [14]. Адсорбцию хлорбензолов рассчитывали методом, описанным в работе [4], при этом численное интегрирование вели с использованием метода Монте-Карло (случайный равномерно распределенный набор углов Эйлера). Для хлордифенилов использовали сочетание метода [4] с интегрированием по всем возможным углам между бензольными кольцами. Взаимодействие сорбата и однородной поверхности графита описывали атом-атомными потенциалами в форме Леннард-Джонса с параметрами из набора GAFF [15] с использованием указанных в этой работе правил смешивания. Для атомов графита с поверхности ГТС использовали параметры из работы [6] (ε = 0.295 кДж/моль; r = 1.908 Å).
Для оценки зависимости внутренней энергии от угла между бензольными кольцами использовали методы классической молекулярной динамики: GAFF [15] и MMFF94 [16], полуэмпирический метод AM1 [17] и неэмпирический квантово-химический метод (теория функционала плотности, ТФП, базис 6-31G) для сравнения. Связи и углы предполагали жесткими и неизменными, расчет вели с точностью до 0.02 единиц логарифма константы Генри адсорбционного равновесия. Предварительную оптимизацию геометрии молекулы выполняли методом сопряженных градиентов.
Молекулярно-статистические расчеты и все расчеты с использованием GAFF проводили с помощью собственного программного обеспечения. Расчеты по методу MMFF94 проводили с помощью программного пакета OpenBabel [18], метода AM1 – с помощью программы MOPAC 7.1 [19], с помощью ТФП – nwchem [20]. Данные приложения вызывали из программы молекулярно-статистических расчетов в автоматическом режиме.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
В табл. 1 представлены результаты молекулярно-статистического расчета логарифмов констант Генри адсорбционного равновесия для всех хлорбензолов и соответствующие экспериментальные значения при 200°С. Случайная погрешность экспериментального определения составляет 2%, погрешность при интегрировании методом Монте-Карло составляет 0.02 единицы логарифма константы Генри адсорбции (lnK1). Существенное различие экспериментальных и теоретически рассчитанных констант Генри может быть обусловлено такими факторами, как неверная оценка параметров колонки при экспериментальном определении или неверные параметры взаимодействия для атома графита, что приводит к значительной систематической погрешности. Экспериментальные данные в разных источниках также существенно различаются, например, значения lnK1 равны –0.44 и 0.93 [12] или 0 и 1.67 [13] соответственно для хлорбензола и 1,3-дихлорбензола, для других соединений наблюдается примерно такое же различие. Однако на практике важны не абсолютные значения времен удерживания и тем более не значения констант Генри адсорбции, а порядок элюирования соединений и соотношение хроматографических параметров удерживания. В связи с этим в данной работе рассматривали коэффициенты корреляции между экспериментальными и рассчитанными значениями. Соответствующая корреляционная зависимость изображена на рис. 1.
Таблица 1.
Экспериментальные и вычисленные молекулярно-статистическим методом константы Генри адсорбции на графитированной термической саже для всех возможных полихлорбензолов
| Соединение | lnK1, см3/м2 | |
|---|---|---|
| эксперимент | расчет | |
| Хлорбензол | –0.44 | 0.19 |
| 1,3-Дихлоpбензол | 0.93 | 1.97 |
| 1,2-Дихлоpбензол | 1.01 | 2.01 |
| 1,4-Дихлоpбензол | 1.06 | 2.09 |
| 1,3,5-Трихлорбензол | 2.23 | 3.76 |
| 1,2,4-Трихлорбензол | 2.41 | 3.91 |
| 1,2,3-Трихлорбензол | 2.42 | 3.97 |
| 1,2,3,5-Тетрахлорбензол | 3.86 | 5.83 |
| 1,2,4,5-Тетрахлорбензол | 3.89 | 5.89 |
| 1,2,3,4-Тетрахлорбензол | 4.24 | 6.02 |
| Пентахлоpбензол | 5.51 | 7.99 |
| Гексахлоpбензол | 7.07 | 10.14 |
Рис. 1.
Корреляционная зависимость между рассчитанными и экспериментальными значениями логарифмов констант Генри адсорбции хлорбензолов на графитированной термической саже.

Для всех изомеров хлорбензолов предсказание порядка элюирования изомеров без введения поправок на “орто-эффект” оказалось правильным. Одним из объяснений несовпадения, которое привело к необходимости введения таких поправок в работе [12], может служить неточность значений параметров потенциалов в форме Бакингема−Корнера. Верификацию этих параметров проводили только путем сопоставления данных по удерживанию на ГТС с результатами молекулярно-статистического расчета.
Для хлордифенилов чрезвычайно важно точно оценивать внутреннюю энергию молекулы при разных углах между бензольными кольцами. Если рассмотреть молекулы дифенила (рис. 2), ориентированные относительно поверхности сорбента энергетически наиболее выгодным образом с углом между плоскостями бензольных колец 44.4° и 0°, то разница внутренних энергий составит 25.2 кДж/моль, а разница энергий взаимодействия с графитом – около 21 кДж/моль. Заслоненные конформации вносят большой вклад в величину, характеризующую адсорбцию, ввиду значительно большей потенциальной энергии взаимодействия с сорбентом, поэтому оценка их внутренней энергии влияет на результат расчета.
Рис. 2.
Взаимодействие молекулы дифенила с однородной поверхностью адсорбента при разных углах между плоскостями бензольных колец (стрелками показана часть дисперсионных взаимодействий между поверхностью сорбента и атомами сорбата).
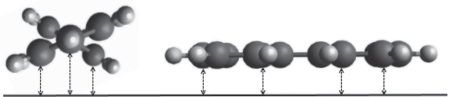
Не все методы вычислительной химии способны одинаково точно и эффективно оценивать зависимость внутренней энергии молекулы дифенила от угла между плоскостями бензольных колец. Так, в работе [21] показано, что из трех полуэмпирических квантовых методов (AM1, MNDO, PM3) только первый позволяет правильно предсказать равновесное значение угла между бензольными кольцами и величины барьеров вращения, в то время как два других дают качественно совершенно неверную информацию. В ходе предварительных расчетов (путем сравнения с энергиями, рассчитанными с помощью ТФП) показано, что метод AM1 позволяет быстро и адекватно оценивать внутреннюю энергию для бифенильных молекул, а также таких молекул, как дифенилметан.
В табл. 2 представлены значения логарифмов факторов удерживания для хлордифенилов на ГТС, полученные в режиме программирования температуры от 200 до 350°C в течение 15 мин и рассчитанные значения константы Генри адсорбции. Для расчета использовали различные методы оценки внутренней энергии при 300 K. Так как в этом случае экспериментальные данные констант Генри отсутствуют, рассчитанные константы используют в качестве условной меры, характеризующей взаимодействие сорбат–сорбент. В этом случае можно предсказать лишь относительное расположение пиков, а не абсолютные значения характеристик. Ранее в работе [6] использовали данные по удерживанию, приведенные к одной температуре, и продемонстрировали работоспособность такого подхода.
Таблица 2.
Экспериментально измеренные коэффициенты удерживания и рассчитанные с использованием различных методов оценки внутренней энергии константы Генри адсорбции дифенила, хлордифенилов и дихлордифенилов
| № | Соединение | ln k (эксперимент) | ln K1, см3/м2 (расчет) | ||
|---|---|---|---|---|---|
| GAFF | MMFF94 | AM1 | |||
| 1 | Дифенил | 0.75 | 18.4 | 18.3 | 19.1 |
| 2 | 2-Хлордифенил | 0.37 | 17.1 | 14.9 | 16.6 |
| 3 | 2,2'-Хлордифенил | 0.01 | 14.8 | 13.1 | 15.0 |
| 4 | 2,3'-Хлордифенил | 1.13 | 19.1 | 16.2 | 18.0 |
| 5 | 2,4'-Хлордифенил | 1.56 | 20.7 | 17.6 | 19.4 |
| 6 | 3-Хлордифенил | 1.73 | 21.6 | 21.4 | 22.3 |
| 7 | 3,3'-Хлордифенил | 2.26 | 24.1 | 24.3 | 25.5 |
| 8 | 3,4'-Хлордифенил | 2.37 | 25.1 | 25.0 | 25.8 |
| 9 | 4-Хлордифенил | 1.91 | 22 | 21.8 | 22.7 |
| 10 | 4,4'-Хлордифенил | 2.51 | 25.5 | 25.5 | 26.2 |
На рис. 3 приведены корреляционные зависимости между логарифмом фактора удерживания на ГТС и логарифмом рассчитанной константы Генри. Методы классической молекулярной динамики GAFF и MMFF94 (рис. 3а) не позволяют полностью правильно предсказать порядок удерживания соединений. Хотя для большинства пар изомеров эти подходы верно предсказывают, какое из соединений будет элюировано раньше, для отнесения пиков на хроматограммах сложных систем они недостаточно точны. Молекулярно-статистический метод с использованием полуэмпирического метода AM1 (рис. 3б) для оценки внутренней энергии позволяет надежно предсказать порядок элюирования всех рассмотренных соединений. Данный подход можно использовать для предсказания удерживания таких соединений, как дихлордифенилтрихлорэтан (ДДТ), дихлордифенилдихлорэтилен (ДДЕ) и продуктов их трансформации в окружающей среде, а также для полихлордибензодиоксинов. Основным применением этого метода может быть подтверждение или опровержение предполагаемой структуры вещества путем сопоставления рассчитанных величин и экспериментальных данных по удерживанию, в том числе при определении изомеров.
Рис. 3.
Корреляционные зависимости между экспериментальными факторами удерживания и константами Генри адсорбции, рассчитанными с помощью методов классической молекулярной механики MMFF94 (◆) и GAFF (▲) для оценки внутренней энергии молекулы (а) и с помощью полуэмпирического метода AM1 (б). Номера сорбатов соответствуют приведенным в табл. 2.

Работа выполнена при поддержке программы фундаментальных исследований президиума РАН № 14П.
Список литературы
Put R., Vander Heyden Y. Review on modelling aspects in reversed-phase liquid chromatographic quantitative structure–retention relationships // Anal. Chim. Acta. 2007. V. 602. № 2. P. 164.
Hancock T., Put R., Coomans D., Vander Heyden Y., Everingham Y. A performance comparison of modern statistical techniques for molecular descriptor selection and retention prediction in chromatographic QSRR studies // Chemom. Intell. Lab. Syst. 2005. V. 76. № 2. P. 185.
Zellner B.D.A., Bicchi C., Dugo P., Rubiolo P., Dugo, G., Mondello L. Linear retention indices in gas chromatographic analysis: A review // Flavour Fragr. J. 2008. V. 23. № 5. P. 297.
Буряк А.К. Применение молекулярно-статистических методов расчета термодинамических характеристик адсорбции при хромато-масс-спектрометрической идентификации органических соединений // Успехи химии. 2002. Т. 71. № 8. С. 788. (Buryak A.K. The use of molecular-statistical methods for the calculation of thermodynamic characteristics of adsorption for identification of organic compounds by gas chromatography–mass spectrometry // Russ. Сhem. Rev. 2002. V. 71. № 8. P. 695.)
Kiselev A.V., Poshkus D.P., Grumadas A.J. Statistical molecular calculation of thermodynamic parameters of adsorption of aromatic hydrocarbons on graphite. Part 1. – Condensed aromatic hydrocarbons // J. Chem. Soc. Faraday Trans. 1. 1979. № 75. P. 1281.
Матюшин Д.Д., Буряк А.К. Моделирование адсорбции нежестких органических молекул на графитированной термической саже методом Монте-Карло для предсказания их характеристик удерживания // Сорбционные и хроматографические процессы. 2017. Т. 17. № 2. С. 204.
Милюшкин А.Л., Лактюшина А.А., Буряк А.К. Хроматографическая, масс-спектрометрическая и молекулярно-статистическая идентификация пептидов с защищенными функциональными группами // Изв. АН. Сер. хим. 2017. Т. 66. № 1. С. 56. (Milyushkin A.L., Laktyushina A.A., Buryak A.K. Identification of peptides with protected functional groups by chromatography, mass spectrometry, and molecular statistics. // Russ. Chem. Bull. 2017. V. 66. № 1. P. 56.)
Fuoco R., Colombini M.P., Samcova E. Individual determination of ortho and non-ortho substituted polychlorobiphenyls (PCBs) in sediments by high performance liquid chromatographic pre-separation and gas chromatography/ECD detection // Chromatographia. 1993. V. 36. № 1. P. 65.
Pietrogrande M. C., Benvenuti A., Previato S., Dondi F. HPLC analysis of PCBs on porous graphitic carbon: Retention behavior and gradient elution // Chromatographia. 2000. V. 52. № 7-8. P. 425.
Zagorevskaya E.V., Ishchenko N.V., Kiselev A.V., Kovaleva N.V. Studies of adsorption of polychlorocarbons on carbon blacks by gas chromatography // Adsorpt. Sci. Technol. 1985. V. 2. № 4. P. 219.
Glausch A., Hirsch A., Lamparth I., Schurig V. Retention behaviour of polychlorinated biphenyls on polysiloxane-anchored C60 in gas chromatography // J. Chromatogr. A. 1998. V. 809. № 1–2. P. 252.
Белякова Л.Д., Буряк А.К., Ларионов О.Г. Хроматография-метод исследования химии поверхности и процессов на межфазных границах // Физикохимия поверхности и защита материалов. 2013. Т. 49. № 6. С. 551. (Belyakova L.D., Buryak A.K., Larionov O.G. Chromatography: Method of investigation of surface chemistry and interface processes. // Protection of Metals and Physical Chemistry of Surfaces. 2013. V. 49. № 6. P. 605.)
Vidal-Madjar C., Guiochon G., Dondi F. Prediction of retention data of polychlorobenzenes and-naphthalenes on graphitized carbon black // J. Chromatogr. A. 1984. V. 291. P. 1.
Буряк А.К., Федотов А.Н., Киселев А.В. Связь структуры хлорированных дифенилов с их удерживанием на графитированной термической саже // Вестн. Моск. ун-та. Сер 2. Химия. 1985. Т. 26. № 6. С. 568.
Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general AMBER force field // J. Comput. Chem. 2004. V. 25. P. 1157.
Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94 // J. Comput. Chem. 1996. V. 17. № 5–6. P. 490.
Dewar M.J., Zoebisch E.G., Healy E.F., Stewart J.J. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model // J. Am. Chem. Soc. 1985. V. 107. № 13. P. 3902.
O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: An open chemical toolbox // J. Cheminform. 2011. V. 3 № 1. P. 33.
Stewart J.J.P. MOPAC: A semiempirical molecular orbital program // J. Computer-aided Molecular Design. 1990. V. 4. № 1. P. 1.
Valiev M., Bylaska E.J., Govind N., Kowalski K., Straatsma T.P., Hubertus J.J., Dunyou W., Nieplocha J., Apra E., Windus T.L., De Jong W.A. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations // Comput. Phys. Commun. 2010. V. 181. № 9. P. 1477.
Mulholland J.A., Sarofim A.F., Rutledge G.C. Semiempirical molecular orbital estimation of the relative stability of polychlorinated biphenyl isomers produced by o-dichlorobenzene pyrolysis // J. Phys. Chem. 1993 V. 97. № 26. P. 6890.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии