

Журнал аналитической химии, 2019, T. 74, № 9, стр. 710-720
Спектрометрия ядерного магнитного резонанса в сочетании с хемометрическими методами обработки данных как способ изучения состава и свойств препаратов гепарина
Ю. Б. Монахова a, b, c, *, Е. М. Рубцова a, Б. Диль c, С. П. Муштакова a
a Саратовский государственный университет им. Н.Г. Чернышевского, Институт химии
410012 Саратов, ул. Астраханская, 83, Россия
b Санкт-Петербургский государственный университет, Институт химии
199034 Санкт-Петербург, Университетская набережная, 7–9, Россия
c Spectral Service AG
50996 Cologne, Emil-Hoffmann-Straße 33, Germany
* E-mail: yul-monakhova@mail.ru
Поступила в редакцию 27.08.2018
После доработки 02.11.2018
Принята к публикации 11.03.2019
Аннотация
Изучены состав и свойства препаратов гепарина методами ЯМР-спектрометрии. Рассмотрены подходы к установлению важнейших качественных показателей (биологическое происхождение и фирма-производитель) гепарина на основе многомерного моделирования ЯМР-профиля образца. Для повышения эффективности анализа проведено хемометрическое моделирование двумерных (2D) спектров ЯМР (DOSY, HSQC, HMBC) методами главных компонент и дискриминантного анализа. Впервые построены многомерные регрессионные модели (метод проекции на латентные структуры) для определения средней молекулярной массы гепарина. Метод количественного ЯМР применен для определения воды, катионов кальция и натрия, а также хлорид- и ацетат-анионов в препаратах гепарина. Для определения всех характеристик необходимо пять последовательных измерений методом ЯМР (1Н, 2D, 35Cl, 23Na и 2D DOSY) и одна пробоподготовка. Продолжительность анализа одного образца, включая измерение и обработку сигналов, не более 20 мин. Предлагаемый подход носит универсальный характер и может быть применен для анализа других лекарственных препаратов.
Известно, что при проведении хирургических операций и гемодиализа необходимо максимально замедлить свертываемость крови. Веществ, которые вырабатываются в тучных клетках организма человека, не хватает для поддержания его жизнеспособности и снижения риска побочных эффектов в подобных ситуациях. Одним из наиболее часто используемых в медицинской практике антикоагулянтов является гепарин [1]. Он представляет собой линейный гликозаминогликан со средней молекулярной массой от 10 до 20 кДа и состоит в основном из трисульфированных дисахаридных звеньев (остатки уроновой кислоты и гликозамина, соединенные 1–4 гликозидными связями) [2]. Известны также низкомолекулярные гепарины с молекулярной массой около 4.5 кДа, характеризующиеся повышенной биодоступностью при подкожном введении и более продолжительным периодом полураспада по сравнению с “нефракционированным” гепарином [3].
В международной практике аналитический контроль качества лекарственного сырья и готовых препаратов гепарина проводят в соответствии с требованиями международного стандарта GMP (Good Manufacturing Practice) и на основе стандартизированных фармакопейных методик анализа. Однако, несмотря на широкий спектр применения гепарина в медицинской практике, в Государственной Фармакопее РФ XIII 2015 г., не прописано статей на субстанцию и лекарственные препараты, содержащие его в качестве действующего вещества. Согласно американской и европейской Фармакопеи гепарин определяют с использованием набора взаимодополняющих фармакологических и физико-химических методов, которые длительны, дороги и требуют наличия специализированного оборудования [4, 5]. Актуален переход к определению всех необходимых параметров на основе одного спектрометрического метода с простой пробоподготовкой. Подобный подход несомненно облегчит рутинное определение стандартизованных характеристик (идентичность, молекулярная масса, антитромбозная активность), а также даст возможность определять дополнительные, не включенные в фармакопейный список, показатели качества препаратов гепарина (биологическое происхождение, фирма-производитель, содержание органических и неорганических примесей и др.).
Развитие спектрометрических методов исследования и новых вариантов многомерного анализа данных создает мощную платформу для разработки надежных подходов к исследованию свойств и рациональному выбору наиболее эффективных медицинских препаратов. Одним из перспективных с этой точки зрения методов является спектрометрия ЯМР [6]. Методика на основе этого метода включена в Европейскую Фармакопею (EP) и Фармакопею США (USP) в качестве обязательного теста на идентичность гепарина и наличия в нем примесей [4, 5]. Так, спектрометрию ЯМР используют для обнаружения токсичных примесей в образцах гепарина, например хондроитинсульфата и дерматансульфата с пределом обнаружения менее 0.5% [7, 8].
До настоящего времени спектрометрию ЯМР не использовали для определения важнейших с точки зрения клинической практики параметров, например антитромбозной активности, молекулярной массы, а также содержания активного ингредиента, воды и неорганических анионов. В связи с этим перспективно расширение круга определяемых качественных и количественных характеристик гепарина на основе спектров ЯМР на различных ядрах (1Н, 13С, 2D, 35Cl, 15N, 23Na и др.) и двумерных спектров ЯМР. Это позволит не прибегать к серии альтернативных трудоемких и дорогостоящих методов для определения каждого конкретного физико-химического или фармакологического параметра. Применение хемометрической обработки данных к профилям ЯМР серии образцов позволит решить проблему низкой специфичности (перекрывания сигналов) [6].
Цель настоящей работы продемонстрировать возможности спектрометрии ЯМР как способа исследования состава и свойств препаратов гепарина и его низкомолекулярных аналогов различного биологического происхождения (из крупного рогатого скота, КРС, свиней и овец) с использованием хемометрических методов обработки данных (главных компонент, многомерной регрессии, дискриминантного анализа и др.).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Пробоподготовка и измерения методом ЯМР. В исследование включено более ста образцов нефракционированного и низкомолекулярного гепарина, полученных из внутренних органов КРС, овец и свиней. Пробоподготовка заключалась в растворении 70 мг образца или их смесей в 0.7 мл D2O, содержащей 0.1% триметилсилилпропановой кислоты в качестве внутреннего стандарта.
Измерения проводили при 297 К на спектрометре Bruker Avance III 600 МГц (Bruker Biospin, Rheinstetten, Германия) с криозондом BBO, оснащенным автоматическим дозатором B-ACS 120. Параметры используемых импульсных программ приведены в публикациях [9–14].
В работе использовали протонный магнитный резонанс (ПМР) с подавлением сигналов сателлитов 13С (стандартный протокол USP), а также нестандартные варианты спектрометрии ЯМР на ядрах натрия и хлора. Также применяли методы двумерной спектрометрии ЯМР, такие как диффузионноупорядоченная спектроскопия (Diffusion-Ordered Spectroscopy, DOSY) и гетероядерная многоквантовая корреляция (Heteronuclear Multiple-Quantum Correlation spectroscopy, HMQC). Методика DOSY позволяет получать спектры индивидуальных соединений в смеси без их физического разделения и основана на различии в скоростях диффизии молекул с различной молекулярной массой. Метод DOSY использовали для установления молекулярной массы гепарина. Метод HMQC перспективен для повышения эффективности определения качественных характеристик сложных полимерных медицинских препаратов при сильном перекрывании контуров ЯМР [12].
Хемометрические методы. Для хемометрического моделирования спектрометрических данных использовали методы главных компонент (МГК), дискриминантного анализа (ДА) и проекции на латентные структуры (ПЛС). Хемометрические вычисления проводили в программных комплексах Matlab 2015a (The Math Works, Natick, MA, USA) и SAISIR (Statistics Applied to the Interpretation of Spectra in the InfraRed) для Matlab [15].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Качественные характеристики гепарина. Определение биологического происхождения и производителя гепарина. Практически все медицинские препараты на основе гепарина, использующиеся в фармацевтической практике в Европе и России, выделяют только из внутренних органов свиней. Альтернативные источники гепарина, например, кишечник и легкие коров или овец, известны давно, но после выявления случаев загрязнения гепарина в Китае в 2007–2008 гг., рассматриваются только как потенциальные заменители свиного гепарина [8, 16]. Однако, как показали результаты исследований, гепарин, выделенный из внутренних органов овец или КРС, также перспективен для применения в медицинской практике [17, 18]. Несомненно, необходима разработка методов определения биологического происхождения гепарина в медицинских препаратах на его основе.
Для физико-химического исследования подлинности и биологического происхождения гепарина существуют методы, основанные на количественной полимеразной цепной реакции, ВЭЖХ и изотопном анализе [19–21]. К сожалению, все они обладают серьезными недостатками, связанными с окислением маркеров гепарина, низкой чувствительностью и сложностью аппаратурного оформления. ЯМР ранее использовали для определения биологического происхождения гепарина с помощью небольшой выборки образцов без применения хемометрических методов [22–24]. Очевидна необходимость расширения возможностей спектрометрии ЯМР для определения этого параметра.
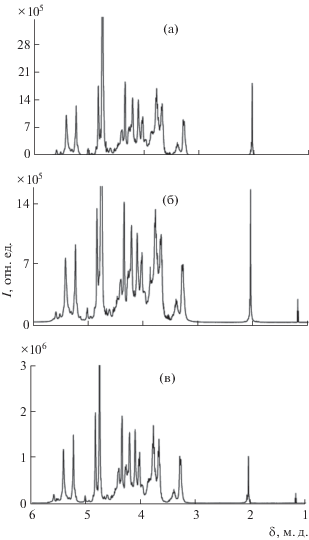
На рис. 1 представлены спектры 1Н ЯМР типичных образцов гепарина, выделенных из внутренних органов КРС, овцы и свиньи. Очевидно, что, несмотря на некоторые различия в спектрометрических профилях, классический одномерный анализ спектров не позволяет однозначно охарактеризовать биологическое происхождение конкретного образца. Следует отметить, что еще большее сходство в спектрах ЯМР наблюдали для низкомолекулярных гепаринов, полученных из овечьего и свиного материалов [11].
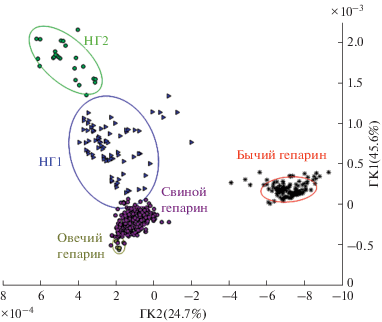
Для оценки возможности дифференциации биологического происхождения гепарина на основе многомерного анализа использовали МГК (рис. 2). Различия между кластерами образцов бычьего, свиного и овечьего гепарина очевидны при рассмотрении графика счетов на первые три главные компоненты (ГК1−ГК2−ГК3) после моделировании 88 спектров ЯМР гепарина. Аналогичные результаты получены на основе исследовательского анализа 26 образцов низкомолекулярного свиного и овечьего гепарина [11]. Кроме того, были построены дискриминантные модели (наилучшие результаты показал метод линейного ДА), на основе которых все образцы из тестовых наборов (30% от полного набора данных) для двух типов образцов (“нефракционированный” и низкомолекулярный гепарин) были правильно распознаны.
Рис. 2.
Трехмерный график счетов МГК в координатах ГК1–ГК2–ГК3 для кластеризации совокупности спектров ЯМР в соответствии с различным биологическим происхождением образцов гепарина: B – бычий (bovine), P – свиной (porcine), O – овечий (ovine) гепарин.

Многомерная обработка спектров ЯМР двухкомпонентных смесей гепарина различного происхождения показала, что качественное выявление преднамеренного добавления бычьего или овечьего (низкомолекулярного) гепарина в препарат свиного происхождения возможно, начиная с содержания чужеродного компонента 2 мас. % [11]. Кроме того, разработана регрессионная модель ПЛС для определения содержания бычьего гепарина в фальсифицированном препарате, заявленном как “Свиной гепарин”, с пределом определения и среднеквадратичной погрешностью валидации менее 2 и 1 мас. % соответственно [10].
В литературе имеется лишь упоминание об использовании различных вариантов двумерной спектрометрии ЯМР (гетероядерная корреляция через несколько связей, гетероядерная одноквантовая когерентность, HMQC и др.) для получения более детальной информации о биологическом происхождении препаратов гепарина [25]. Гипотетически спектры двумерного ЯМР, имеющие дополнительную ось химических сдвигов (13С, 15N, 16О и др.), при определенных условиях содержат дополнительный объем структурной информации по сравнению со спектрами ПМР. Нами впервые обработаны двумерные спектры HMQC для репрезентативной выборки из 79 образцов методами многомерного моделирования. Результаты показали эффективную дискриминацию спектров индивидуальных препаратов “нефракционированного” и низкомолекулярного гепарина различного биологического происхождения, а также их смесей (рис. 3) [12]. Недостатком данного подхода является низкая чувствительность двумерной спектрометрии ЯМР, так как единичное измерение занимает 4 ч машинного времени (2 мин для одномерного ЯМР-спектра).
Рис. 3.
Трехмерный график счетов МГК в координатах ГК1–ГК2–ГК3 для кластеризации совокупности спектров двумерного ЯМР (HMQC) в соответствии с различным биологическим происхождением образцов гепарина: BO – бычий, PO – свиной, OV – овечий гепарин, а также смеси свиного с бычьим (M1) и овечим гепарином (M2).

Другим аспектом проверки подлинности образцов гепарина является определение его изготовителя или процесса производства. По сравнению с биологическим происхождением, незначительные различия между брендовым продуктом и дженериками или образцами, полученными с использованием различных процессов деполимеризации, выявить сложнее. В нашем исследовании МГК показал различия в спектрах ЯМР для 34 образцов бычьего гепарина двух производителей, хотя их значения счетов расположены в одном кластере на основной модели (рис. 2) [11].
Таким образом, наши результаты показали, что хемометрика является полезным инструментом для дискриминации свиного, бычьего и овечьего “нефракционированного” и низкомолекулярного гепарина с эффективностью, близкой к 100%. Кроме того, показана дифференциация партий бычьих гепаринов, изготовленных двумя различными производителями. Хемометрические модели на основе измерений HMQC рекомендованы в исключительных случаях при возникновении сомнений в результатах дискриминации на основе одномерного измерения ЯМР.
Ретроспективный многомерный анализ спектров образцов гепарина. Спектрометрия ЯМР является важным инструментом для рутинного определения подлинности гепарина. При этом пробоподготовка и параметры импульсной программы для измерений ЯМР гепарина строго регламентированы международными директивами EP и USP [4, 5]. Таким образом, представляет интерес ретроспективная обработка большой серии спектров ЯМР препаратов гепарина, полученных в течение нескольких лет в одной лаборатории. Подобные спектрометрические данные за период 2012–2017 гг. были предоставлены фирмой “Spectral Service“ (Кельн, Германия), занимающейся заказным анализом фармацевтических препаратов, в том числе гепарина. Набор данных включал в себя 990 спектров ЯМР гепарина, из которых 759 представляли собой очищенные гепарины (134 бычьего, 20 овечьего и 605 свиного происхождения) и 111 необработанных образцов свиного гепарина (первичный продукт, выделяемый из животного сырья и содержащий дополнительные гликозаминогликаны). Кроме того, в исследование были включены 120 измерений двух стандартных образцов USP.
Метод главных компонент использовали для исследовательского анализа полного набора данных ЯМР. График счетов в координатах ГК1–ГК2 показал присутствие пяти кластеров (рис. 4). Различия между группами бычьего и свиного/овечьего гепарина проявляются вдоль оси ГК2. Дополнительная дифференциация овечьего и свиного гепарина возможна на основе графика счетов в пространстве ГК2–ГК3. В области положительных значений счетов по оси ГК1 обнаружены два кластера НГ1 и НГ2, которые представляют собой образцы неочищенного материала для производства гепарина с различным качественным и количественным составом гликозаминогликанов (рис. 4). Шесть образцов, находящихся вне эллипса для 95%-ной вероятности для кластера НГ1, содержали экстремально большие концентрации метанола (растворитель, использующийся при производстве гепарина). Таким образом, МГК можно использовать для проверки биологического происхождения гепарина и его химической чистоты. Образцы, содержащие персульфатированный хондроитинсульфат или другие, еще не обнаруженные загрязнители, можно различить с помощью исследовательского анализа, что предотвратит попадание опасных для здоровья лекарственных препаратов на фармацевтический рынок.
Рис. 4.
График счетов МГК для 870 спектров гепарина различного биологического происхождения и 111 спектров неочищенного гепарина (НГ1 и НГ2). Эллипсы ограничивают 95%-ную вероятность распределения для каждой группы.
Представляет интерес также дифференциация внутри отдельных групп образцов, выделенных на рис. 4, в соответствии с производителем и близостью к спектру стандарта гепарина, распространяемого USP. Анализ МГК был проведен для 605 спектров свиного гепарина, предоставленных шестью компаниями, и 120 спектров стандарта USP (рис. 5). Образцы гепарина, принадлежащие различным дистрибьюторам, разделены на графике счетов ГК2–ГК3. Две группы образцов (П1 и П6) наиболее обособлены и разнесены по оси ГК2, тогда как значения счетов для кластера П3 сильно разбросаны по графику счетов. Значения счетов для образцов из групп П2 и П4 значительно перекрывались вследствие близости их профилей ЯМР. Значения счетов для стандарта гепарина (группа USP) плотно сгруппированы вокруг начала координат на графике счетов МГК. Cпектры образцов свиного гепарина, чьи счета расположены около начала координат, наиболее близки к “усредненному” профилю ЯМР гепарина и стандарту USP (рис. 5).
Рис. 5.
График счетов МГК для 605 спектров свиного гепарина от 6 производителей (П1−П6) и 120 спектров стандартного гепарина USP. Эллипсы ограничивают 95%-ную вероятность распределения для каждой группы.
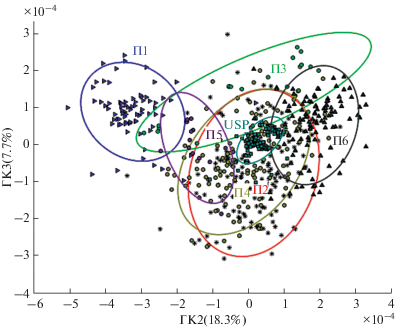
Наиболее важным практическим результатом данного ретроспективного исследования является возможность быстрой проверки подлинности новых образцов, используя проекцию их спектров на многомерные модели. Прежде всего, можно дифференцировать неочищенный материал и обработанный продукт, а затем выявить биологическое происхождение нового образца (рис. 4). Критерием отнесения образца к конкретной группе является попадание проекции спектра в эллипс, ограниченный 95%-ной вероятностью, на соответствующем графике счетов. Затем следует использовать подмодели для каждой конкретной группы гепарина (например, свиного) для оценки близости спектра нового образца к спектрам препаратов, произведенных конкретным производителем, а также к стандарту USP (рис. 5).
В сочетании с априорными знаниями результаты исследовательского анализа могут быть использованы для оценки подлинности заявленной информации об образце в течение нескольких секунд. Для большинства образцов наличие опасных примесей и несоответствие маркировке можно исключить. Более детальный анализ необходим только для небольшой выборки “подозрительных” образцов.
Количественные характеристики. Определение молекулярной массы гепарина. Одной из важнейших количественных характеристик препаратов гепарина, тесно связанной с его антитромбозной активностью, является молекулярная масса. Например, установлено влияние молекулярной массы препаратов нефракционированного гепарина, низкомолекулярного гепарина (гаммапарина) и олигосахаридной смеси на скорость свертывания крови у приматов [26]. Таким образом, быстрое определение этого параметра необходимо для эффективного скрининга полимерных препаратов относительно их фармакологической активности и соответствию международному стандарту.
Наиболее распространенным инструментальным методом определения молекулярной массы гепарина является гель-проникающая хроматография (ГПХ) [27]. Основной ее недостаток − необходимость набора дорогостоящих и труднодоступных эталонных стандартов с четко выраженным средним значением и узким распределением молекулярной массы [2, 9]. Предпринимались попытки использовать и 13С ЯМР-спектрометрию для определения молекулярной массы гепарина без использования стандартов [28]. Однако время измерения, требуемое для подобного анализа с достаточной точностью даже при использовании современного ЯМР-оборудования, оказалось неприемлемым для рутинного исследования гепарина [28].
В качестве альтернативы нами разработана экспрессная методика определения средней молекулярной массы гепарина на основе DOSY, которая представляет собой метод дискриминации веществ с различающейся молекулярной массой путем измерения коэффициентов диффузии (lg D), дополнительно к профилю 1Н ЯМР. Известно, что градуировка с использованием серии стандартных полимеров с заданной молекулярной массой позволяет определить молекулярную массу соединения с неопределенностью ±10% [29]. Однако для гепарина отсутствует набор необходимых стандартов.
Для корреляции данных двумерного ЯМР с данными анализа методом ГПХ мы использовали хемометрический метод ПЛС. Данные двумерного метода DOSY и метода ГПХ обрабатывали методом ПЛС для репрезентативных наборов нефракционированного (n = 32) и низкомолекулярного (n = 30) гепарина. Среднеквадратичная погрешность предсказания составила 498 и 179 Дa для нефракционированного и низкомолекулярного гепарина соответственно. Коэффициенты вариации не превышали 2.1% при пятикратном последовательном измерении одного раствора и 2.5% при повторной пробоподготовке образца (табл. 1). Значения средней молекулярной массы образцов гепарина для тестовой выборки (три образца для каждой группы гепaрина) методами ЯМР и ГПХ хорошо сопоставимы между собой (табл. 1). В целом результаты показали, что ПЛС подходит для определения средней молекулярной массы нефракционированного и низкомолекулярного гепарина, выделенного из органов различных животных.
Таблица 1.
Результаты валидации моделей ПЛС для определения молекулярной массы нефракционированного и низкомолекулярного гепарина
| Параметр | Нефракционированный гепарин (n = 32) | Низкомолекулярный гепарин (n = 30) | |
|---|---|---|---|
| Диапазон, Да | 12 130–15 090 | 3858–4778 | |
| Число факторов ПЛС | 5 | 6 | |
| RMSECVа, Да | 443 | 159 | |
| RMSEPб, Да | 498 | 179 | |
| Коэффициент вариациив (n = 5), % | |||
| свиной | 2.1 | 1.8 | |
| овечий | 2.0 | 1.3 | |
| бычий | 1.2 | –г | |
| Среднее значение относительного расхождения между результатами ЯМР и ГПХ, % | |||
| свиной | 0.26 | 1.9 | |
| овечий | 1.4 | 2.5 | |
| бычий | 1.6 | –б | |
а RMSECV – Root Mean Squared Error Cross Validation. бRMSEP – Root Mean Square Error of Prediction. вПараметр определяли после проведения пяти параллельных измерений с использованием трех образцов из каждой группы (свиной, овечий и бычий гепарин). гОбразцы низкомолекулярного бычьего гепарина в исследование не включены.
Показано, что свиной гепарин обладает самой высокой средней молекулярной массой (14.5 кДа), второе место занимает бычий гепарин (13.8 кДа), в то время как для овечьего характерны более низкие значения молекулярной массы около 12.5 кДа. Противоположная тенденция обнаружена для образцов низкомолекулярных гепаринов.
Таким образом, разработанные на основе метода DOSY многомерные модели для определения молекулярной массы нефракционированного и низкомолекулярного гепарина различного биологического происхождения позволили в течение 5 мин определять этот параметр для новых образцов. На этой основе можно предсказать антитромбозную активность препарата, не прибегая к трудоемким фармакологическим методам. В будущем для рутинного анализа предполагается разработать алгоритмы переноса многомерной модели ПЛС на основе экспериментов двумерного ЯМР на однотипные спектрометры.
Определение неорганических соединений. До настоящего времени спектрометрию ЯМР не использовали для количественного определения других важных с точки зрения клинической практики параметров фармацевтических препаратов (и гепарина, в частности), например содержания воды и различных ионов.
Содержание воды является ключевым параметром при исследовании фармацевтических продуктов, так как ее избыток может быть причиной низкой стабильности и пониженной фармацевтической активности препарата. Содержание воды в гепарине определяют с помощью термогравиметрического анализа и титрования по Фишеру, которые требуют дополнительного оборудования и не подходят для определенных матриц [30].
Количественное определение воды в гепарине с помощью ЯМР возможно, благодаря стабильной химической структуре его полимерной молекулы, которая состоит из дисахаридных звеньев, имеющих определенную массу и соответствующее количество протонов. Известно, что сигналы ЯМР гепарина расположены в трех спектрометрических диапазонах (5.0−5.7, 2.9−4.5 и 1.9−2.2 м. д.), а полоса при δ = 4.7 м. д. представляет собой сумму лабильных OH-протонов гепарина (например, группы −OH и –COOH), воды, содержащейся в растворителе D2O, и влаги препарата (рис. 1). Учитывая, что к анализируемому гепарину в процессе стандартной пробоподготовки добавляют постоянное количество дейтерированного растворителя, сумма первых двух перечисленных выше слагаемых также является постоянной.
Можно предположить, что существует корреляция между содержанием воды в гепарине и относительным значением интеграла при δ = 4.7 м. д. (сумму трех остальных интегральных интенсивностей приравнивали к 50). Действительно, на основании значений влажности, полученных для 26 образцов гепарина методом титрования по Фишеру, получена линейная зависимость в диапазоне содержания воды между 2.4 и 18.9 мас. % с коэффициентом корреляции 0.99 (табл. 2). Уравнение линейной регрессии Y = (33.4 ± 0.9) + + (3.2 ± 0.1)X можно использовать для быстрого прогнозирования содержания воды в гепарине на основе стандартного измерения методом ЯМР. Важной особенностью разработанной методики является ее универсальность, поскольку полученная корреляция применима для образцов бычьего, свиного и овечьего гепарина, гепарина натрия, гепарина кальция и низкомолекулярного гепарина.
Таблица 2.
Результаты валидации и количественного определения воды и ионов в образцах гепарина методом ЯМР
| Параметр | Вода | Na+ | Cl– | CH3COO– | Ca2+ |
|---|---|---|---|---|---|
| Необходимые ЯМР-измерения | Протокол USP | USP ЯМР 23Na |
USP ЯМР 35Cl |
USP | USP |
| Количество образцов | 26 | 88 | 90 | 131 | 10 |
| Предел определения, мас. % | –a | 0.05 | 0.05 | 0.002 | 0.5 |
| Диапазон линейности, мас. % | 2.4–18.9 | 0.05–30 | 0.05–30 | 0.002–15 | 0.5–20 |
| Коэффициент вариации, %б | 0.7 | <3 | <2 | <3 | <2 |
| Открываемость добавки, % | 95–105 | 92–104 | 94–109 | 96–102 | 97–107 |
Катионы (Na+, Ca2+) и анионы (Cl–, СH3CОO–). В промышленном производстве гепарина используют различные соли и буферные растворы [31]. В результате в матрице гепарина могут присутствовать нежелательные свободные анионы, включая хлорид и ацетат. Положительно заряженные ионы, такие как катионы кальция и натрия, являются важными составляющими гепарина. Эти противоионы имеют решающее значение для фармакологического действия антикоагулянтов. Предложены различные методы определения ионов в гепарине, но большинство из них трудоемки и требуют дополнительных измерений для комплексного исследования всех интересующих ионов [13, 31].
Очевидно, что хлорид-ионы и катионы натрия нельзя определить с помощью стандартного 1H ЯМР. Мы показали, что для этой цели можно использовать дополнительные эксперименты ЯМР на других ядрах, в частности, 35Cl (δ − 3.6 м. д.) и 23Na (δ − 0.2 м. д.) [31]. Результаты валидации представлены в табл. 2. В качестве примера в табл. 3 представлены данные, характеризующие правильность ЯМР-методики определения хлорид-ионов способом введено−найдено.
Таблица 3.
Результаты определения хлорид-ионов в образце гепарина методом введено–найдено (n = 5, P = 0.95)
| Введено, мг | Найдено, мг | Отн. погрешность определения массы добавки, % |
|---|---|---|
| 0 | 0.35 ± 0.01 | – |
| 0.35 | 0.71 ± 0.04 | 1.4 |
| 0.5 | 0.84 ± 0.03 | 1.2 |
| 0.75 | 1.0 ± 0.1 | 9.1 |
| 1.0 | 1.3 ± 0.1 | 3.7 |
| 1.2 | 1.6 ± 0.1 | 3.2 |
Разработанная методика использована для анализа 71 реального образца гепарина, включая гепарин натрия и гепарин кальция различного биологического происхождения. В 62 образцах (87%) содержание хлорид-иона было выше предела определения 0.05 мас. %. Экстремально высокое содержание хлорида (2.4 мас. %) наблюдали только в одном образце гепарина. Среднее значение и медиана составили 0.33 и 0.20 мас. % соответственно. С другой стороны, содержание Na+ варьировалось незначительно (средние значения составили 11.5, 11.3 и 11.7 мас. % для бычьего, овечьего и свиного гепарина соответственно). Очевидно, что содержание этого катиона нельзя использовать в качестве дополнительного фактора для оценки биологического происхождения гепарина.
Что касается катиона Ca2+, то для его определения уже предложены методики 1Н ЯМР, основанные на интегрировании сигнала комплекса Са–ЭДТА при добавлении ЭДТА к минеральной воде и препаратам Алое [32, 33]. Аналогично ионам натрия содержание кальция в исследуемых образцах гепарина кальция различалось незначительно, средняя концентрация составила 10.1 ± ± 0.2 мас. % (n = 10). Таким образом, образцы гепарина демонстрируют постоянное содержание противоионов.
Ацетат-ион может образовываться в результате неустойчивости N-ацетилированных полисахаридных звеньев гепарина и/или может быть преднамеренно введен в процессе производства. Метильная группа свободного ацетат-иона наблюдается при δ 1.9 м. д. в стандартном спектре 1Н ЯМР гепарина [16]. Ацетат натрия в концентрации до 0.5 мас. % обнаружили в партиях гепарина, загрязненных хондроитин сульфатом в 2008 г. [16]. В настоящем исследовании свободный ацетат-ион не обнаружен в более чем 100 исследованных образцах гепарина (предел обнаружения 0.0002 мас. %, табл. 2).
* * *
Отсутствие спектрометрических методик для надежной оценки антикоагулянтной активности препаратов гепарина делает необходимым регулярное использование трудоемких фармакологических методик. На основе предварительных исследований установлено, что на значения антикоагулянтной активности влияют различные факторы, в том числе ЯМР-профиль органической составляющей гепарина, его биологическое происхождение, наличие нежелательных компонентов (избыточная влажность, содержание ацетат- и хлорид-ионов), а также молекулярная масса препарата. Эти факторы в разной степени влияют на конечное значение активности. Молекулярная масса, по-видимому, является основным фактором внутри каждой подгруппы препаратов, выделенных из органов животных одного типа (бычий, свиной и овечий гепарин). Разработанную модель можно использовать для установления фармакологических характеристик новых препаратов в скрининговом режиме и по результатам анализа судить о перспективности применения данного препарата в медицинской практике. Целесообразно разработать алгоритмы переноса хемометрических моделей на однотипные спектрометры (в том числе, впервые на основе экспериментов двумерного ЯМР) и пакеты программ для автоматической компьютерной обработки спектрометрических профилей гепарина. Это позволит применять разработанный подход для рутинного анализа в любом научно-исследовательском учреждении в режиме реального времени.
Исследование выполнено за счет гранта Российского научного фонда (проект № 18-73-10009).
Список литературы
Gómez-Outes A., Suárez-Gea M.L., Calvo-Rojas G., Lecumberri R., Rocha E., Pozo-Hernández C., Terleira-Fernández A.I., Vargas-Castrillón E. Discovery of anticoagulant drugs: A historical perspective // Curr. Drug Discov. Technol. 2012. V. 9. P. 83.
Fu L., Li G., Yang B., Onishi A., Li L., Sun P., Zhang F., Linhardt R.J. Structural characterization of pharmaceutical heparins prepared from different animal tissues // J. Pharm. Sci. 2013. V. 102. P. 1447.
Ingle R.G., Agarwal A.S. A world of low molecular weight heparins (LMWHs) enoxaparin as a promising moiety–a review // Carbohydr. Polymers. 2014. V. 106. P. 148.
US Pharmacopoeia (2012) United States Pharmacopoeia heparin sodium stage 3 monograph. US Pharmacopoeia, Rockville. http://www.usp.org/sites/default/files/usp_pdf/EN/USPNF/ key-issues/m36690_pf386.pdf (01.08.2018).
European Pharmacopoeia (2010) European Pharmacopoeia heparin sodium monograph PA/PH/Exp. 6/T(0) 42 PUB monograph number 333 (2010). EDQM, Strasbourg. http://www.edqm.eu/medias/fichiers/NEW_Heparin_sodium_0820100333.pdf (01.08.2018).
Монахова Ю.Б., Кубалла Т., Лахенмайер Д.В. Хемометрические методы в ЯМР-спектроскопическом анализе пищевых продуктов // Журн. аналит. химии. 2013. Т. 68. № 9. С. 837.
Beyer T., Diehl B., Randel G., Humpfer E., Schäfer H., Spraul M., Schollmayer C., Holzgrabe U. Quality assessment of unfractionated heparin using 1H nuclear magnetic resonance spectroscopy // J. Pharm. Biomed. Anal. 2008. V. 48. P. 13.
Alban S., Lühn S., Schiemann S., Beyer T., Norwig J., Schilling C., Rädler O., Wolf B., Matz M., Baumann K., Holzgrabe U. Comparison of established and novel purity tests for the quality control of heparin by means of a set of 177 heparin samples // Anal. Bioanal. Chem. 2011. V. 399. P. 605.
Monakhova Y.B., Diehl B.W.K., Do T.X., Schulze M., Witzleben S. Novel method for the determination of average molecular weight of natural polymers based on 2D DOSY NMR and chemometrics: Example of heparin // J. Pharm. Biomed. Anal. 2018. V. 149. P. 128.
Monakhova Y.B., Diehl B.W.K. Combining 1H NMR spectroscopy and multivariate regression techniques to quantitatively determine falsification of porcine heparin with bovine species // J. Pharm. Biomed. Anal. 2015. V. 115. P. 543.
Monakhova Y.B., Diehl B.W.K., Fareed J. Authentication of animal origin of heparin and low molecular weight heparin including ovine, porcine and bovine species using 1D NMR spectroscopy and chemometric tools // J. Pharm. Biomed. Anal. 2018. V. 149. P. 114.
Monakhova Y.B., Yao Y., Fareed J., Diehl B.W.K. Improving reliability of chemometric models for authentication of species origin of heparin by switching from 1D to 2D NMR experiments // J. Pharm. Biomed. Anal. 2018. V. 153. P. 168.
Monakhova Y.B., Diehl B.W.K. Nuclear magnetic resonance spectroscopy as a tool for the quantitative analysis of water and ions in pharmaceuticals: Example of heparin // J. Pharm. Biomed. Anal. 2018. V. 154. P. 332.
McEwen I., Mulloy B., Hellwig E., Kozerski L., Beyer T., Holzgrabe U., Wanko R., Spieser J.M., Rodomonte A. Determination of oversulphated chondroitin sulphate and dermatan sulphate in unfractionated heparin by (1)H-NMR – Collaborative study for quantification and analytical determination of LoD // Pharmeuropa Bio. 2008. V. 1. P. 31.
Cordella C.B.Y., Bertrand D. SAISIR: A new general chemometric toolbox // Trends Anal. Chem. 2014. V. 54. № 2. P. 75.
Beyer T., Matz M., Brinz D., Rädler O., Wolf B., Norwig J., Baumann K., Alban S., Holzgrabe U. Composition of OSCS-contaminated heparin occurring in 2008 in batches on the German market // Eur. J. Pharm. Sci. 2010. V. 40. P. 297.
Gerotziafas G.T., Van Dreden P., Jeske W., Walenga J.M., Hoppensteadt D., Larsen A., Elalamy I., Yao Y., Fareed J. Comparison of Ufh and enoxaparin originated from bovine, ovine and porcine mucosa with functional coagulation assays // Blood. 2016. V. 128. P. 5020.
Bouchard O., Abro S., Kahn D., Iqbal O., Hoppensteadt D., Yao Y., Fareed J. A comparison of ovine and porcine heparins and enoxaparins: a case for an alternative source of heparin products // The FASEB J. 2016. V. 30. P. 1177.5.
Auguste C., Dereux S., Rousset M., Anger P. Validation of quantitative polymerase chain reaction methodology for monitoring DNA as a surrogate marker for species material contamination in porcine heparin // Anal. Bioanal. Chem. 2012. V. 404. P. 43.
Huang Q., Xu T., Wang G.Y., Huang J.F., Xia H., Yin R., Tang A., Fu W.L. Species-specific identification of ruminant components contaminating industrial crude porcine heparin using real-time fluorescent qualitative and quantitative PCR // Anal. Bioanal. Chem. 2012. V. 402. P. 1625.
Houstine C., Auguste C., Macrez C., Dereux S., Derouet A., Anger P. Quantitative PCR and disaccharide profiling to characterize the animal origin of low-molecular-weight heparins // Clin. Appl. Thromb. Hemost. 2009. V. 15. P. 50.
Watt D.K., Yorke S.C., Slim G.C. Comparison of ovine, bovine and porcine mucosal heparins and low molecular weight heparins by disaccharide analyses and 13C NMR // Carbohydr. Polymers. 1997. № 1. V. 33. P. 5.
Ruiz-Calero V., Saurina J., Galceran M.T., Hernández-Cassou S., Puignou L. Estimation of the composition of heparin mixtures from various origins using proton nuclear magnetic resonance and multivariate calibration methods // Anal. Bioanal. Chem. 2002. V. 373. P. 259.
Guerrini M., Bisio A., Torri G. Combined quantitative (1)H and (13)C nuclear magnetic resonance spectroscopy for characterization of heparin preparations // Semin. Thromb. Hemost. 2001. V. 27. № 5. P. 473.
Beni S., Limtiaco J.F.K., Larive C.K. Analysis and characterization of heparin impurities // Anal. Bioanal. Chem. 2011. V. 399. P. 527.
Ma Q., Tobu M., Schultz C., Jeske W., Hoppensteadt D., Walenga J., Cornelli U., Lee J., Linhardt R., Hanin I., Fareed J. Molecular weight dependent tissue factor pathway inhibitor release by heparin and heparin oligosaccharides // Thromb. Res. 2007. V. 119. № 5. P. 653.
Mulloy B., Heath A., Shriver Z., Jameison F., Al Hakim A., Morris T.S., Szajek A.Y. USP compendial methods for analysis of heparin: chromatographic determination of molecular weight distributions for heparin sodium // Anal. Bioanal. Chem. 2014. V. 406. № 20. P. 4815.
Desai U., Linhardt R.J. Molecular weight of heparin using 13C nuclear magnetic resonance spectroscopy // J. Pharm. Sci. 1995. V. 84. № 2. P. 212.
Groves P., Rasmussen M.O., Molero M.D., Samain E., Cañada F.J., Driguez H., Jiménez-Barbero J. Diffusion ordered spectroscopy as a complement to size exclusion chromatography in oligosaccharide analysis // Glycobiol. 2004. V. 14. № 5. P. 451.
European Pharmacopoeia Commission, European Pharmacopeia, 2008b. Microdetermination of water. Council of Europe, Strasbourg. P. 184.
van der Meer J.Y., Kellenbach E., van den Bos L.J. From farm to pharma: an overview of industrial heparin manufacturing methods // Molecules. 2017. V. 22. № 6. P. 1025.
Monakhova Y.B., Randel G., Diehl B.W. Automated control of the organic and inorganic composition of A-loe vera extracts using (1)H NMR spectroscopy // J. AOAC Int. 2016. V. 99. P. 1213.
Monakhova Y.B., Kuballa T., Tschiersch C., Diehl B.W.K. Rapid NMR determination of inorganic cations in food matrices: Application to mineral water // Food Chem. 2017. V. 221. P. 1828.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии