

Журнал аналитической химии, 2019, T. 74, № 9, стр. 703-709
Определение метаболитов нитрофуранов в мышечной ткани методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием
А. В. Куликовский a, b, *, И. Ф. Горлов a, М. И. Сложенкина a, Н. Л. Вострикова b, А. Н. Иванкин b, О. А. Кузнецова b
a Поволжский Научно-исследовательский институт производства и переработки мясомолочной продукции
400131 Волгоград, ул. им. Маршала Рокоссовского, 6, Россия
b Федеральный научный центр Пищевых систем им. В.М. Горбатова Российской академии наук
109316 Москва, ул. Талалихина, 26, Россия
* E-mail: a.kulikovskii@fncps.ru
Поступила в редакцию 14.03.2018
После доработки 03.10.2018
Принята к публикации 29.01.2019
Аннотация
Разработана методика определения в мышечной ткани остаточных количеств 4 метаболитов нитрофуранов (3-амино-2-оксазолидинона, 3-амино-5-метилморфолино-2-оксазолидинона, 1-аминогидантоина гидрохлорида и семикарбазида гидрохлорида) в виде 2-нитрофенильных производных методом ВЭЖХ с масс-спектрометрическим детектированием в режиме мониторинга заданных реакций. Показано влияние подавления ионизации матрицей, повышена селективность экстракции за счет оптимизации процедуры подготовки проб, минимизировано влияние органических примесей на результат измерений. Нижняя граница определяемых содержаний составила 10 мкг/кг для семикарбазида гидрохлорида и 1 мкг/кг для остальных метаболитов нитрофуранов.
Среди ветеринарных препаратов достаточно широкое распространение получили нитрофураны, которые используют для лечения и профилактики заболеваний в свиноводстве, птицеводстве и промышленном разведении рыбы и креветок [1]. Большинство этих препаратов используют как антибактериальные, однако некоторые из них, например нифуртимокс, проявляют антипаразитарные свойства. Основную часть нитрофуранов (фуразолидон, нитрофурантоин и др.) не относят к антибиотикам, поскольку они проявляют бактериостатический эффект. Однако в больших количествах нитрофураны могут обладать и бактерицидными свойствами. Нитрофураны также использовали, а в некоторых странах и продолжают использовать в ветеринарии в качестве опосредованных стимуляторов роста животных [2].
Проблема антимикробной резистентности обусловлена применением в животноводстве тех же групп антимикробных препаратов, что и в медицине. В связи с возрастающей актуальностью данной проблемы ряд стран предпринимает активные законодательные усилия по ограничению применения антимикробных средств. В РФ и на территории Таможенного союза действует регламент ТР ТС 034/2013, согласно которому в продукции животного происхождения не допускается наличие остатков 51 ветеринарного препарата, в том числе производных нитрофуранов. В странах Евросоюза нитрофураны запрещены к использованию в животноводстве с 1995 г. [3]. Это связано с тем, что у метаболитов нитрофуранов – семикарбазида (производное нитрофуразона), 3-амино-2-оксазолидинона (метаболит фуразолидона) и 1-аминогидантоина (образуется из нитрофурантоина) обнаружили мутагенные и канцерогенные свойства. Семикарбазид и 3-амино-2-оксазолидинон способствуют образованию опухолей сосудов и легких, a 1-аминогидантоин связывают с новообразованиями в почках, костях и яичниках [4].
В странах Южной Америки (Аргентина, Бразилия и др.), поставляющих животноводческую продукцию в РФ, разрешено использование синтетических ветеринарных препаратов при выращивании сельскохозяйственных животных. В связи с этим необходим строгий контроль импортной продукции [5].
В организме животного нитрофураны распадаются быстро, что затрудняет их обнаружение. Но образующиеся при этом метаболиты сохраняются в тканях в течение длительного времени [2]. В связи с этим методы определения нитрофуранов в тканях животных и продуктах животного происхождения основаны на контроле содержания метаболитов.
Существующие методы определения антибиотиков микробиологическим и иммуноферментным анализом (ИФА) недостаточно чувствительны и селективны по отношению к тканеспецифическим метаболитам антибиотиков [6, 7]. Микробиологические методы часто не позволяют выявить группу антибиотиков или антимикробных препаратов, их целесообразно применять как скрининговые. Методы ИФА позволяют селективно тестировать пробы на наличие антибиотиков, однако вероятность ложноположительного или ложноотрицательного результата составляет 5%. Следует учитывать, что высокоспецифическая иммунная реакция белкового коньюгата и вторичная реакция со специфичными к определяемому веществу антителами, фиксированными на плашке, требует строгого соблюдения сроков и условий хранения тест-систем ИФА. Для ИФА необходимы подготовка проб и очистка на сорбентах, что достаточно сложно при работе с белковыми матрицами животного происхождения [8].
Капиллярный электрофорез и ВЭЖХ хорошо зарекомендовали себя при работе с фармацевтическими субстанциями. Однако чувствительность данных методов недостаточна для определения антибиотиков в пищевых матрицах [9], поэтому они не получили широкого распространения при анализе ветеринарных препаратов. Наиболее достоверным является метод ВЭЖХ–тандемной масс-спектрометрии (ВЭЖХ–МС/МС), который дает возможность селективно определять любые группы веществ с пределом обнаружения на уровне 1 нг/кг [10–13] даже в условиях мешающего влияния липидных компонентов [14, 15].
Цель настоящей работы заключалась в разработке методики определения нитрофуранов в органах и тканях животных методом ВЭЖХ–МС/МС и установлении особенностей идентификации основных метаболитов нитрофуранов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты. Использовали следующие реагенты: ацетонитрил для ВЭЖХ (Panreac, Франция), метанол для ВЭЖХ (Merck, США), гексан (Panreac, Франция), этилацетат (Sigma-Aldrich, США), муравьиную кислоту (Merck, США), 2-нитробензальдегид (Acros Organics, Бельгия). Деионизованную воду получали в системе MilliQDirect 8 (Франция). В качестве стандартных образцов использовали 3-амино-2-оксазолидинон (АОЗ), 3-амино-5-метилморфолино-2-оксазолидинон (АМОЗ), 1-аминогидантоина (АГД) гидрохлорид, семикарбазида (СЕМ) гидрохлорид, 3-[(2-нитрофенил)метилен]амино-2-оксазолидинон (2-НФ-АОЗ), 5-метилморфолино-3-[(2-нитрофенил)метилен]-3-амино-2-оксазолидинон (2-НФ-АМОЗ), 1-[(2-нитрофенил)метилен]аминогидантоин (2-НФ-АГД), (2-нитрофенил)метиленсемикарбазид (2-НФ-СЕМ) с содержанием основного вещества не менее 95.0% (Sigma-Aldrich, США). В качестве внутренних стандартов метаболитов нитрофуранов использовали d5-3-амино-5-метилморфолино-2-оксазолидинон (d5-АМОЗ), 13С3-аминогидантоин (13С3-АГД), d4-3-амино-2-оксазолидинон (d4-АОЗ), 15N,13C-семикарбазид (15N,13C-СЕМ) с содержанием основного вещества не менее 99.0% (Sigma-Aldrich, США).
Подготовка проб к анализу. Образцы мышечной ткани измельчали с использованием гомогенизатора BUCHI B-400. Гомогенизированную пробу массой 1 г помещали в центрифужную пробирку, добавляли 1 мл воды и 4 мл метанола, перемешивали и центрифугировали при 2000 g 10 мин. После центрифугирования надосадочный слой отбрасывали, образец вновь промывали, добавляя 4 мл метанола, и удаляли органический слой. К пробе добавляли 50 мкл внутреннего стандарта c концентрацией 100 нг/мл, 5 мл 0.1 М соляной кислоты и 100 мкл раствора, содержащего 100 ммоль 2-нитробензальдегида (НБА) в метаноле, перемешивали и помещали в водяную баню с температурой 37°С на 16 ч. Гидролизат охлаждали, для нейтрализации добавляли 0.5 мл 0.3 М раствора фосфата натрия и доводили pH до значения 7.0 10 М раствором гидроксида натрия. Для экстракции нитрофенильных производных к нейтрализованному образцу добавляли 4 мл этилацетата, перемешивали и центрифугировали 5 мин при 4000 g. Экстракцию повторяли дважды. Органический слой, содержащий этилацетат, упаривали на роторном испарителе Heidolph Laborota 4003 при 40°С. Сухой остаток растворяли с использованием ультразвуковой ванны в 1 мл смеси 0.1%-ного раствора муравьиной кислоты и ацетонитрила (85 : 15, по объему). Для удаления жировых фракций к экстракту добавляли 2 мл гексана и центрифугировали 1 мин при 2000 g, гексановый слой отбрасывали. Раствор пропускали через мембранный фильтр с диаметром пор 0.45 мкм и анализировали методом ВЭЖХ–МС/МС.
Условия определения метаболитов нитрофуранов методом ВЭЖХ–МС/МС. Использовали систему для ВЭЖХ Agilent1200 (США) с трехквадрупольным масс-спектрометром Agilent 6410B. Разделение проводили на колонке (50 × 4.6 мм) Agilent XDB-C18 (1.8 мкм) при 40°С в режиме градиентного элюирования. Расход подвижной фазы 0.4 мл/мин. Объем вводимой пробы 10 мкл. Условия хроматографического определения представлены в табл. 1.
Использовали ионизацию распылением в электрическом поле и следующие оптимизированные параметры масс-спектрометрического детектирования: температура источника 100°С, температура газа для десольватации 350°С, расход газа для десольватации 8 л/мин, давление иглы распылителя 30 psi.
Условия детектирования оптимизировали в ручном режиме. Напряжение фрагментации определяли, варьируя с шагом 10 В, по максимальному отклику протонированной молекулы, энергию диссоциации оптимизировали с шагом 2 В по максимальному отклику характерного иона-продукта, при этом соотношение сигнал/шум иона-продукта должно быть не менее 1 : 10. Условия регистрации аналитических сигналов в режиме мониторинга заданных реакций представлены в табл. 2.
Таблица 2.
Условия детектирования в режиме мониторинга заданных реакций (ионизация электрораспылением с регистрацией положительных ионов)
| Производное | Ион-предшественник, m/z | Ион-продукт, m/z | Потенциал фрагментации, В | Энергия диссоциации, В |
|---|---|---|---|---|
| 2-НФ-АМОЗ | 335.1 | 291.2 | 130 | 10 |
| 262.0 | 10 | |||
| 128.1 | 20 | |||
| 2-НФ-d5-АМОЗ | 340.3 | 296.4 | 140 | 12 |
| 265.2 | 12 | |||
| 2-НФ-АГД | 249.1 | 178.0 | 120 | 10 |
| 134.0 | 8 | |||
| 104.0 | 20 | |||
| 2-НФ-13С3-АГД | 251.9 | 133.9 | 120 | 10 |
| 179.1 | ||||
| 2-НФ-АОЗ | 236.0 | 149.0 | 90 | 10 |
| 134.0 | ||||
| 2-НФ-d4-АОЗ | 240.3 | 134.1 | 100 | 10 |
| 149.1 | ||||
| 2-НФ-СЕМ | 209.1 | 192.0 | 120 | 5 |
| 166.0 | ||||
| 2-НФ-15N,13C-СЕМ | 212.2 | 168.3 | 120 | 5 |
| 195.2 |
Метаболиты нитрофуранов идентифицировали по абсолютному времени удерживания хроматографических пиков целевых веществ, регистрируемых в режиме мониторинга заданных реакций. С использованием средств программного обеспечения Mass Hunter Workstation (Agilent, США) строили градуировочные зависимости площади пика от концентрации вещества в пробе. Хроматограммы стандартных образцов метаболитов нитрофуранов и пробы, содержащей АОЗ, представлены на рис. 1, 2.


РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Сущность методики определения метаболитов нитрофуранов заключается в предварительном гидролизе пробы для разрушения белковых связей с остатками метаболитов, последующей дериватизации НБА и определении нитрофенильных производных метаболитов нитрофуранов методом ВЭЖХ–МС/МС. Молекулярные структуры нитрофуранов и их метаболитов приведены на схеме 1 .
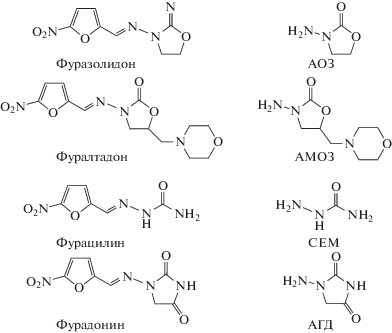
Схема 1 . Структурные формулы нитрофуранов и их метаболитов.
Схемы гидролиза и дериватизации связанных с белками остатков нитрофурановых препаратов под действием соляной кислоты приведены ниже.
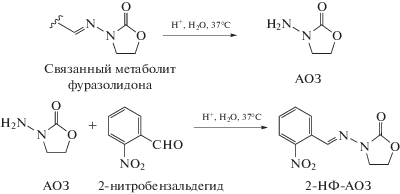
Схема 2 . Схемы гидролиза связанного с белком метаболита фуразолидона (АОЗ) в кислой среде и последующей его дериватизации 2-нитробезальдегидом.
Попытка обнаружения метаболитов нитрофуранов методом ВЭЖХ с УФ-детектированием при 275 нм показала, что спектрофотометрические детекторы неселективны по отношению к нитрофенильным производным метаболитов нитрофуранов и не способны обнаружить следовые количества в сложных матрицах. Идентификация с использованием масс-селективного детектора позволяет исключить ошибки, связанные с веществами, присутствующими в матрице.
Для повышения чувствительности в ходе подготовки проб увеличили навеску образца до 5 г. Это привело к появлению матричных эффектов и подавлению ионов. Для снижения подавления ионизации матрицей использовали предварительную промывку метанолом не связанных с белками метаболитов. Для учета матричного эффекта применяли матричную градуировку. Для этого обрабатывали холостые пробы, приготовленные и проанализированные ранее и не содержащие метаболитов нитрофуранов. Нитрофенильные производные метаболитов нитрофуранов вносили на стадии растворения сухого остатка. Для каждой концентрации растворов (10 и 100 нг/мл) выполняли по три параллельных измерения. Степень подавления ионизации матрицей мышечной ткани показана на рис. 3. Как видно, оптимизация условий определения позволила для ряда аналитов снизить матричный эффект более чем на 16%, что положительно влияет на общий уровень шума детектора. Так, соотношение сигнал/шум для нитрофенильных производных метаболитов нитрофуранов составляло не менее 100 : 1. При этом увеличение напряжения на электронном умножителе от 250 до 700 В позволило добиться соотношения сигнал/шум до 150 : 1 при концентрации аналитов 1 нг/мл.
Рис. 3.
Степень подавления (%) ионизации матрицей мышечной ткани до (◼) и после (◻) оптимизации условий экстракции.

Гидролиз и дериватизация являются ключевыми этапами извлечения. Эндогенные метаболиты нитрофуранов существуют у животных в связанных с белками формах и высвобождаются в кислой среде, образуя свободные соединения с низкой молекулярной массой. Относительные молекулярные массы АОЗ, АМОЗ, АГД и СЕМ составляют соответственно 102.1, 201.2, 115.1 и 75.1, характерный разброс ионов в условиях масс-спектрометра не наблюдается, что затрудняет качественный и количественный анализ. Дериватизация увеличивает относительные молекулярные массы метаболитов нитрофуранов до 248.2, 334.3, 235.2 и 208.2 соответственно. Метаболиты одновременно обрабатывали реагентами для кислотного гидролиза и дериватизации для увеличения относительных молекулярных масс целевых соединений. В соответствующих условиях в ячейке соударений эти производные дают не менее двух характерных ионов, что позволяет проводить количественное определение.
Основной задачей при разработке методики являлась минимизация потерь метаболитов нитрофуранов, связанных непосредственно с процедурой пробоподготовки. Для исключения возможности присутствия в анализируемой пробе органических загрязнителей оптимизировали условия жидкостно–жидкостной экстракции. Основной проблемой при подготовке проб являлись матричные эффекты, связанные с взаимодействием веществ матрицы с дериватизирующим агентом. Уменьшение количества вносимого дериватизирующего агента до 80 мкл привело к снижению степени дериватизации метаболитов нитрофуранов до 70–75%. Для уменьшения матричных эффектов избыток НБА и жировые фракции пробы удаляли гексаном.
Достоверность методики проверяли в соответствии с Регламентом Комиссии ЕС 2002/657. Для этого оценивали специфичность, линейность, правильность (степень извлечения), предел обнаружения, предел количественного определения. Для подтверждения специфичности методики исследовали 10 образцов тканей животных, не содержащих метаболиты нитрофураны. Во всех 10 образцах отсутствовали хроматографические пики, мешающие определению метаболитов нитрофуранов. Коэффициенты корреляции полученных градуировочных зависимостей составляли не менее 0.98 в диапазоне концентраций 1.0–100.0 нг/мл. За счет использования колонок с мелкозернистым сорбентом (1.8 мкм) продолжительность анализа не превышала 12 мин. Определили степени дериватизации и извлечения в ходе подготовки проб методом введено–найдено. Степень дериватизации определяли внесением в пробу, не содержащую метаболиты нитрофуранов, смеси стандартных растворов АОЗ, АМОЗ, АГД, СЕМ и 100 мкл раствора, содержащего 100 ммоль НБА в метаноле. Степень извлечения определяли внесением в пробу смеси нитрофенильных производных метаболитов нитрофуранов. Данные полученные при анализе 25 проб с концентрацией метаболитов нитрофуранов 1, 10, 20, 50, 100 мкг/кг представлены в табл. 3.
Таблица 3.
Степени дериватизации и извлечения метаболитов нитрофуранов из мышечной ткани (n = 25, P = 0.95)
| Аналит | Степень дериватизации, % | Степень извлечения, % |
|---|---|---|
| АМОЗ | 96 ± 2 | 73 ± 8 |
| АГД | 98 ± 2 | 65 ± 10 |
| АОЗ | 97 ± 3 | 76 ± 8 |
| СЕМ | 94 ± 3 | 68 ± 6 |
Пределы обнаружения и количественного определения рассчитывали при соотношении сигнал/шум для ионов-продуктов не менее 1 : 3 и 1 : 10 соответственно. Результаты получены с использованием 12 проб мышечной ткани с внесенными метаболитами нитрофуранов в концентрации 0.2, 0.5, 1.0 и 10.0 мкг/кг. Пределы обнаружения (мкг/кг) составили 0.2 для АОЗ, 0.5 для АМОЗ и АГД, 1.0 для СЕМ. Пределы количественного определения (мкг/кг) составили 1.0 для АОЗ, АМОЗ и АГД, 10.0 для СЕМ.
По данным проведенного в 2017 г. мониторинга отмечается значительный рост положительной идентификации остатков ветеринарных препаратов. Метаболиты нитрофураны обнаружены более чем в 5% случаев анализа мышечной ткани животных, причем в 95% положительных проб идентифицирован АОЗ. Фактическое содержание АОЗ составило от 1.4 до 78.2 мкг/кг. В 76% проб, содержащих АОЗ, концентрации варьировались в диапазоне 4.58–16.54 мкг/кг.
Разработанную методику определения метаболитов нитрофуранов в мышечной ткани и пищевой продукции на основе животного сырья методом ВЭЖХ с масс-спектрометрическим детектированием можно использовать для целей практической сертификации.
Работа выполнена в рамках гранта РНФ № 15-16-10000, ГНУ НИИММП.
Список литературы
Vass M., Hruska K., Franek M. Nitrofuran antibiotics: A review on the application, prohibition and residual analysis // Vet. Med. Czech. 2008. V. 53. № 9. P. 469.
Vroomen L.H., Berghmans M.C., Van Bladeren P.J., Groten J.P., Wissink C.J., Kuiper H.A. In vivo and in vitro metabolic studies of furazolidone: A risk evaluation // Drug Metab. Rev. 1990. V. 22. № 6–8. P. 663.
Commission Regulation (EC) 1995/1442/EC (1995) // Off. J. Eur. Commun. 1995. V. 38. № 143. p. 26.
Bogialli S., Di Corcia A. Recent applications of liquid chromatography–mass spectrometry to residue analysis of antimicrobials in food of animal origin // Anal.-Bioanal. Chem. 2009. V. 395. № 4. P. 947.
Points J., Burns D.T., Walker M.J. Forensic issues in the analysis of trace nitrofuran veterinary residues in food of animal origin // Food Control. 2015. V. 50. № 4. P. 92.
Cooper M.K., Samsonova J.V., Plumpton L., Elliott C.T., Kennedy D.G. Enzyme immunoassay for semicarbazide. Thenitrofuran metabolite and food contaminant // Anal. Chim. Acta. 2007. V. 592. № 1. P. 64.
ISO 18330:2003 (IDF 188:2003) Milk and milk products – Guidelines for the standardized description of immunoassays or receptor assays for the detection of antimicrobial residues.
Li J., Liu J., Zhang H.C., Li H., Wang J.P. Broad specificity indirect competitive immunoassay for determination of nitrofurans in animal feeds // Anal. Chim. Acta. 2010. V. 678. № 1. P. 1.
Fernando R., Munasinghe D.M.S., Gunasena A.R.C., Abeynayake P. Determination of nitrofuran metabolites in shrimp muscle by liquid chromatography-photo diode array detection // Food Control. 2017. V. 72. P. 300.
Park M.S., Kim K.T., Kang J.S. Development of an analytical method for detecting nitrofurans in bee pollen by liquid chromatography – electrospray ionization tandem mass spectrometry // J. Chromatogr. B. 2017. V. 1046. P. 172.
Kaufmann A., Butcher P., Maden K., Walker S., Widmer M. Determination of nitrofuran and chloramphenicol residues by high resolution mass spectrometry versus tandem quadrupole mass spectrometry // Anal. Chim. Acta. 2015. V. 862. P. 41.
Qi J., Yu S., Cheng N., Wu L., Cao W. Stability of nitrofuran residues during honey processing and nitrofuran removal by macroporous adsorption resins // Food Chem. 2014. V. 162. № 11. P. 110.
Thongsrisomboon P., Liawruangrath B., Liawruangrath S., Satienperakul S. Determination of nitrofurans residues in animal feeds by flow injection chemiluminescence procedure // Food Chem. 2010. V. 123. № 3. P. 834.
Radovnikovic A., Moloney M., Byrne P., Danaher M. Detection of banned nitrofuran metabolites in animal plasma samples using UHPLC–MS/MS // J. Chromatogr. B. 2011. V. 879. № 2. P. 159.
Barbosa J., Moura S., Barbosa R., Ramos F., da Silveira M.I.N. Determination of nitrofurans in animal feeds by liquid chromatography-UV photodiode array detection and liquid chromatography-ionspray tandem mass spectrometry // Anal. Chim. Acta. 2007. V. 586. № 1–2. P. 359.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии