

Журнал аналитической химии, 2019, T. 74, № 9-app, стр. 21-27
Методика хроматографического определения продуктов прямого синтеза диметилового эфира
П. В. Самохин a, И. А. Белостоцкий a, И. И. Маршев a, М. А. Кипнис a, *
a Институт нефтехимического синтеза им. А.В. Топчиева Российской академии наук
119991 Москва, Ленинский просп., 29, Россия
* E-mail: kipnis@ips.ac.ru
Поступила в редакцию 12.04.2018
После доработки 12.07.2018
Принята к публикации 05.03.2019
Аннотация
Разработана оригинальная методика газохроматографического определения продуктов и реагентов каталитического синтеза диметилового эфира (ДМЭ) из синтез-газа, позволяющая одновременно детектировать СН3ОН, ДМЭ, СО, СО2, Н2, Н2О, N2, углеводороды до С6. Газовая схема включает четыре детектора, три насадочных колонки, две предколонки (для предотвращения попадания воды и органических соединений в колонки с молекулярными ситами) в сочетании с двумя клапанами сброса и двумя регуляторами давления после себя для реализации обратной продувки предколонок. Схема смонтирована на базе хроматографа Хроматэк-Кристалл 5000. Определение проводят при постоянной температуре 140°С. Продолжительность анализа не более 12 мин. Благодаря наличию пламенно-ионизационного детектора возможно детектирование следовых количеств углеводородов, а также этана в присутствии больших количеств СО2.
Несмотря на существующие предположения о неисчерпаемости запасов нефти и газа, устойчивым спросом пользуются технологии переработки различного ненефтяного сырья (природный газ, попутный нефтезаводской газ, бытовые отходы, биомасса, уголь и т.д.). Например, с помощью окислительной конверсии (окислитель – водяной пар, воздух, кислород) ненефтяное сырье по известным промышленным технологиям переводят в синтез-газ, из которого затем синтезируют метанол и/или диметиловый эфир [1–5]. Интерес к ДМЭ связан с известными возможностями его применения. Так, ДМЭ может быть использован как пропеллент (в устройствах для распыления жидких веществ), хладоагент [6], моторное топливо (альтернатива дизельному топливу) [7]. Кроме того, ДМЭ рассматривают как исходное сырье для получения ряда углеводородов (олефинов, бензина, ароматических соединений и триптана) [8–11].
Экспериментальное исследование прямого синтеза ДМЭ из синтез-газа предполагает расчет состава и расхода конвертированного газа, содержащего легко конденсирующиеся компоненты (вода, метанол) и компоненты, способные растворяться под давлением в конденсате в заметных количествах (ДМЭ, СО2). В связи с этим необходим способ прямого газохроматографического анализа горячего газа из реактора до момента конденсации его компонентов. Такой анализ используют в недавних работах [12–17], однако условия его проведения и необходимое аппаратурное оформление, как правило, подробно не описаны. Сложность хроматографического анализа при каталитических исследованиях связана с необходимостью одновременного определения разнородных групп веществ, традиционно разделяемых на адсорбентах, работающих при отличающихся температурах. При этом анализ ограничен во времени, а диапазон определяемых концентраций значителен. Дополнительную сложность вносит отрицательное влияние некоторых компонентов реакционного газа на адсорбционную способность используемых адсорбентов.
При каталитических исследованиях, связанных с синтезом ДМЭ или метанола, проблемой является расчет расхода горячего конвертированного газа на выходе из реактора. Часто расчет выполняют, исходя из сохранения материального баланса по инертному компоненту синтез-газа, например азоту. При прямом хроматографическом определении всех компонентов газового потока на выходе из реактора появляется дополнительная возможность контроля расхода горячего газа после реактора, исходя из баланса по углероду.
Цель настоящей работы – разработка методики газохроматографического определения в изотермическом режиме продуктов и реагентов каталитического синтеза ДМЭ с использованием колонок, заполненных Porapak T и молекулярными ситами 13Х.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Хроматограф Хроматэк-Кристалл 5000 (ЗАО СКБ “Хроматэк”, Йошкар-Ола) собран производителем для анализа горячего газа в процессе синтеза ДМЭ в соответствии с предварительным заказом.
Исходный хроматограф включал обогреваемую линию ввода анализируемого газа, один термостат, три детектора по теплопроводности (ДТП), пламенно-ионизационный детектор (ПИД), обогреваемые автоматические краны-дозаторы на 6 и 10 входов, две идентичные насадочные колонки с молекулярными ситами 13X (цеолит NaX) длиной 3 м и внутренним диаметром 2 мм (одна для работы с газом-носителем гелием для определения простых газов, кроме водорода, вторая для работы с газом-носителем аргоном для определения водорода), две предколонки перед ними с адсорбентом HayeSep R (для сорбции воды и органических соединений из пробы, вредных для молекулярных сит), колонку с HayeSep Q длиной 3 м и внутренним диаметром 2 мм, три дозирующие петли, два клапана сброса для обратной продувки предколонок (для удаления из них воды и органических соединений в ходе каждого анализа), регуляторы расхода и давления (массовые электронные). Определение выполняли в режиме программируемого нагрева.
Схема доработанного нами хроматографа с узлом ввода пробы горячего газа из реактора для анализа представлена на рис. 1. По обогреваемой линии 27 (80°С) анализируемый газ поступает в горячую зону хроматографа, где разделяется на два равных потока, проходя через два крана-дозатора 1, 2 и омывая три дозирующие петли 3, 4, 5. На выходе из хроматографа потоки анализируемого газа из кранов 1, 2 объединяются, проходят ловушку 21, поддерживаемую при температуре 0°С, где конденсируются вода и метанол, и направляются на реометр 22 для контроля расхода анализируемого газа. Прочность адсорбции воды и органических соединений на адсорбенте HayeSep R выше, чем простых газов, поэтому предколонки 9, 10 поглощают воду и органические соединения и пропускают в основные колонки с молекулярными ситами 11, 12 только простые газы. Во время каждого анализа после выхода последнего целевого компонента из предколонок 9, 10 осуществляется их обратная продувка для десорбции и выведения из предколонок воды и органических соединений. Для обратной продувки открываются клапаны 17, 18 и потоки газов-носителей (аргон, гелий) из регуляторов давления 19, 20 направляются через предколонки в обратном направлении. Расход газа определяется пневмосопротивлениями, которые расположены в линиях после клапанов 17, 18. Потоки через основные колонки 11, 12 при этом не изменяются благодаря поддержанию постоянного давления перед ними регуляторами давления “после себя” 19, 20 (метод полуобратной продувки). Использование схемы полуобратной продувки позволило применить молекулярные сита в качестве адсорбента для определения простых газов в сложных, многокомпонентных пробах; исключить регулярное кондиционирование колонок с молекулярными ситами (нагрев до 350°С); размещать вместе с ними в одном термостате колонки с предельно допустимыми температурами ниже 350°С.
Рис. 1.
Принципиальная газовая схема хроматографа с узлом ввода пробы горячего газа для анализа (положение кранов-дозаторов 1, 2 соответствует вводу пробы в колонки): 1 – 10-канальный кран-дозатор; 2 – 6-канальный кран-дозатор; 3, 4, 5 – дозирующие петли; 6 – колонка с Porapak T (колонка с HayeSep Q в заводской поставке); 7 – ДТП-1; 8 – ПИД; 9, 10 – предколонки с HayeSep R; 11 – колонка с молекулярными ситами 13X (газ-носитель гелий); 12 – колонка с молекулярными ситами 13X (газ-носитель аргон); 13 – ДТП-2; 14 – ДТП-3; 15, 16, 17, 18 – клапаны сброса обратной продувки предколонок (положение 15, 16 – заводская поставка); 19, 20 – регуляторы давления “после себя”; 21 – ловушка (0°С); 22 – реометр; 23 – фильтр; 24 – трехходовой кран; 25 – дозирующий вентиль с микрорегулировкой; 26 – запорный вентиль; 27 – обогреваемая линия ввода.

В качестве газа-носителя использовали гелий (марка “А”, ТУ 0271-135-31323949-2005, 99.995 об. %) и аргон (высший сорт ГОСТ 10157-79, 99.993 об. %). Для ПИД использовали водород марки “А”, ГОСТ 3022-80, 99.99 об. %.
Поверочные газовые смеси. Для проведения абсолютной калибровки хроматографа использовали поверочные газовые смеси, изготовленные сертифицированными производителями, а также смеси, составленные нами с использованием специально сконструированной установки путем смешения индивидуальных газов по давлению. Использовали азот 99.999 об. % (ТУ 6-21-39-96), водород 99.99 об. % (ГОСТ 3022-80), оксид углерода 99.98 об. %, диоксид углерода 99.5 об. % (ГОСТ 8050-85), ДМЭ (ОАО НАК АЗОТ) 99.4 об. % или (Akzo Nobel) 99.99 об. %. Газы последовательно добавляли в предварительно вакуумированный баллон в заданной пропорции при контролируемых давлении в баллоне и температуре баллона. Предотвращение попадания паров масла из вакуумного насоса в баллон при вакуумировании обеспечивали с помощью ловушки, поддерживаемой над поверхностью жидкого азота.
Абсолютные коэффициенты по воде и метанолу. Мольные коэффициенты для воды и метанола определяли на независимой лабораторной установке, оборудованной плунжерным насосом для подачи жидкости и хроматографом Кристалл Люкс 4000М. Жидкие смеси воды и метанола известного состава подавали насосом в испаритель (145°С) с контролем подачи массовым измерителем жидкости (Bronkhorst, Нидерланды). Смеси разбавляли потоком чистого азота, ДМЭ или смеси азота и ДМЭ известного состава. Скорость потока варьировали массовым регулятором расхода газа (Bronkhorst, Нидерланды). Данные по содержанию и скорости подаваемых смесей позволяли рассчитать состав конечной смеси. По обогреваемой (145°С) линии смеси подавали в кран-дозатор хроматографа для анализа. Сопоставление данных по ДМЭ для двух хроматографов позволило рассчитать соответствующие коэффициенты для воды и метанола применительно к хроматографу Хроматэк-Кристалл 5000.
Все расходы ниже приведены к условиям 25°С, 101.3 кПа.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
При изучении кинетики синтеза ДМЭ требуется выполнять серию анализов с минимальными промежутками времени между ними. Однако анализ в режиме программированного нагрева, предложенный производителем, занимал значительное время. В связи с этим оптимизировали условия хроматографического анализа в режиме стационарной температуры термостата. Для эффективного выбора стационарной температуры решали необходимые методические задачи.
Газовая схема отбора пробы. Для отбора пробы газа была смонтирована газовая схема (рис. 1, 23–26). Чтобы избежать конденсации воды и метанола на выходе из реактора, все элементы схемы вплоть до обогреваемой линии ввода пробы анализируемого газа в хроматограф размещены в обогреваемой электропечи (120°С). Использовали газовую арматуру с предельно допустимой рабочей температурой 145°С (Swagelok, HOKE).
Лабораторный синтез диметилового эфира обычно проводят при давлении до 3 МПа. Опыт работы показал, что обеспечить плавную регулировку расхода газа, направляемого на хроматограф, при перепаде давления 3 МПа на регулировочном элементе можно только при использовании регулировочных вентилей с коэффициентом пропускной способности (Cv) не более 0.001. Мы использовали дозирующий вентиль 25 с микрорегулировкой Micromite 1600, (HOKE) с Cv, равным 0.0008. Для надежной работы вентиля после реактора, из которого возможен вынос катализаторной пыли, перед вентилем установлен противопылевой фильтр 23 с металлокерамическим фильтрующим элементом (диаметр пор 0.5 мкм). Расход анализируемого газа поддерживали постоянным (контроль по реометру 22) и настолько малым (менее 2 л/ч), что он не создавал избыточного давления в дозирующих петлях.
Основная часть потока из реактора через фильтр 23 и вентиль 26 поступает в сепаратор каталитической установки, охлаждаемый проточной водой, и далее через регулятор давления “до себя” после сброса давления до атмосферного на массовый измеритель расхода. Трехходовой вентиль 24 позволяет направлять на анализ либо газ после реактора, либо исходный синтез-газ.
Коррекция газовой схемы хроматографа. По результатам тестовых анализов, выполненных на исходной комплектации хроматографа, было отмечено, что пики имеют затянутый задний фронт, а базовая линия долго выходит на горизонтальный уровень после ввода пробы.
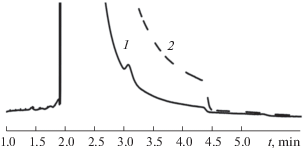
Как выяснилось, это связано с тем, что, поскольку в заводском варианте газовой схемы прибора клапаны сброса для полуобратной продувки (линии ДТП-2, ДТП-3) находятся перед предколонками после кранов-дозаторов (рис. 1, 15, 16), в газовых линиях, подходящих к ним, создаются застойные зоны, влияющие на качество записи хроматограммы. Разветвление газовой линии по ходу следования пробы приводит к уменьшению газовой пробы из-за ее частичного застаивания, последующему выходу пробы из зоны застоя и разбавлению ею газа-носителя после прохождения основного фронта пробы. Чтобы избежать указанных проблем, клапаны сброса были переустановлены до кранов-дозаторов 17, 18. Это позволило добиться стабильности горизонтальной базовой линии и снижения ширины пиков (рис. 2).
Рис. 2.
Влияние расположения клапана сброса для полуобратной продувки на форму пиков и базовой линии: 1 – до крана-дозатора, 2 – после крана-дозатора. Условия: пик аргона, 80°С, газ-носитель He, расход 14.6 мл/мин, колонка с молекулярными ситами 13Х (длина 5 м, внутренний диаметр 2 мм).
Выбор интервала времени от начала анализа до момента открытия клапана сброса для полуобратной продувки. Выше отмечено, что качество проб для колонок с молекулярными ситами обеспечивали две предколонки в совокупности системой полуобратной продувки. Эффективность работы предколонок зависит от правильности выбранного времени открытия клапанов сброса. Момент времени открытия клапана выбирали, исходя из величин площадей пика последнего целевого компонента, разделяемого в основной колонке, полученных в ходе последовательных анализов с разным временем открытия клапана от начала анализа. При раннем открытии клапана только часть этого компонента успевает выйти из предколонки и площадь пика занижена. При запаздывании открытия в основную колонку попадают компоненты, элюируемые сразу после последнего целевого компонента и негативно влияющие на способность колонки к разделению веществ.
Стабильность расхода газа-носителя через основные колонки в момент открытия клапанов сброса зависит от реакции регуляторов давления 19, 20 на возникающее изменение давления. Для уменьшения времени реакции регуляторов задавали постоянный минимальный перепад давления на них и, как следствие, минимальный расход так, чтобы в ходе анализа стабилизация давления происходила без заметных изменений базовой линии в основной колонке.
Выбор условий хроматографического анализа в режиме стационарной температуры термостата. Решали задачу выбора удовлетворительной стационарной температуры для колонок с молекулярными ситами 13Х и с Porapak T, которые традиционно работают в разных температурных диапазонах.
Для разделения диоксида углерода, ДМЭ, метанола и воды широко применяют адсорбенты HayeSep и Porapak при стационарной температуре около 100°С. Сложность выбора оптимального адсорбента для разделения этих веществ связана с возможным присутствием в пробе компонентов с близкими временами удерживания. Колонка с HayeSep Q (3 м × 2 мм), предложенная производителем для работы в режиме программированного нагрева, не удовлетворила требованиям по разделению пиков компонентов и временам их удерживания при стационарной температуре. Окончательный выбор сделан нами в пользу колонки с Porapak T (4 м × 2 мм) вместо колонки с HayeSep Q. Пример хроматограммы представлен на рис. 3. Нестабильность базовой линии после ввода пробы в течение первых 0.5 мин связана с различием давления в линии анализа и в линии газа-носителя. При использовании Porapak T времена удерживания ДМЭ (рис. 3) и н-бутана (рис. 4) совпадают, а пик метанола частично накладывается на пики пентана и изопентана (рис. 4).
Рис. 3.
Разделение CO2, ДМЭ, воды и метанола на колонке (4 м × 2 мм) с Porapak T при 140°С (расход газа-носителя гелия 30 мл/мин): 1 – H2 + O2 + N2 + CH4 + + CO; 2 – CO2 (1.19 об. %); 3 – ДМЭ (9.61 об. %); 4 – H2O (2.26 об. %); 5 – CH3OH (3.55 об. %).

Рис. 4.
Разделение смеси углеводородов с водой и метанолом на колонке (4 м × 2 мм) с Porapak T при 140°С (расход газа-носителя гелия 30 мл/мин): 1 – H2 + O2+ + N2 + CH4 + CO; 2 – CO2 + этан; 3 – пропан; 4 – н‑бутан (0.10 об. %); 5 – изобутан (0.107 об. %); 6 – вода; 7 – метанол; 8 – н-пентан (0.058 об. %); 9 – изопентан (0.052 об. %). (Содержание углеводородов приведено в пересчете на сухой газ.)

Качество разделения пиков в ходе выбора параметров разделения проверяли путем анализа многокомпонентной газовой смеси, насыщаемой водой и метанолом при барботировании через водно-метанольный раствор. Заводские колонки с молекулярными ситами 11, 12 (рис. 1) показали неудовлетворительное качество разделения пиков; их длина была увеличена нами самостоятельно: колонки 11 на 2 м, колонки 12 на 1 м.
Таким образом, заменив колонку с HayeSep Q, предложенную производителем для работы в режиме программированного нагрева, на колонку с Porapak T, увеличив длину колонoк молекулярными ситами 13Х, варьируя фиксированную температуру термостата, расходы газов-носителей, нам удалось подобрать удовлетворительную температуру разделения (140°С) и прочие условия для детектирования всех реагентов и продуктов прямого синтеза ДМЭ. Качество разделения удовлетворительно для всех компонентов в широком диапазоне концентраций, с пределами обнаружения 0.0005–0.01 об. %. Продолжительность анализа не превышает 12 мин.
Условия выполнения определения. Анализ проводили при фиксированной температуре термостата колонок 140°С. Температура детекторов – 200°С, кранов-дозаторов – 150°С.
Углекислый газ, ДМЭ, воду, метанол и углеводороды С1–С6 разделяли на колонке Porapak T (4 м × 2 мм), детектор ДТП-1. Газ-носитель гелий, расход 30 мл/мин, объем петли-дозатора 1 мл. Последовательно расположенный ПИД позволяет детектировать следовые количества углеводородов, а также этан в присутствии СО2. Азот, СО, СН4 (при необходимости, О2) разделяли на колонке с молекулярными ситами 13X (5 м × 2 мм) (газ-носитель гелий, расход 15 мл/мин, петля-дозатор объемом 1 мл) и определяли детектором ДТП-2 с пределом детектирования 0.0005 об. %. Предколонка HayeSep R (1 м × 2 мм). Избыточное давление перед основной колонкой 330 кПа. Расход газа-носителя по линии поддержания давления для реализации полуобратной продувки 0.8 мл/мин. Суммарный поток газа-носителя через колонку 15.8 мл/мин. Клапан полуобратной продувки открывается через 65 с после поворота крана дозатора. Расход через предколонку в обратном направлении 52 мл/мин.
Водород определяли на колонке с молекулярными ситами 13X с детектором ДТП-3 с пределом детектирования 0.0005 об. %, (газ-носитель аргон, расход 15 мл/мин, петля-дозатор 0.25 мл). Предколонка HayeSep R (1 м × 2 мм). Избыточное давление перед колонкой 335 кПа, расход газа-носителя по линии поддержания давления 1.4 мл/мин. Суммарный поток газа-носителя через колонку 16.4 мл/мин. Клапан полуобратной продувки открывается через 60 с после поворота крана дозатора. Расход через предколонку в обратном направлении 41 мл/мин.
Величины откликов ДТП-3 к N2, СН4 и CO с газом-носителем аргоном значительно меньше (рис. 5, пики 10, 11, 12), чем отклики ДТП-2 к тем же компонентам (в том же анализе) с газом-носителем гелием (рис. 5, пики 5, 6, 7), даже с учетом уменьшенной в четыре раза пробы на ДТП-3. Аналогичный вывод можно сделать, сравнивая пики 10, 11 с пиками 7, 8 на рис. 6.
Рис. 5.
Хроматограммы, регистрируемые детекторами ДТП-1 (сплошная тонкая линия), ДТП-2 (точечная линия) и ДТП-3 (сплошная толстая линия): 1 – N2 + CO; 2 – CO2; 3 – H2 (49.9 об. %); 4 – O2; 5 – N2; 6 – CH4 (0.51 об. %); 7 – CO (4.9 об. %); 8 – He; 9 – H2; 10 – N2; 11 – CH4; 12 – CO. Условия: температура термостата колонок 140°С, детекторов 200°С; расход газа-носителя гелия 30 мл/мин (ДТП-1) и 15.8 мл/мин (ДТП-2), аргона 16.4 мл/мин (ДТП-3).

Рис. 6.
Хроматограммы продуктов синтеза ДМЭ из синтез-газа на основе СО2 (а) или СО (б), полученные с ДТП-1 (сплошная толстая линия), ДТП-2 (точечная линия) и ДТП-3 (сплошная тонкая линия): 1 – N2 + CO; 2 – CO2; 3 – ДМЭ; 4 – H2O; 5 – CH3OH; 6 – H2, 7 – N2; 8 – CO; 9 – H2; 10 – N2; 11 – CO. Условия хроматографического анализа: температура термостата колонок 140°С, детекторов 200°С; расход газа-носителя гелия 30 мл/мин (ДТП-1) и 15.8 мл/мин (ДТП-2), аргона 16.4 мл/мин (ДТП-3). Состав синтез-газа (об. %): (а) – N2 2.1; CO2 26.9; H2 71; (б) – N2 5.3; CO 21.8; CO2 5.2; H2 67.7.

Количественное определение выполняли с применением поправочных коэффициентов, полученных методом абсолютной градуировки. Для этого использовали 32 варианта многокомпонентных градуировочных газовых смесей и индивидуальных газов, что обеспечило необходимый уровень точности определения абсолютных коэффициентов в требуемом диапазоне содержаний ряда веществ, которые могут быть использованы для решения задач, отличных от синтеза ДМЭ, но связанных с использованием синтез-газа.
Результаты типовых экспериментов прямого синтеза диметилового эфира. На рис. 6 в одном масштабе представлены хроматограммы реакционного газа в экспериментах с синтез-газом, богатым СО2 (а) и СО (б). Сумма экспериментально определенных концентраций компонентов, как правило, около 100 об. % (отклонение не более чем на 3–4%), что требует проведения процедуры нормировки. В табл. 1 в качестве примера приведены экспериментальные данные, полученные в процессе синтеза ДМЭ из синтез-газа состава, об. %: СО – 21.4, СО2 – 6.5, Н2 – 66.5, N2 – 5.6. Использовали бифункциональный катализатор CuO/ZnO/Al2O3 [18] (масса 1.6 г). Данные получены при давлении 3 МПа, температуре 260°С. Исходя из расхода синтез-газа на входе в реактор (VВХ) и отношения концентраций азота на входе и выходе из реактора (КN), можно рассчитать расход газа на выходе из реактора. Характерно, что отношение концентраций азота на входе и выходе из реактора практически совпадает с отношением суммарных концентраций углеродсодержащих компонентов на входе и выходе из реактора (КС), что говорит о корректности результатов хроматографических анализов.
Таблица 1.
Состав конвертированного газа (об. %) при синтезе диметилового эфира из синтез-газа
| VВХ, л/ч | СО2 | ДМЭ | Н2О | СН3ОН | N2 | CO | H2 | КC | КN |
|---|---|---|---|---|---|---|---|---|---|
| 9.8 | 8.7 | 1.97 | 0.70 | 2.5 | 6.2 | 16.5 | 63.5 | 0.88 | 0.90 |
| 14.8 | 7.8 | 1.22 | 0.61 | 2.1 | 6.0 | 18.3 | 64.0 | 0.91 | 0.92 |
| 19.7 | 7.3 | 0.85 | 0.58 | 1.8 | 5.9 | 19.2 | 64.4 | 0.93 | 0.94 |
| 24.6 | 7.0 | 0.59 | 0.56 | 1.6 | 5.8 | 19.9 | 64.6 | 0.94 | 0.95 |
Выражаем благодарность сотрудникам ЗАО СКБ “Хроматэк” К.В. Перепелкину, Е.И. Токтаеву, А.Г. Семенову, А.Г. Слепневу за плодотворное сотрудничество.
Работа выполнена в рамках Государственного задания ИНХС РАН.
Список литературы
Haro P., Trippe F., Stahl R., Henrich E. Bio-syngas to gasoline and olefins via DME – A comprehensive techno-economic assessment // Appl. Energy. 2013. V. 108. P. 54.
Fornell R., Berntsson T., Åsblad A. Techno-economic analysis of a kraft pulp-mill-based biorefinery producing both ethanol and dimethyl ether // Energy. 2013. V. 50. P. 83.
Bhattacharya S., Kabir K.B., Hein K. Dimethyl ether synthesis from Victorian brown coal through gasification - Current status, and research and development needs // Progr. Energy Combustion Sci. 2013. V. 39. P. 577.
Lv P., Chang J., Wang T., Fu Y., Chen Y., Zhu J. Hydrogen-Rich gas production from biomass catalytic gasification // Energ. Fuel. 2004. V. 18. P. 228.
Lv P., Chang J., Xiong Z., Huang H., Wu C., Chen Y., Zhu J. Biomass air–steam gasification in a fluidized bed to produce hydrogen-rich gas // Energ. Fuel. 2003. V. 17. P. 677.
Архаров А.М., Глухов С.Д., Грехов Л.В., Жердев А.А., Иващенко Н.А., Калинин Д.Н. Применение диметилового эфира в качестве топлива и хладоагента для городских автомобилей. // Ползуновский вестник. 2003. № 1–2. С. 9.
Розовский А.Я. Диметиловый эфир и бензин из природного газа // Рос. хим. журн. 2003. Т. 47. № 6. С. 53.
Ilias S., Bhan A. Mechanism of the catalytic conversion of methanol to hydrocarbons // ACS Catal. 2013. V. 3. P. 18.
Mokrani T., Scurrell M. Gas conversion to liquid fuels and chemicals: the methanol route-catalysis and processes development // Catal. Rev. 2009. V. 51. P. 1.
Хаджиев С.Н., Магомедова М.В., Пересыпкина Е.Г. Выбор схемного решения GTL-процесса для технологии ИНХС РАН // Нефтехимия. 2016. Т. 56. № 6. С. 567.
Маркова Н.А., Букина З.М., Ионин Д.А., Колесниченко Н.В., Хаджиев С.Н. Диметиловый эфир в процессе переработки попутного нефтяного газа в смесь синтетических углеводородов // Нефтехимия. 2016. Т. 56. № 6. С. 639.
García-Trenco A., Valencia S., Martínez A. The impact of zeolite pore structure on the catalytic behavior of CuZnAl/zeolite hybrid catalysts for the direct DME synthesis // Appl. Catal. A: Gen. 2013. V. 468. P. 102.
Dadgar F., Myrstad R., Pfeifer P., Holmen A., Venvik H.J. Direct dimethyl ether synthesis from synthesis gas: The influence of methanol dehydration on methanol synthesis reaction // Catal. Today. 2016. V. 270. P. 76.
Cai M., Palčić A., Subramanian V., Moldovan S., Ersen O., Valtchev V., Ordomsky V.V., Khodakov A.Y. Direct dimethyl ether synthesis from syngas on copper–zeolite hybrid catalysts with a wide range of zeolite particle sizes // J. Catal. 2016. V. 338. P. 227.
Asthana S., Samanta C., Bhaumik A., Banerjee B., Voolapalli R.K., Saha B. Direct synthesis of dimethyl ether from syngas over Cu-based catalysts: Enhanced selectivity in the presence of MgO // J. Catal. 2016. V. 334. P. 89.
Erenã J., Garoña R., Arandes J.M., Aguayo A.T., Bilbao J. Effect of operating conditions on the synthesis of dimethyl ether over a CuO–ZnO–Al2O3/NaHZSM-5 bifunctional catalyst // Catal. Today. 2005. V. 107–108. P. 467.
Le Valant A., Comminges C., Tisseraud C., Canaff C., Pinard L., Pouilloux Y. The Cu–ZnO synergy in methanol synthesis from CO2. Part 1: Origin of active site explained by experimental studies and a sphere contact quantification model on Cu + ZnO mechanical mixtures // J. Catal. 2015. V. 324. P. 41.
Кипнис М.А., Самохин П.В., Белостоцкий И.А., Туркова Т.В. Синтез диметилового эфира из синтез-газа на катализаторе Мегамакс 507/γ-Al2O3 // Катализ в промышленности. 2017. Т. 17. С. 442.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии