

Журнал аналитической химии, 2020, T. 75, № 10, стр. 931-941
Электроокисление сульфит-ионов на композитном углеродсодержащем электроде, модифицированном субмикронными частицами золота
С. В. Ковалева a, О. С. Аксиненко a, А. В. Коршунов b, *
a Томский государственный педагогический университет
634061 Томск, ул. Киевская, 60, Россия
b Московский государственный строительный университет
129337 Москва, Ярославское шоссе, 26, Россия
* E-mail: androkor@mail.ru
Поступила в редакцию 30.12.2019
После доработки 20.02.2020
Принята к публикации 20.02.2020
Аннотация
С использованием циклической вольтамперометрии изучены закономерности электроокисления сульфит-ионов в щелочной среде (рН ≥ 8; LiClO4, KNO3, Na2SO4, NaOH) на композитном углеродсодержащем электроде, модифицированном субмикронными (300 нм и менее) частицами золота. Обоснована электрокаталитическаяó природа аналитического сигнала, предложена схема протекания процесса окисления. Оптимизирована процедура предварительной электрохимической обработки модифицированного электрода, позволяющая повысить величину тока окисления субстрата и заключающаяся в анодной поляризации электрода в интервале потенциалов 0.9–1.4 В (электрод сравнения Ag/AgCl) в фоновом электролите. Сущность активирования заключается в увеличении доли поверхности частиц золота, принимающей участие в каталитическом цикле генерации/восстановления (гидр)оксидов золота и обеспечивающей повышение величины аналитического сигнала. Предложенный подход позволяет существенно повысить чувствительность вольтамперометрического метода определения сульфита. Концентрационная зависимость аналитического сигнала линейна в диапазоне концентраций ${\text{SO}}_{3}^{{2 - }}$-ионов 1 × 10–7–2 × 10–4 М (предел обнаружения 5.5 × 10–8 М). Результаты определения сульфита в винах, пиве и соках свидетельствуют о более высокой точности вольтамперометрического метода по сравнению со стандартной иодометрической методикой.
Основными источниками поступления неорганических соединений серы(IV) (диоксид, сернистая кислота и сульфиты) в организм человека являются техногенные выбросы (теплоэнергетика, нефтепереработка, металлургия, химическая промышленность, транспорт [1]) и продукты питания, в которые SO2 и сульфиты вводят в качестве консервантов Е220–Е228 (вина, пиво и безалкогольные напитки; мясные и рыбные полуфабрикаты; фасованные скоропортящиеся продукты; фрукты, овощи и продукты их переработки – до 3 г/кг [2]). Сульфиты относятся к третьему классу опасности [3]. При попадании в организм человека их действие проявляется в форме аллергических дерматитов, желудочно-кишечных расстройств, астматических приступов [4]. Всемирной организацией здравоохранения рекомендовано низкое значение суточного поступления сульфитов в организм человека (0.7 мг/кг массы тела в пересчете на SO2 [5]), в связи с чем содержание SO2 и сульфитов в продуктах питания на уровне 10 мг/кг и выше должно быть отражено в составе продуктов [6].
Принятые в химико-аналитической практике методики определения сульфитов в пищевых продуктах, в том числе внесенные в национальные стандарты, основаны на классическом методе Монье-Вильямса и включают отгонку SO2 в ходе кислотного разложения анализируемого объекта и последующее титриметрическое, кулонометрическое или фотометрическое определение продукта взаимодействия сернистого газа с поглотительными растворами [5, 7, 8]. В напитках свободный и связанный SO2 определяют йодометрическим титрованием [9]. Эти методы имеют много недостатков: неполное выделение и улавливание SO2; влияние тиосульфатов, политионатов, меркаптанов, тиоэфиров [7, 8, 10], а также высокие трудозатратность и материалоемкость. В связи с этим совершенствование известных и разработка новых методов определения SO2 и сульфитов в объектах окружающей среды и в продуктах питания – актуальная задача.
Применение электроаналитических методов для определения SO2 и сульфитов в продуктах питания позволит избежать стадии отгонки SO2, минимизировать число операций и существенно сократить продолжительность анализа [11]. Наибольшей селективностью и чувствительностью характеризуются методы с использованием электрохимических сенсоров с иммобилизованными ферментами (сульфитоксидаза [12, 13], полифенолоксидаза [14]) или бактериями [15], обеспечивающие предел обнаружения порядка 1 × 10–7 М ${\text{SO}}_{3}^{{2 - }}$. Внедрение таких сенсоров в аналитическую практику ограничено сложной процедурой их изготовления, необходимостью строгого контроля условий применения и хранения. Высокой чувствительностью характеризуются амперометрические и вольтамперометрические методы определения, основанные на электрокаталитическом окислении SO2 и сульфитов с использованием рабочих электродов, модифицированных различными медиаторами (неорганические и органические комплексы Cu, Fe, Со, Ni, производные антрахинона, порфирина и др. [16–26]). Определению сульфита мешают аскорбиновая кислота, полифенолы, сульфиды; не мешают определению тиосульфат, бромид, углеводы, гидроксикарбоновые кислоты [11, 16, 20, 26, 27]. Недостатками применения модифицированных электродов при анализе реальных объектов являются многостадийная и длительная процедура приготовления электродов, нестабильность медиаторов при хранении, блокирование активной поверхности загрязнителями. Анализ литературы показал, что достаточно простые по составу и процедуре приготовления электроды, модифицированные частицами благородных металлов с высокой каталитической активностью, для определения сульфита изучены недостаточно.
Цель настоящей работы – оптимизация условий вольтамперометрического определения сульфит-ионов с использованием композитных углеродсодержащих электродов, модифицированных субмикронными частицами золота.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Электроокисление сульфит-ионов изучали методом циклической вольтамперометрии (ЦВА) с линейной разверткой потенциала (вольтамперометрический анализатор TA-Lab, НПП “Томьаналит”, Томск, Россия). Использовали кварцевую трехэлектродную ячейку емк. 20 мл без разделения электродного пространства, вспомогательным электродом являлся платиновый проволочный электрод, электродом сравнения служил хлоридсеребряный электрод (х. с. э.) Ag/AgCl/1 M KCl. Все потенциалы в работе приведены относительно х. с. э. В качестве рабочего электрода использовали композитный углеродсодержащий электрод (КУЭ), состоящий из трубчатого полиэтиленового корпуса (d = 5 мм), заполненного при помощи литья под давлением смесью высокодисперсного углерода марки N220 с размером частиц 24–33 нм (30 мас. %) и термостабилизированного полиэтилена высокого давления (НПП “Томьаналит”, Томск, Россия). Рабочую поверхность электрода (∼12 мм2) обновляли путем срезания тонкого (0.1–0.3 мм) слоя гексанитовым резцом. Выбор электрода такого типа обусловлен простотой подготовки активной поверхности и высокой воспроизводимостью ее характеристик. Перед проведением измерений рабочую поверхность электрода промывали в конц. HNO3 и бидистиллированной воде, затем электрод подвергали многократной циклической поляризации в интервале потенциалов от –0.5 до +1.5 В в деаэрированном фоновом электролите 0.1 М растворе LiClO4 до получения стабильной вольт-амперной кривой. Поверхность электрода модифицировали частицами золота методом катодного осаждения в растворах HAu-Cl4 (готовили путем разбавления аттестованного раствора ГСО 8429-2003). Морфологию и состав поверхности электрода Au-КУЭ изучали с использованием растрового электронного микроскопа Philips SEM 515, оборудованного рентгеноспектральным микроанализатором EDAX ECON IV.
В работе использовали реактивы Na2SO3, KNO3, LiClO4, NaOH, Na2SO4, HNO3 х. ч. и ч. д. а. без дополнительной очистки. Исходные 0.1 М растворы готовили растворением точных навесок реагентов в бидистиллированной воде, рабочие растворы с меньшими концентрациями готовили разбавлением исходных. Растворы сульфита натрия готовили непосредственно перед измерениями растворением безводной соли в предварительно деаэрированной воде, фоновые электролиты перед смешиванием с раствором сульфита также деаэрировали пропусканием азота высокой чистоты. рН растворов корректировали добавлением растворов NaOH и HNO3, для измерения рН применяли рН-метр рН-410 “Аквилон” с комбинированным электродом.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Морфологическими характеристиками модифицированного рабочего электрода Au-КУЭ, определяющими параметры аналитического сигнала, являются дисперсность частиц золота и особенности их распределения по поверхности углеродсодержащей подложки. Предварительные эксперименты показали, что условиями формирования мелкодисперсных равномерно распределенных по поверхности катодных осадков золота являются потенциал осаждения Еэ = –0.1 В, концентрация раствора НАuCl4 2–3 мМ (на фоне 0.1 М HCl), перемешивание раствора, продолжительность осаждения 60–80 с. При указанных условиях золото осаждается в виде субмикронных частиц размером не более 300 нм с относительно равномерным распределением (1–2 частицы на 1 мкм2) по поверхности подложки (рис. 1а, 1б).
Рис. 1.
Микрофотографии поверхности модифицированного композитного углеродсодержащего электрода при продолжительности катодного осаждения частиц золота 40 (а), 60 (б), 80 (в), 100 с (г). с(НАuCl4) = 2.5 мМ, Еэ = –0.1 В. На вставке (а) – характеристический рентгеновский спектр поверхности электрода; на вставке (г) – участок поверхности агрегированных частиц золота.

Условия формирования активной поверхности Au-КУЭ проверяли по результатам измерения максимального тока (Iр при Ер = 0.4–0.5 В) окисления ${\text{SO}}_{3}^{{2 - }}$-ионов: наибольшая величина аналитического сигнала достигается в случае высокой поверхностной плотности единичных субмикронных и наночастиц золота, а не их агрегатов (рис. 2а). Использование более отрицательных потенциалов осаждения, более концентрированных растворов НАuCl4 и большей продолжительности осаждения приводит к формированию грубодисперсных неоднородных катодных осадков (рис. 1г). При использовании более разбавленных растворов, а также при малых временах осаждения площадь активной поверхности электрода существенно уменьшается.
Рис. 2.
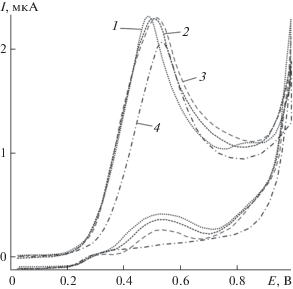
(а) Зависимость величины максимума тока анодного окисления (Iр при Ер = 0.52 В) сульфит-ионов в растворе 2 × 10–4 М Na2SO3 + 0.1 М KNO3 (рН 8) на модифицированном частицами золота композитном углеродсодержащем электроде (Au-КУЭ) от продолжительности предварительного катодного осаждения частиц золота (см. условия в подписи к рис. 1). (б) Циклические вольтамперограммы для различных электродов: 1 – КУЭ (5 × 10–5 М Na2SO3 + 0.1 М KNO3); 2 – Au-КУЭ (0.1 М KNO3); 3 – Au-КУЭ (5 × 10–5 М Na2SO3 + 0.1 М KNO3); 4 – Au-КУЭ (5 × 10–5 М Na2SO3 + 0.1 М KNO3) после предварительной анодной поляризации в 0.1 М растворе KNO3 в интервале потенциалов 0.9–1.4 В. Условия регистрации циклических вольтамперограмм: рН 8, Енач = 0.0 В, Екон = 0.8 В, V = 100 мВ/с.

Анодное окисление сульфита на немодифицированном КУЭ протекает в интервале потенциалов 0.6–0.8 В, максимум тока при низких концентрациях сульфит-ионов (менее 0.1 мМ) плохо выражен и растянут по шкале потенциалов (рис. 2б). В отличие от растворов с более высокой концентрацией ${\text{SO}}_{3}^{{2 - }}$-ионов, для которых характерна выраженная концентрационная зависимость аналитического сигнала [27, 28], использование углеродсодержащих электродов для определения сульфита на уровне микромолярных концентраций нецелесообразно.
На модифицированном Au-КУЭ ток окисления ${\text{SO}}_{3}^{{2 - }}$-ионов отображается в виде выраженного максимума, потенциал которого смещен на ∼0.2 В в область отрицательных потенциалов по сравнению с КУЭ и практически совпадает с потенциалом максимума тока окисления частиц золота (Ер = 0.52 В) в фоновом электролите (рис. 2б). Известно [29, 30], что значения потенциалов электроокисления субстрата на Au-электроде связаны с потенциалом образования метастабильных поверхностных (гидр)оксидов золота. Формирование окисленных слоев в условиях анодной поляризации золота начинается при сравнительно низких потенциалах 0.2–0.3 В за счет реструктуризации поверхностного слоя металла и внедрения атомов кислорода [30, 31]. Варьирование состава и сплошности этих слоев позволяет изменять условия электроокисления деполяризатора [32]. Следовательно, предварительное окисление частиц золота в составе Au-КУЭ в контролируемых условиях может стать одним из факторов регулирования эффективности электрокатализа и повышения величины аналитического сигнала при окислении сульфита. Очевидно, что развитая поверхность электрода в отношении возможности регулирования электрокаталитического эффекта имеет существенное преимущество перед макроэлектродами [33].
Из полученных данных следует, что предварительная анодная поляризация Au-КУЭ в фоновом электролите приводит к повышению тока окисления Iр сульфита (рис. 2б). Наибольший эффект наблюдали при предварительной анодной поляризации Au-КУЭ в растворе фонового электролита в интервале потенциалов 0.9–1.4 В в режиме линейной развертки потенциалов (v = 100 мВ/с). Активирование Au-КУЭ при более высоких потенциалах приводит к снижению величины Iр, что можно объяснить образованием пассивирующей пленки на поверхности частиц золота [34, 35]. Потенциалы Е < 0.0 В не использовали в работе вследствие возможного блокирования активной поверхности электрода продуктами глубокого восстановления сульфита [34, 35].
В различных фоновых электролитах (LiClO4, KNO3, Na2SO4, NaOH) электроокисление сульфита на предварительно активированном Au-КУЭ характеризуется наличием на вольтамперограммах одного выраженного максимума анодного тока в области потенциалов Ер = 0.45–0.55 В (рис. 3). Аналогичные значения Ер характерны для электроокисления сульфита на Au-макроэлектроде в среде 0.1 М H2SO4 [36]. В фоновых электролитах со слабо адсорбирующимися анионами (KNO3, LiClO4 [31]) значения Ер и Iр практически не зависят от типа аниона; на фоне Na2SO4Ер смещается в область положительных потенциалов, при этом величина аналитического сигнала Iр понижается (рис. 3).
Рис. 3.
Циклические вольтамперограммы для модифицированного частицами золота композитного углеродсодержащего электрода (предварительно активирован путем анодной поляризации в интервале потенциалов 0.9–1.4 В) в 1 × 10–4 М растворах Na2SO3 в 0.1 М фоновых электролитах: 1 – NaOH, 2 – LiClO4 (рН 8), 3 – KNO3 (рН 8), 4 – Na2SO4 (рН 8); условия регистрации ЦВА: Енач = 0.0 В, Екон = 1.0 В, v = = 100 мВ/с.
Количественные характеристики процесса электроокисления сульфита приведены в табл. 1. Степень возрастания каталитической составляющей тока Iкат по сравнению с током окисления медиатора (I0) оценивали по соотношению (Iкат – I0)/I0. Анализ значений Ер и Iр (табл. 1) показал, что предварительное активирование Au-КУЭ приводит к существенному повышению величины аналитического сигнала в разбавленных растворах сульфита.
Таблица 1.
Параметры анодного процесса на немодифицированном и модифицированном частицами золота композитном углеродсодержащем электроде в деаэрированных растворах по данным циклической вольтамперометрии (Енач = 0.0 В, Екон = 1.0 В, v = 100 мВ/с, рН 8)
| Электрод | Раствор | Ер, В | Iр, мкА | (Iкат – I0)/I0 |
|---|---|---|---|---|
| КУЭ | 0.1 M KNO3 | 0.68 | 0.08 | – |
| Au-КУЭ | 0.52 | 0.18 | ||
| Au-КУЭ активированный | 0.52 | 0.16 | ||
| КУЭ | 0.1 M KNO3 + 5 × 10–5 М Na2SO3 | 0.68 | 0.30 | |
| Au-КУЭ | 0.52 | 0.54 | 2.0 | |
| Au-КУЭ активированный | 0.48 | 1.33 | 7.3 |
Особенностью электроокисления сульфита на неактивированном Au-КУЭ в щелочной среде, в отличие от процесса на активированном электроде (рис. 3), является сигмоидная форма зависимости аналитического сигнала от рН Iр = f(рН) (рис. 4а): в интервале рН 8.0–9.5 величина Iр практически не изменяется, угловой коэффициент ∂Ер/∂рН → 0; при рН ≥ 10 Iр существенно возрастает (в 6–7 раз при рН 13).
Рис. 4.
(а) Зависимость величины Iр окисления сульфита в растворе 1 × 10–4 М Na2SO3 + 0.1 М KNO3 от рН на неактивированном модифицированном частицами золота композитном углеродсодержащем электроде по данным циклической вольтамперометрии (Енач = 0.0 В, Екон = 1.0 В, v = 100 мВ/с). (б) Расчетные зависимости логарифмов равновесных активностей ионно-молекулярных форм в системе Au3+–H2O от рН при Е = 0.7 В (отн. станд. водородного электрода) ([Au3+]0 = 1 × 10–5 М, 25°С) [37].
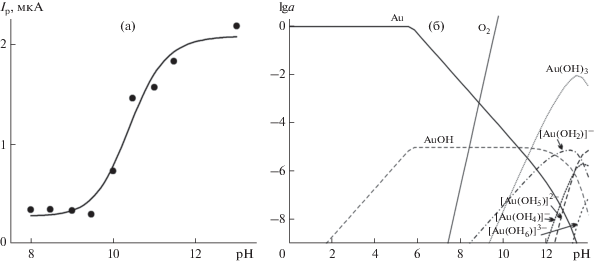
Наблюдаемый характер зависимости Iр = f(pH) может определяться как соотношением равновесных форм ${{{\text{HSO}}_{3}^{ - }} \mathord{\left/ {\vphantom {{{\text{HSO}}_{3}^{ - }} {{\text{SO}}_{3}^{{2 - }}}}} \right. \kern-0em} {{\text{SO}}_{3}^{{2 - }}}}$, так и состоянием частиц золота в условиях анодной поляризации в щелочной среде. Преобладающей формой S(IV) в растворах при рН > 8 являются ионы ${\text{SO}}_{3}^{{2 - }}$ (рКа2 = 7.2), при рН > 10 концентрация ионов ${\text{SO}}_{3}^{{2 - }}$ практически постоянна [34]. Следовательно, возрастание доли ${\text{SO}}_{3}^{{2 - }}$ с ростом рН не определяет форму зависимости Iр = f(pH) (рис. 4а). Расчет равновесных активностей компонентов системы Au3+–H2O с использованием программы [37] показал (рис. 4б), что в области потенциала максимума тока окисления ионов ${\text{SO}}_{3}^{{2 - }}$ (0.7 В отн. стандартного водородного электрода, ст. в. э.) при рН > 10 термодинамически более устойчивы окисленные формы золота (гидроксиды и гидроксокомплексы). В этой области рН также возможно анодное окисление воды с образованием кислорода (рис. 4б), но вследствие высокого перенапряжения этой реакции на золоте [30] влиянием кислорода на изучаемый процесс в рассматриваемой области потенциалов можно пренебречь.
Близость значений Iр окисления сульфита на неактивированном электроде при рН ≥ 12 (рис. 4а) и на активированном электроде при рН 8 (рис. 3) при одинаковых концентрациях ионов ${\text{SO}}_{3}^{{2 - }}$ свидетельствует об определяющем влиянии состояния частиц золота на величину аналитического сигнала. Следовательно, процедура предварительного активирования рабочего электрода позволяет повысить вклад каталитической составляющей аналитического сигнала за счет контролируемого формирования (гидр)оксидов в составе частиц золота. Поскольку величина Iр на активированном электроде практически не зависит от рН среды (рис. 3), сульфит можно определять в растворах при различных значениях в области рН ≥ 8 без существенного изменения чувствительности метода.
Характерной особенностью циклических вольтамперограмм ионов ${\text{SO}}_{3}^{{2 - }}$ является отсутствие на катодных участках ЦВА тока, соответствующего восстановлению продуктов окисления сульфита (рис. 2б, рис. 3). Напротив, в области потенциалов 0.4–0.5 В наблюдаются “обратные” максимумы анодного тока (рис. 5а). Даже в разбавленных 1 × 10–7– 1 × 10–6 М растворах ${\text{SO}}_{3}^{{2 - }}$ токи на катодных участках в области 0.2–0.6 В соответствуют восстановлению оксидов золота. Очевидно, что продукты анодного окисления сульфита на Au-электроде (${{{\text{S}}}_{2}}{\text{O}}_{6}^{{2 - }}$ и ${\text{SO}}_{4}^{{2 - }}$ [38]) не участвуют в катодном процессе. Такие “обратные” анодные токи зафиксированы в ряде работ по изучению механизма окисления SO2 и сульфитов на Pt- и Au-электродах [34, 39, 40]. В работе [39] эффект появления анодного пика при катодной развертке потенциалов объяснен депассивацией электрода, который пассивируется при анодной поляризации до высоких положительных потенциалов 1.75 В (ст. в. э.). Вместе с тем, в настоящей работе такие “обратные” анодные максимумы присутствуют на ЦВА после предшествующей анодной поляризации до сравнительно низких потенциалов 0.8 В (рис. 2б), при которых полная пассивация частиц золота не достигается.
Рис. 5.
(а) Циклические вольтамперограммы для модифицированного частицами золота композитного углеродсодержащего электрода, предварительно активированного путем анодной поляризации в интервале потенциалов 0.9–1.4 В, в растворе 1.5 × 10–4 М Na2SO3 + 0.1 М KNO3 (рН 8) от скорости развертки потенциалов V; 1 – 40, 2 – 60, 3 – 100, 4 – 140, 5 – 160 мВ/с. (б) Зависимости величин Ip и Ip/v½ от v½ по данным циклических вольтамперограмм рис. 5а; Енач = = 0.0 В, Екон = 0.9 В.

Учитывая влияние процедуры предварительного активирования Au-КУЭ на величину Iр окисления сульфита, более вероятной причиной появления анодного тока при катодной развертке потенциалов является протекание каталитической реакции окисления сульфита (гидр)оксидами золота при том же значении Ер. Такие токи подтверждают наличие каталитического цикла, включающего генерацию медиатора и его взаимодействие с субстратом при близких потенциалах, независимо от направления развертки потенциала. Показана возможность использования таких “обратных” максимумов тока в аналитических целях [41].
Ряд признаков (линейные зависимости Ер = = f(lg v) и Ip = f(v½), значение углового коэффициента ∂ lg Ip/∂ lg v = 0.34 [42]) указывает на то, что процесс электроокисления ${\text{SO}}_{3}^{{2 - }}$-ионов на активированном электроде Au-КУЭ является необратимым со смешанным контролем диффузии субстрата и переноса электрона в области Ер и не осложнен адсорбцией (рис. 5). Угловой коэффициент тафелевского участка вольтамперограмм ba = ∂Е/∂ lg I для токов I < 0.1Ip равен 0.23, что в случае одноэлектронного процесса (na = 1) соответствует низкому значению кажущегося коэффициента переноса αа ≈ 0.25. Следует отметить, что опубликованные данные касаются в основном механизма окисления SO2 на Au-электроде в кислой среде [34, 36, 39], количественные характеристики электродного процесса в щелочной среде не описаны. Низкие значения α < 0.5 подтверждают каталитическую природу анодного процесса [42] и характеризуют его протекание на наноструктурированных участках активной поверхности электрода с неоднородным распределением тока [43], что согласуется с особенностями строения частиц золота (рис. 1г). Отрицательный угловой коэффициент зависимости Iр/v½ = f(v) (рис. 5б) свидетельствует о протекании стадии химической реакции, следующей за стадией переноса электрона [42]. Порядок реакции по ионам ${\text{SO}}_{3}^{{2 - }}$ r = ∂ lg I/∂ lg c в интервале концентраций сульфита 1 × 10–5–1 × 10–4 М составляет 0.5–0.7, что свидетельствует об участии сульфит-ионов, помимо стадии разряда, в побочной химической реакции, вклад которой в кинетику процесса увеличивается при снижении концентрации сульфита.
С учетом полученных формально-кинетических данных, характеризующих процесс электроокисления сульфита в щелочной среде на активированном Au-КУЭ, а также данных о составе продуктов окисления сульфита [38, 44] и составе поверхностных окисленных слоев на Au-электроде [29–32], электродный процесс можно представить в виде следующей последовательности реакций:
(1)
$\begin{gathered} \underline {{\text{электродная реакция}}} \,{\text{:}} \\ {\text{SO}}_{3}^{{2 - }} \to {\text{SO}}_{3}^{--} + {{{\text{e}}}^{--}}\left( {{\text{медленная}}} \right), \\ \end{gathered} $(2)
$\begin{gathered} \underline {{\text{химическая}}\,\,{\text{реакция}}} {\kern 1pt} : \\ 2{\text{SO}}_{3}^{--} \to {{{\text{S}}}_{2}}{\text{O}}_{6}^{{2--}}\left( {{\text{быстрая}}} \right), \\ \end{gathered} $(3)
$\begin{gathered} \underline {{\text{генерация}}\,\,{\text{медиатора}}} \,:\,\, \\ {\text{Au}} + x{\text{O}}{{{\text{H}}}^{--}} \to {\text{Au}}{{({\text{OH}})}_{x}} + x{{{\text{e}}}^{--}}\left( {{\text{быстрая}}} \right), \\ \end{gathered} $(4)
$\begin{gathered} \underline {{\text{химическая реакция}}} \,:x{\text{SO}}_{3}^{{2--}} + 2{\text{Au}}{{({\text{OН}})}_{x}} \to \\ \to 2{\text{Au}} + x{\text{SO}}_{4}^{{2--}} + х{{{\text{Н}}}_{2}}{\text{О }}\left( {{\text{быстрая}}} \right). \\ \end{gathered} $При низких концентрациях ${\text{SO}}_{3}^{{2 - }}$ вклад каталитических процессов (3), (4) существенно повышается, что объясняет целесообразность использования процедуры активирования электрода при анализе разбавленных растворов сульфита.
Полученные результаты позволили обосновать условия регистрации аналитического сигнала окисления сульфита методом ЦВА: деаэрированный пропусканием азота фоновый электролит – 0.05–0.1 М растворы KNO3, LiClO4 или NaOH; рН ≥ 8; предварительное активирование Au-КУЭ в фоновом электролите путем линейной анодной поляризации в интервале потенциалов 0.9–1.4 В в течение 5 с; скорость развертки потенциалов 100 мВ/с. Эти условия использовали при получении концентрационной зависимости величины анодного тока Iр = f[с(${\text{SO}}_{3}^{{2 - }}$)], которая является линейной в двух интервалах концентраций (табл. 2). По диапазону определяемых концентраций ${\text{SO}}_{3}^{{2 - }}$ предлагаемый метод не уступает наиболее чувствительным вольтамперометрическим методам определения сульфита с использованием ферментных сенсоров [12–14].
Таблица 2.
Параметры линейного уравнения, аппроксимирующего зависимость Iр от концентрации ионов ${\text{SO}}_{3}^{{2 - }}$ по данным циклической вольтамперометрии с модифицированным частицами золота композитным углеродсодержащем электродом, предварительно активированным анодной поляризацией в интервале потенциалов 0.9–1.4 В (фон 0.1 М KNO3, Енач = 0.0 В, Екон = 1.0 В, v = 100 мВ/с; P = 0.95; для 10 измерений t(0.95;8) = 2.3)
| Диапазон c(${\text{SO}}_{3}^{{2 - }}$), М | Iр (нА) = a + bc (М) | Коэффициент корреляции R | |
|---|---|---|---|
| a | b | ||
| 1 × 10–7–1 × 10–6 | 201.95 ± 7.36 | (3.0 ± 0.1) × 108 | 0.9994 |
| 1 × 10–6–2 × 10–4 | 327.29 ± 34.80 | (2.00 ± 0.08) × 107 | 0.9977 |
Примечание: предел обнаружения 5.5 × 10–8 М ${\text{SO}}_{3}^{{2 - }}$; t(0.99;8) = 3.36; nj = 2 [45].
Стабильность Au-КУЭ при непрерывной работе без обновления модифицированной поверхности изучена путем 100-кратного повторения процедуры регистрации аналитического сигнала при указанных выше условиях в 1 × 10–4 М растворе сульфита. Относительная погрешность определения концентрации сульфита не превышала 3–5%. При большем числе измерений наблюдалось понижение величины аналитического сигнала, связанное, по-видимому, с деградацией структуры частиц золота в условиях многократного циклического наложения потенциалов.
На практике одним из основных источников погрешности при анализе объектов с низким содержанием сульфитов является кислород воздуха, оказывающий мешающее влияние на всех стадиях анализа от пробоподготовки до измерения аналитического сигнала. Вместе с тем, в подавляющем большинстве работ этим влиянием пренебрегают, ограничиваясь указанием о необходимости использования свежеприготовленных растворов. Влияние условий приготовления и хранения сульфитсодержащих растворов на величину сигнала Au-КУЭ изучали следующим образом. Приготовили серию разбавленных 1 × 10–5–1 × 10–4 М растворов Na2SO3 в фоновом электролите 0.1 М KNO3 (без предварительного деаэрирования), которые хранили в условиях естественной аэрации. В течение 3–5 суток с интервалом 12 ч в этих растворах определяли сульфит по принятому методу. На рис. 6 приведены типичные зависимости Iр–t, из которых следует, что в течение первых суток после приготовления раствора концентрация сульфита в нем снижается в среднем на 75% от первоначальной, т.е. на ∼3% в течение каждого часа. Через 48 ч содержание сульфит-ионов в исследуемых растворах снизилось до уровня предела обнаружения. Таким образом, все операции с сульфитсодержащими растворами, включая их приготовление, хранение и анализ, необходимо проводить с использованием деаэрированной воды в атмосфере инертного газа.
Рис. 6.
Зависимости величины аналитического сигнала Iр окисления сульфита на модифицированном частицами золота композитном углеродсодержащем электроде, предварительно активированном анодной поляризацией в интервале 0.9–1.4 В, в 1 × 10–4 М растворе Na2SO3 (1) и 5 × 10–5 М растворе Na2SO3 (2) (фон 0.1 М раствор KNO3 без предварительного деаэрирования, рН 8) от времени их хранения в условиях естественной аэрации по данным циклической вольтамперометрии. Енач = 0.0 В, Екон = 1.0 В, v = 100 мВ/с.

Полученные в работе результаты использовали для определения сульфита в винах, соках, пиве (табл. 3). Пробоподготовка заключалась во введении 1–2 мл анализируемого объекта в мерную колбу емк. 25 мл, добавлении 2.5 мл 1 М раствора NaOH и доведении общего объема раствора до метки деаэрированной дистиллированной водой. Выбор щелочного фона обусловлен необходимостью выделения связанного сульфита из органических производных, содержащихся в объектах исследования [9]. Полученный раствор переносили в электрохимическую ячейку и в течение 15 мин пропускали через него азот. Затем регистрировали вольтамперограмму на активированном Au-КУЭ в интервале потенциалов 0.1–1.0 В и измеряли величину анодного тока при Е 0.45–0.55 В. Для определения концентрации сульфита в анализируемый раствор вводили добавки 1 × 10–3 М раствора Na2SO3 при помощи микродозатора и после каждой добавки регистрировали вольтамперограмму. Использование метода добавок позволило минимизировать влияние состава анализируемого многокомпонентного объекта на величину аналитического сигнала. Для контроля точности вольтамперометрического метода применяли стандартную иодометрическую методику определения сульфита в напитках [9].
Таблица 3.
Результаты (мг/л) определения сульфита (в расчете на SO2 [2, 9]) в растворах и напитках (n = 10; t(0.95; 9) = 2.26 [45])
| Объект | Настоящая работа | Стандартная методика [9] | |||
|---|---|---|---|---|---|
| общий SO2 | sr | свободный SO2 | общий SO2 | sr | |
| 1 × 10–4 М раствор Na2SO3 (6.4 мг/л SO2) | 6.4 ± 0.3 | 0.12 | 6.3 ± 0.7 | 6.3 ± 0.8 | 0.34 |
| Вино белое сухое (тетрапак, РФ) | 112 ± 1 | 0.55 | 4 ± 1 | 111 ± 2 | 0.95 |
| Вино белое сухое (стеклянная бутылка, РФ) | 102 ± 1 | 0.54 | 40 ± 2 | 102 ± 2 | 0.86 |
| Пиво светлое пастеризованное (алюминиевая банка, РФ) | 36.5 ± 1.5 | 0.68 | 7.7 ± 2.5 | 35 ± 3 | 1.51 |
| Пиво светлое непастеризованное фильтрованное (стеклянная бутылка, РФ) | 31 ± 1 | 0.57 | – | 29 ± 4 | 1.73 |
| Сок яблочный прямого отжима (тетрапак, РФ) | 18 ± 1 | 0.53 | – | 16 ± 2 | 0.89 |
| Сок яблочный восстановленный (тетрапак, РФ) | 15 ± 1 | 0.52 | – | 13 ± 2 | 0.96 |
Из результатов анализа (табл. 3) следует, что данные вольтамперометрического метода характеризуются высокой точностью (раствор Na2SO3), высокой сходимостью результатов и низкими значениями относительных стандартных отклонений при анализе реальных объектов. Повышение качества измерений по сравнению с контрольной методикой достигается за счет использования щелочной среды (предотвращение эмиссии SO2 в ходе анализа; повышение полноты выделения связанного сульфита за счет гидролитического разложения органических производных; повышение селективности вследствие смещения потенциалов окисления мешающих компонентов – аскорбата и производных других оксикислот, спиртов – в направлении отрицательных потенциалов); деаэрирования растворов; существенного уменьшения используемых объемов анализируемого объекта, числа операций и продолжительности анализа (для получения одного результата в соответствии со стандартной методикой необходимо четырехкратное повторение титрования аликвоты 50 мл). Таким образом, вольтамперометрический метод с Au-КУЭ позволяет определять сульфит в объектах сложного состава с большей точностью и при существенно меньших материалоемкости и трудозатратности, чем стандартные методики.
Предложенный метод может быть использован для усовершенствования существующих и разработки новых методик определения сульфита в объектах окружающей среды, технологических средах и продуктах питания.
Список литературы
Vallero D. Fundamentals of Air Pollution. Elsevier, Academic Press, 2014. 996 p.
Технический регламент Таможенного союза (ТР ТС 029/2012) “Требования безопасности пищевых добавок, ароматизаторов и технологических вспомогательных средств” (официальный сайт Росстандарта http://old.gost.ru/wps/portal/pages/main) (27.12.2019 г.).
ГОСТ 5644-75 Межгосударственный стандарт. Сульфит натрия безводный. Технические условия (с Изменениями № 1, 2, 3, 4). М.: Издательство стандартов, 2004. 13 с.
Sulfur dioxide and sulfites (addendum): Fifty-first meeting of the Joint FAO/WHO Expert Committee on Food Additives. WHO food additives series No. 42. Geneva: World Health Organization, 1999.
Evaluation of certain food additives: Sixty-ninth report of the Joint FAO/WHO Expert Committee on Food Additives. WHO Technical Report Series No. 952. Geneva: World Health Organization, 2009. 209 p.
Codex Alimentarius Commission. General Standard for the Labelling of Prepackaged Foods, 1991 (CODEX STAN 1-1985, Rev. 1-1991; http://www.codexalimentarius.net/download/standards/32/CXS_001e.pdf) (18.11.2019 г.).
AOAC 990.28 Sulfites in Foods, Optimized Monier-Williams Method. AOAC Official Methods of Analysis. Sec. 47.3.43, 2000.
ГОСТ 25555.5-2014. Продукты переработки фруктов и овощей. Методы определения диоксида серы. М.: Издательство стандартов, 2014. 24 с.
ГОСТ 32115-2013 Продукция алкогольная и сырье для ее производства. Метод определения массовой концентрации свободного и общего диоксида серы. М.: Стандартинформ, 2014. 6 с.
Шендерюк В.В., Сташко А.В., Бычковская А.А. Сравнительная оценка методик определения сульфитов в пищевой продукции, в том числе из водных биологических ресурсов // Труды АтлантНИРО. 2017. Новая серия. Т. 1. № 2. С. 32.
Isaac A., Livingstone C., Wain A.J., Compton R.G., Davis J. Electroanalyical methods for the determination of sulfite in food and beverages // Trends Anal. Chem. 2006. V. 25. № 6. P. 589.
Rawal R., Pundir C.S. Development of electrochemical sulfite biosensor based on SOX/PBNPs/PPY modified Au electrode // Biochem. Eng. J. 2013. V. 71. P. 30.
Molinero-Abad B., Alonso-Lomillo M., Domínguez-Renedo O., Arcos-Martínez M.J. Sulfite oxidase biosensors based on tetrathiafulvalene modified screen-printed carbon electrodes for sulfite determination in wine // Anal. Chim. Acta. 2014. V. 812. P. 41.
Sartori E.R., Vicentini F.C., Fatibello-Filho O. Indirect determination of sulfite using a polyphenol oxidase biosensor based on a glassy carbon electrode modified with multi-walled carbon nanotubes and gold nanoparticles within a poly(allylamine hydrochloride) film // Talanta. 2011. V. 87. P. 235.
Suzuki M., Lee S., Fujii K., Arikawa Y., Kubo I., Kanagawa T., Mikami E., Karube I. Determination of sulfite ion by using microbial sensor // Anal. Lett. 1992. V. 25. № 6. P. 973.
Pournaghi-Azar M.H., Hydarpour M., Dastangoo H. Voltammetric and amperometric determination of sulfite using an aluminum electrode modified by nickel pentacyanonitrosylferrate film. Application to the analysis of some real samples // Anal. Chim. Acta. 2003. V. 497. P. 133.
Lowinsohn D., Alipazaga M.V., Coichev N., Bertotti M. Indirect FIA amperometric determination of sulphite based on the autocatalytic generation of Cu3+ complexes // Microchim. Acta. 2004. V. 144. P. 57.
Raoof J.B., Ojani B., Karimi-Maleh H. Electrocatalytic determination of sulfite at the surface of a new ferrocene derivative-modified carbon paste electrode // Int. J. Electrochem. Sci. 2007. V. 2. P. 257.
Ardakani M.M., Habibollahi F., Zare H. R., Naeimi H. Electrocatalytic oxidation of sulfite by quinizarine at carbon paste electrode // Int. J. Electrochem. Sci. 2008. V. 3. P. 1236.
Dadamos T., Teixeira M. Electrochemical sensor for sulfite determination based on a nanostructured copper-salen film modified electrode // Electrochim. Acta. 2009. V. 54. P. 4552.
Alamo L.S., Tangkuaram T., Satienperakul S. Determination of sulfite by pervaporation-flow injection with amperometric detection using copper hexacyanoferrate-carbon nanotube modified carbon paste electrode // Talanta. 2010. V. 81. P. 1793.
Ensafi A.A., Karimi-Maleh H. Ferrocenedicarboxylic acid modified multiwall carbon nanotubes paste electrode for voltammetric determination of sulfite // Int. J. Electrochem. Sci. 2010. V. 5. P. 392.
Majidi M.R., Asadpour-Zeynali K., Shahmoradi K., Shivaeefar Y. Electrochemical characteristics of a copper hexacyanoferrate (CuHCNF) modified composite carbon electrode and its application toward sulfite oxidation // J. Chin. Chem. Soc. 2010. V. 57. P. 391.
Vélez J.H., Muena J.P., Aguirre M.J., Ramírez G., Herrera F. Electrochemical oxidation of sulfite in aqueous solution by glassy carbon electrode modified with polymeric Co(II) meso-tetrakis (2-thienyl)porphyrin // Int. J. Electrochem. Sci. 2012. V. 7. P. 3167.
Amatatongchai M., Sroysee W., Chairam S., Nacapricha D. Simple flow injection for determination of sulfite by amperometric detection using glassy carbon electrode modified with carbon nanotubes–PDDA–gold nanoparticles // Talanta. 2015. V. 133. P. 134.
Beitollahi H., Mahmoudi-Moghaddam H., Tajik S., Jahani S. A modified screen printed electrode based on La3+-doped Co3O4 nanocubes for determination of sulfite in real samples // Microchem. J. 2019. V. 147. P. 590.
Makhotkina O., Kilmartin P.A. The use of cyclic voltammetry for wine analysis: determination of polyphenols and free sulfur dioxide // Anal. Chim. Acta. 2010. V. 668. P. 155.
Zelinsky A.G., Pirogov B.Ya. Electrochemical oxidation of sulfite and sulfur dioxide at a renewable graphite electrode // Electrochim. Acta. 2017. V. 231. P. 371.
Burke L.D., Ryan T.G. The role of incipient hydrous oxides in the oxidation of glucose and some of its derivatives in aqueous media // Electrochim. Acta. 1992. V. 37. P. 1363.
Burke L.D., Nugent P.F. The electrochemistry of gold. I. The redox behaviour of the metal in aqueous media // Gold Bull. 1997. V. 30. P. 43.
Orlik M., Galus Z. Electrochemistry of Gold // Inorganic Electrochemistry. Encyclopedia of Electrochemistry. V. 7b. / Eds. Bard A.J., Stratman M., Scholz F., Pickett C.J. Weinheim: Wiley-VCH, 2006. P. 839.
Burke L.D., Hurley L.M. An investigation of the electrochemical responses of superactivated gold electrodes in alkaline solution // J. Solid State Electrochem. 2002. V. 6. P. 101.
Manikandan V.S., Liu Z., Chen A. Simultaneous detection of hydrazine, sulfite, and nitrite based on a nanoporous gold microelectrode // J. Electroanal. Chem. 2018. V. 819. P. 524.
O’Brien J.A., Hinkley J.T., Donne S.W., Lindquist S.-E. The electrochemical oxidation of aqueous sulfur dioxide: A critical review of work with respect to the hybrid sulfur cycle // Electrochim. Acta. 2010. V. 55. P. 573.
Zelinsky A.G. Features of sulfite oxidation on gold anode // Electrochim. Acta. 2016. V. 188. P. 727.
Cai X., Lin C., Foord J.S., Compton R.G. The electrochemical oxidation of sulphite on gold electrodes // Electroanalysis. 2019. V. 31. P. 1783.
Felmy A.R., Girvin D., Jenne E.A. MINTEQ: Calculating Aqueous Geochemical Equilibria. Washington: US Environmental Protection Agency, 1984. 98 p.
Glasstone S., Hickling A. Studies in electrolytic oxidation. Part III. The formation of dithionate by the electrolytic oxidation of potassium sulphite // J. Chem. Soc. 1933. P. 829.
Seo E.T., Sawyer D.T. Electrochemical oxidation of dissolved sulphur dioxide at platinum and gold electrodes // Electrochim. Acta. 1965. V. 10. P. 239.
Quijada C., Morallon E., Vazquez J.L., Berlouis L.E.A. Electrochemical behaviour of aqueous SO2 at polycrystalline gold electrodes in acidic media. A voltammetric and in-situ vibrational study. Part II. Oxidation of SO2 on bare and sulphur-modified electrodes // Electrochim. Acta. 2000. V. 46. P. 651.
Perevezentseva D.O., Korshunov A.V., Gorchakov E.V., Bimatov V.I., Phedorov I.E. Au-nanoparticles based sensors for voltammetric determination of glutathione // Curr. Anal. Chem. 2017. V. 13. P. 225.
Pletcher D., Greff R., Peat R., Peter L.M., Robinson J. Instrumental Methods in Electrochemistry. Oxford: Woodhead Publishing, 2001. 442 p.
Soderberg J.N., Co A.C., Sirk A.H.C., Birss V.I. Impact of porous electrode properties on the electrochemical transfer coefficient // J. Phys. Chem. B. 2006. V. 110. P. 10 401.
Neta P., Huie R.E. Free-radical chemistry of sulfite // Environ. Health Perspec. 1985. V. 64. P. 209.
Doerffel K. Statistik in der Analytischen Chemie. Leipzig: Deutscher Verlag für Grundstoffindustrie, 1990. 256 s.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии