

Журнал аналитической химии, 2020, T. 75, № 11, стр. 980-996
Повышение эффективности разделения смесей веществ методами жидкостно-жидкостной хроматографии
А. Е. Костанян *
Институт общей и неорганической химии им. Н.С. Курнакова Российской академии наук
119991 Москва, Ленинский просп., 31, Россия
* E-mail: kost@igic.ras.ru
Поступила в редакцию 10.12.2019
После доработки 27.02.2020
Принята к публикации 27.04.2020
Аннотация
Статья посвящена краткому обзору методов жидкостно-жидкостной хроматографии, таких как циклическая с дискретным движением подвижной фазы, рециркуляционная, противоточная, рециркуляционно-противоточная, элюентно-экструзивная и стационарная. Рассмотрены также методы препаративного разделения, методы выделения концентрированных фракций целевых компонентов из смесей и вопросы аппаратурного оформления процессов жидкостно-жидкостной хроматографии. Более подробно освещены работы в области математического моделирования процессов разделения методами жидкостно-жидкостной хроматографии, выполненные автором с сотрудниками в ИОНХ РАН им. Н.С. Курнакова. Показано, что методы жидкостно-жидкостной хроматографии позволяют многократно повысить эффективность и производительность процессов разделения.
В процессах жидкостно-жидкостной хроматографии, в отличие от классической жидкостной хроматографии, нет твердого носителя неподвижной фазы: обе фазы находятся в свободном жидком состоянии, и так называемая неподвижная фаза обладает подвижностью в пределах аппарата. Перераспределение компонентов пробы происходит между двумя жидкими фазами, одна из которых удерживается в колонке с помощью центробежных сил, а вторая перемещается относительно первой, контактируя с ней и увлекая с собой разделяемую смесь. В отечественной литературе [1–6] процессы жидкостно-жидкостной хроматографии получили название жидкостной хроматографии со свободной неподвижной фазой (ЖХСНФ), в зарубежных публикациях используются названия противоточная хроматография (counter-current chromatography) и центробежная распределительная хроматография (centrifugal partition chromatography) [7–21]. Преимуществами ЖХСНФ по сравнению с обычной жидкостной хроматографией являются большая производительность, широкий выбор и легкость замены экстракционных систем, отсутствие загрязнения и потерь, вызванных взаимодействием с сорбентом.
В ЖХСНФ, как и в жидкостной экстракции, разделение смеси обусловлено различной растворимостью компонентов в двух жидких фазах. Эффективность работы хроматографической колонки, как и экстракционной колонны, определяется скоростью массопередачи (межфазного массообмена) и степенью осевого (продольного) перемешивания в обеих фазах. В отличие от противоточной экстракции, в ЖХСНФ процессы переноса вещества не стационарны, что позволяет в одной технологической операции разделить большое число компонентов. Вклад продольного перемешивания и массопередачи в эффективность процесса разделения в хроматографической колонке, оцениваемую числом теоретических тарелок (равновесных ступеней) N, определяется зависимостью [22]
(1)
${{N}_{{{\text{эф}}}}} = \frac{{nT{{{(1 + K{\kern 1pt} ')}}^{2}}}}{{T{{{(1 + K{\kern 1pt} ')}}^{2}} + 2n{{K}^{{'2}}}}}$Вклад продольного перемешивания в обеих фазах в общий эффект рассеяния (удерживания) пробы в колонке определяется зависимостью [23, 24]:
(2)
$n = \frac{{{{FL} \mathord{\left/ {\vphantom {{FL} {{{V}_{c}}}}} \right. \kern-0em} {{{V}_{c}}}}}}{{2(1 - {{S}_{f}}){{D}_{m}} + 2{{K}_{D}}{{S}_{f}}{{D}_{S}}}},$Формулы (1) и (2) показывают зависимость эффективности хроматографического разделения от термодинамических (коэффициенты распределения), гидродинамических (коэффициенты продольного перемешивания в фазах, фактор удерживания) и кинетических (число единиц переноса) параметров процесса.
В отличие от жидкостной хроматографии с твердым носителем, жидкостно-жидкостная хроматография с точки зрения математического описания является более простым процессом, что во многих случаях позволяет находить аналитическое решение уравнений математической модели при линейной зависимости равновесных концентраций в фазах. В работе [23] на основе неравновесной модели хроматографии получена довольно сложная теоретическая зависимость, описывающая распределение времени удерживания компонента с коэффициентом распределения KD в хроматографической колонке с учетом обоих механизмов (межфазного массообмена и продольного перемешивания). Когда скорость массопередачи велика $\left( {T \to \infty } \right),$ и достигается мгновенное равновесие между фазами – случай равновесной хроматографии, функция распределения упрощается и имеет следующий вид:
(3)
$X = \frac{x}{{\bar {x}}} = \frac{{{{N}^{N}}{{a}^{N}}}}{{(N - 1)!}}{{t}^{{N - 1}}}\exp ( - aNt),$Сопоставлением функций распределения неравновесной модели, рассчитанных по сложной зависимости, и функций распределения, рассчитанных по зависимости (3), в которой число теоретических тарелок N заменено на Nэф, установлено, что, когда Nэф ≥ 30, точный и упрощенный расчеты дают практически одинаковые результаты [22]. Это позволяет при анализе процессов ЖХСНФ исходить из более простой равновесной модели, имея в виду, что при этом используется эффективное число теоретических тарелок, определяемое по формулам (1), (2).
Рассматриваемые ниже методы жидкостно-жидкостной хроматографии позволяют существенно повысить чистоту разделяемых веществ и производительность хроматографических установок. Для их практической реализации и создания соответствующих хроматографических установок необходимо теоретическое описание этих методов. В настоящем обзоре более подробно рассматриваются работы в области математического моделирования процессов разделения методами жидкостно-жидкостной хроматографии, выполненные автором с сотрудниками в ИОНХ РАН им. Н.С. Курнакова.
Циклическая хроматография. В хроматографии часто используется элюентный метод разделения веществ, когда проба вводится в виде импульса в подвижную фазу на входе в колонку, а на выходе фиксируются концентрации компонентов в виде разделенных во времени концентрационных пиков. Нами предложен метод элюентной колоночной хроматографии, отличительной особенностью которого является дискретное (циклическое) движение подвижной фазы в колонке [25, 26]. Для реализации такого процесса необходимо на линии подачи подвижной фазы в колонку установить регулирующий клапан, на который подается сигнал цикличности.
Известно [27–31], что циклический режим движения потоков фаз приводит к увеличению эффективности противоточных массообменных процессов. Переход к циклическому режиму позволяет проводить процессы разделения при более высоких параметрах массообмена и более низких параметрах продольного перемешивания, что существенно повышает их эффективность.
Замена режима движения подвижной фазы через хроматографическую колонку с постоянной скоростью на циклический режим может значительно повысить эффективность разделения компонентов пробы. Это объясняется тем, что в циклическом режиме уменьшается размывание пиков, обусловленное гидродинамическими неоднородностями: движение подвижной фазы ближе к режиму полного (идеального) вытеснения. Понятие циклический режим хроматографии было использовано нами [32, 33] при анализе различных вариантов жидкостно-жидкостной хроматографии на основе распределения Крейга.
Базируясь на модели равновесных ступеней для циклического режима для условий, когда подвижная фаза подается порциями, равными ее объему в одной ступени, и каждая порция переходит из одной ступени в следующую ступень, не смешиваясь с предыдущей порцией, получена формула для расчета концентраций в выходящих порциях подвижной фазы [32]. Установлено [25], что для описания циклического процесса равновесной хроматографии можно использовать уравнение (3) для обычного процесса с непрерывной прокачкой элюента, заменив в нем значение N на Nц по формуле (5):
(5)
${{N}_{{\text{ц}}}} = N\frac{{1 + K{\kern 1pt} '}}{{K{\kern 1pt} '}} = 1 + \frac{{1 - {{S}_{f}}}}{{{{K}_{D}}{{S}_{f}}}}.$Как следует из формулы (5), при малых величинах коэффициентов распределения (KD < 1) и задержки неподвижной фазы (Sf < 0.5) переход к циклическому режиму может существенно повысить эффективность разделения.
Следует отметить, что рассмотренный выше циклический режим хроматографии в каскаде равновесных ступеней соответствует непрерывной работе аппарата Крейга [34, 35].
В работе [26] проведен анализ циклического режима с учетом влияния скорости межфазного массообмена. Сопоставлением расчетных пиков для равновесного и неравновесного процессов установлено, что практическое совпадение пиков достигается при определенной конечной скорости массообмена. При постоянной скорости массообмена различие между равновесной и неравновесной моделями процесса увеличивается с ростом фактора емкости K'.
Препаративная хроматография. При разделении компонентов смесей в препаративных целях стремятся максимально увеличить объем вводимой в колонку пробы. Однако импульсный ввод больших объемов пробы нарушает установившийся гидродинамический режим в колонке, что ухудшает чистоту получаемых фракций компонентов [36–39]. Проблему ввода больших объемов пробы можно решить, заменив технику импульсного впрыскивания пробы на загрузку пробы в колонку в течение определенного интервала времени [40, 41]. При этом раствор пробы подается в колонку с тем же расходом, что и подвижная фаза путем переключения насоса с емкости подвижной фазы на емкость, содержащую раствор разделяемой смеси в подвижной фазе. После завершения загрузки пробы в колонку вновь с тем же расходом подается подвижная фаза. Теоретический анализ влияния условий загрузки пробы на форму хроматографических пиков и процессы разделения проведен в работе [40]. На основе модели равновесных ступеней получено уравнение, описывающее выходной профиль концентраций после завершения загрузки пробы в колонку:
(6)
$\begin{gathered} X(t) = \frac{{x(t)}}{{\bar {x}}} = \frac{1}{{{{t}_{s}}}}{{e}^{{ - aN(t - {{t}_{s}})}}} \times \\ \times \,\,\sum\limits_1^N {\left\{ {\frac{{{{{[aN(t - {{t}_{s}})]}}^{{N - i + 1}}}}}{{(N - i + 1)!}}\left[ {1 - {{e}^{{ - aN{{t}_{s}}}}}\sum\limits_1^i {\frac{{{{{(aN{{t}_{s}})}}^{{i - 1}}}}}{{(i - 1)!}}} } \right]} \right\}} , \\ \end{gathered} $На основе распределения Гаусса получена более простая зависимость [41]:
(7)
$X(t) = \frac{1}{{\sqrt {\frac{{2\pi }}{{N{{a}^{2}}}} + \frac{{\pi t_{s}^{2}}}{6}} }}\exp \left[ { - \frac{{{{{\left( {\frac{1}{a} + \frac{{{{t}_{s}}}}{2} - t} \right)}}^{2}}}}{{\frac{2}{{N{{a}^{2}}}} + \frac{{t_{s}^{2}}}{6}}}} \right].$Показано [41], что, когда объем пробы не превышает 20–30% объема колонки (ts ≤ 0.2–0.3), вместо уравнения (6) можно использовать более простую зависимость (7).
Уравнения (6) и (7) описывают хроматограмму (пик) одного вещества. Если межфазное распределение отдельных компонентов смеси не зависит от ее состава, то для нормированной хроматограммы многокомпонентной смеси справедлива зависимость:
где Xnс(t) – безразмерная (нормированная) суммарная концентрация компонентов; Xj(t) – концентрации отдельных компонентов, которые рассчитываются по уравнению (6) или (7); qj – доли отдельных компонентов во введенной пробе.Как известно, производительность и качество разделения взаимосвязаны: как правило, с увеличением производительности ухудшается чистота разделяемых веществ. Зависимости (4), (6)–(8) позволяют проводить моделирование процесса хроматографического разделения заданной смеси для определения условий, которые обеспечивают разумный компромисс между производительностью и чистотой получаемых продуктов. В качестве примера на рис. 1 приведены результаты численного моделирования процесса разделения двухкомпонентной смеси (KD1 = 0.4 и KD2 = 0.8) в колонке с N = 300, Sf = 0.8 при загрузке различных объемов пробы (при различной длительности периода загрузки ts (τs)). Заметим, что при ts ≤ 0.01 уравнения (6), (7) описывают хроматограмму для условий импульсного ввода пробы.
Рис. 1.
Разделение двухкомпонентной смеси (KD1 = = 0.4, KD2 = 0.8) в колонке с N = 300, Sf = 0.8 при различной длительности периода загрузки пробы: ts = 0.01 (а), 0.1 (б), 0.2 (в).
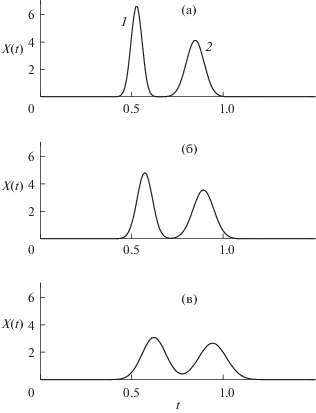
В рассмотренном методе ввода пробы объем подаваемого в колонку раствора смеси компонентов заметно не влияет на гидродинамический режим в колонке. Нарушение гидродинамического режима в колонке в условиях длительного ввода пробы может иметь место лишь при значительном различии физико-химических свойств пробы (раствора разделяемой смеси в подвижной фазе) и чистой подвижной фазы.
Рециркуляционная хроматография. В рециркуляционной хроматографии разделение компонентов смеси проводится в замкнутом циркуляционном контуре путем подачи после ввода пробы выходящего из колонки элюента на ее вход [42–46]. При этом достигается существенное увеличение эффективности процесса разделения. Метод рецикла впервые использован в газовой хроматографии [46]. Метод рецикла в жидкостно-жидкостной хроматографии впервые применен в работах [47, 48]. Экспериментальные исследования по разделению веществ растительного происхождения, в особенности лекарственных продуктов, опубликованы в работах [49–56]. Теория процессов рециркуляционной жидкостно-жидкостной хроматографии разрабатывалась в работах [41, 57–59]. Разделение компонентов в замкнутом контуре определяется двумя противоположно направленными процессами. С увеличением числа циклов прохождения пробы через колонку улучшается качество разделения компонентов: за счет многократного использования колонки увеличивается число теоретических ступеней в процессе разделения. Однако при этом происходит сближение и слияние хроматограмм соседних циклов: вышедшие из колонки, быстро движущиеся компоненты смеси (компоненты с низкими коэффициентами распределения) текущего цикла нагоняют оставшиеся в колонке медленно движущиеся компоненты (компоненты с высокими коэффициентами распределения) предыдущего цикла.
Схема процесса хроматографического разделения смеси компонентов в замкнутом двухфазном жидкостно-жидкостном контуре показана на рис. 2. Разделение осуществляется следующим образом: после ввода пробы выходящий из колонки поток подвижной фазы подается на ее вход, и проба циркулирует в замкнутом контуре до достижения требуемой степени разделения компонентов. Затем контур размыкается, в колонку подается подвижная фаза с расходом, равным скорости ее циркуляции в контуре, и фракции разделенных компонентов выводятся из колонки.
Рис. 2.
Схема процесса хроматографического разделения смеси компонентов в замкнутом контуре: 1 – загрузка пробы, 2 – разделение компонентов при циркуляции пробы в замкнутом контуре, 3 – вывод из колонки разделенных фракций компонентов.
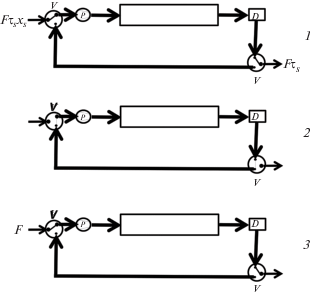
Схема математической модели рециркуляционной хроматографии приведена на рис. 3. Для моделирования различных вариантов процесса разделения можно использовать зависимости [59]:
(8)
$\begin{gathered} X(n,t) = \frac{1}{{\sqrt {\frac{{2\pi }}{{N{{a}^{2}}}} + \frac{{\pi t_{s}^{2}}}{6} + \frac{{2\pi (n - 1)}}{{{{N}_{{{\text{ef}}}}}{{a}^{2}}}}} }} \times \\ \times \,\,\exp \left[ { - \frac{{{{{\left( {\frac{n}{a} + \frac{{{{t}_{s}}}}{2} + b(n - 1) - t} \right)}}^{2}}}}{{\frac{2}{{N{{a}^{2}}}} + \frac{{t_{s}^{2}}}{6} + \frac{{2(n - 1)}}{{{{N}_{{{\text{ef}}}}}{{a}^{2}}}}}}} \right], \\ \end{gathered} $(9)
$\begin{gathered} {{X}_{n}}(t) = \sum\limits_{i = 1}^n {\frac{1}{{\sqrt {\frac{{2\pi }}{{N{{a}^{2}}}} + \frac{{\pi t_{s}^{2}}}{6} + \frac{{2\pi (i - 1)}}{{{{N}_{{{\text{ef}}}}}{{a}^{2}}}}} }}} \times \\ \times \,\,\exp \left[ { - \frac{{{{{\left( {\frac{i}{a} + \frac{{{{t}_{s}}}}{2} + b(i - 1) - t} \right)}}^{2}}}}{{\frac{2}{{N{{a}^{2}}}} + \frac{{t_{s}^{2}}}{6} + \frac{{2(i - 1)}}{{{{N}_{{{\text{ef}}}}}{{a}^{2}}}}}}} \right], \\ \end{gathered} $
где b = Vcр/Vс – отношение объемов системы рецикла Vcр (насос, детектор, клапаны, соединительные трубки) и колонки Vс; Nec – число ячеек полного перемешивания в системе рецикла, характеризующее степень продольного перемешивания (размывания пиков) в системе рецикла; n – номер цикла.
Уравнение (8) описывает пик компонента с коэффициентом распределения KD в отдельных циклах циркуляции пробы через колонку без учета взаимного влияния (возможности наложения) пиков соседних циклов. Уравнение (9) описывает изменение концентрации на выходе из колонки во время всего процесса циркуляции пробы от первого цикла до цикла n.
На рис. 4 показано влияние параметров системы рецикла (b и Nec) на процесс разделения бинарной смеси (KD1 = 0.3 и KD2 = 0.6) в колонке с N = 200, Sf = 0.5 при длительности периода загрузки ts = 0.1.
Рис. 4.
Влияние параметров системы рецикла (b и Nec) на процесс разделения бинарной смеси (KD1 = 0.3, KD2 = 0.6) в колонке с N = 200, Sf = 0.5 при длительности периода загрузки пробы ts = 0.1.
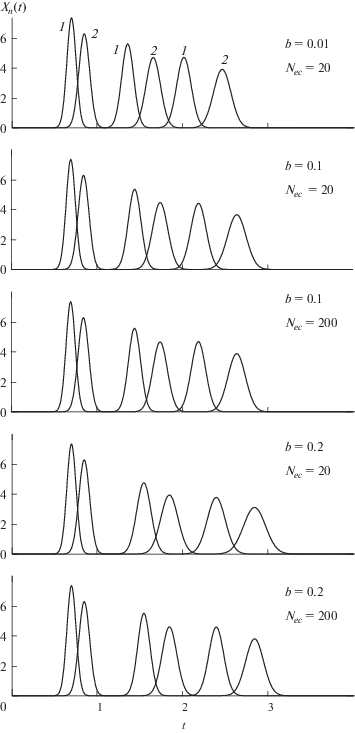
Многократная циркуляция пробы через колонку соответствует эффекту, получаемому от увеличения длины колонки. Однако, как отмечено выше, достигаемый при этом положительный эффект ограничен процессом сближения и наложения хроматограмм соседних циклов. Как видно из рис. 4, снизить отрицательный эффект этого явления, обусловленного расширением пиков компонентов при циркуляции пробы через колонку, можно путем увеличения объема системы рецикла и снижения степени продольного перемешивания в ней. Этого можно достичь, используя в линии рецикла длинные соединительные трубки малого диаметра (обеспечивая большой объем системы рецикла за счет длины соединительных трубок). Таким образом, в отличие от обычного элюентного метода, удлинение соединительных линий в рециркуляционной хроматографии способствует разделению хроматограмм соседних циклов (увеличению расстояния между ними), что повышает эффективность процесса разделения благодаря увеличению числа проходов пробы через колонку.
Противоточная хроматография. Благодаря подвижности обоих жидких фаз в устройствах ЖХ-СНФ появляется возможность создания более эффективных противоточных процессов хроматографического разделения смесей. В этих процессах подвижная и неподвижная фазы периодически меняются местами и направлением движения. Проба вводится непрерывно или в течение короткого времени в среднюю зону (между двумя барабанами с намотанными змеевиками) или в начальное сечение колонки. В зависимости от условий ввода пробы такие процессы в англоязычной литературе получили названия: “dual countercurrent chromatography” (двойная противоточная хроматография); “multiple dual-mode counter-current chromatography” (многорежимная противоточная хроматография); “intermittent counter-current extraction” (прерывистая противоточная экстракция); “trapping multiple dual mode” (улавливающая многорежимная) [60–72]. Каждый цикл противоточного процесса состоит из двух стадий: 1 – стадия движения 1-ой фазы; 2 – стадия движения 2-ой фазы. Режимными параметрами, определяющими качество разделения компонентов и производительность процесса, являются продолжительность периодов подачи (движения) потоков фаз и раствора компонентов.
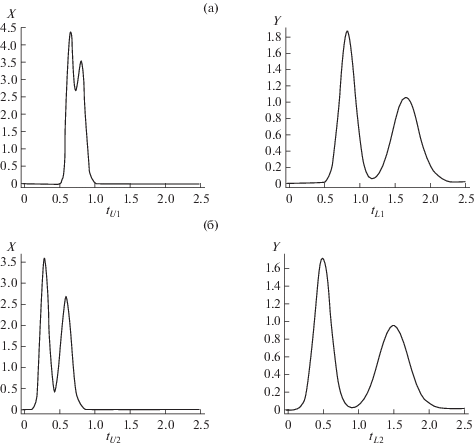
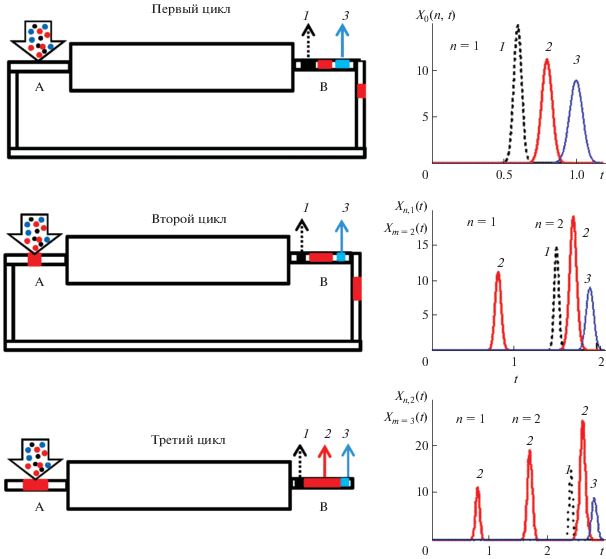
В работах [73, 74] проведен анализ процессов противоточной хроматографии для условий, когда проба подается в среднюю зону колонки или между двумя последовательно соединенными колонками; продолжительность стадий движения фаз сохраняется постоянной во всех циклах процесса. Приведены аналитические зависимости для моделирования и определения оптимальных условий проведения этих процессов. Нами предложен метод противоточной хроматографии с переменной продолжительностью стадий движения фаз [75, 76]. В таком противоточно-циклическом процессе проба совершает возвратно-поступательное движение в колонке до тех пор, пока не будет достигнуто требуемое разделение компонентов. При этом увеличивается путь движения компонентов разделяемой смеси в колонке, что, как и в рециркуляционной хроматографии, увеличивает число теоретических тарелок и повышает эффективность процесса разделения. Поскольку при движении пробы в колонке наряду с разделением происходит расширение пиков компонентов, для удержания пробы в колонке в течение определенного количества циклов необходимо от цикла к циклу регулировать продолжительность стадий движения фаз. Рис. 5 иллюстрирует процесс разделения, в котором каждый компонент смеси полностью выводится из колонки в течение одного цикла. На рис. 5 показаны две схемы такого процесса: А – все компоненты выводятся с одной фазой в определенном цикле; B – отдельные компоненты смеси выводятся с разными потоками фаз. Возможен также вариант противоточного разделения, в котором компоненты смеси выводятся из колонки частями в разных циклах процесса. Здесь важно своевременное переключение потоков фаз, чтобы обеспечить требуемую чистоту собранных фракций индивидуальных компонентов. Для моделирования различных возможных вариантов рассматриваемых процессов разделения получены аналитические зависимости, описывающие профили концентраций в колонке (каскаде равновесных ступеней) и изменение концентраций в выходящих потоках фаз в каждом цикле процесса [76]. На основании этих зависимостей разработана расчетная программа в виде калькулятора для численных исследований. Пример моделирования с помощью калькулятора процесса разделения бинарной смеси (KD1 = 0.3; KD2 = 0.6) в колонке с N = 200, Sf = 0.5 приведен на рис. 6 [76]. Полное разделение компонентов происходит в течение двух циклов. Смесь компонентов вводится с легкой фазой в течение определенного времени (ts = 0.01) в первом цикле. Разделенные компоненты выводятся из колонки с тяжелой фазой во втором цикле. Для разделения данной смеси обычным методом потребуется колонка с N = 1000. На рис. 6 приняты следующие обозначения:
Рис. 5.
Схема метода противоточной хроматографии с переменной продолжительностью стадий движения фаз [76]. Каждый компонент смеси полностью выводится из колонки в течение одного цикла: (а) – все компоненты выводятся с одной фазой в определенном цикле; (б) – отдельные компоненты смеси выводятся с разными потоками фаз.
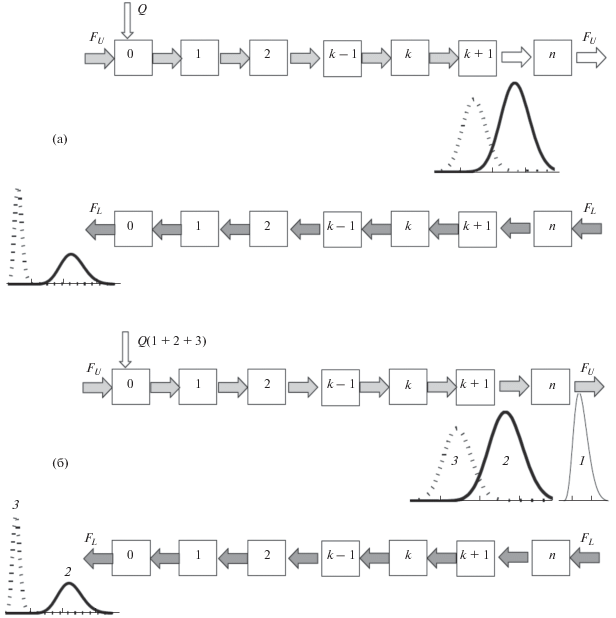
Рис. 6.
Разделение бинарной смеси (KD1 = 0.3, KD2 = 0.6)–в колонке с N = 200, Sf = 0.5 методом противоточной хроматографии [(а) – первый цикл: tL1 = tU1 = 0.5; (б)– второй цикл: tU2 = 0.1] [76].
tU1 = τU1FU / V, tU2 = τU2FU / V – продолжительности стадии движения легкой фазы в первом и втором циклах в безразмерных единицах времени;
tL1 = τL1FL/V, tL2 = τL2FL/V– продолжительности стадии движения тяжелой фазы в первом и втором циклах в безразмерных единицах времени; $X = \frac{x}{{\bar {x}}},$ $Y = \frac{y}{{\bar {x}}}$ – безразмерные выходные концентрации в фазах; $\bar {x} = \frac{Q}{V}$ = $\frac{{{{x}_{s}}{{F}_{U}}{{\tau }_{s}}}}{V}.$
На рис. 6 приведены только выходные профили концентраций с интерфейса калькулятора. Кроме этих графиков, на интерфейсе калькулятора приводятся также кривые распределения концентраций в колонке и выход каждого компонента в конце каждой стадии.
На рис. 7 приведены расчетные и экспериментальные результаты, полученные при разделении смеси аспирина и кумарина за два цикла противоточного процесса [76]. Процесс начинается в режиме движения тяжелой фазы; продолжительность первой стадии t1L = 0.5; продолжительность второй стадии (движение легкой фазы) t1U = 0.3. В течение этого времени около 20% кумарина выводится из установки с легкой фазой. На первой стадии второго цикла с легкой фазой выводятся разделенные фракции обоих компонентов.
Рис. 7.
Расчетные (а) и экспериментальные (б) хроматограммы, полученные при разделении смеси аспирина и кумарина за два цикла противоточного процесса [76].
Рециркуляционно-противоточная хроматография. Рециркуляционные и противоточные методы позволяют многократно повысить эффективность установок жидкостно-жидкостной хроматографии. Путем комбинации этих методов можно создать еще более эффективные процессы разделения. В работах [77, 78] разработана теория процесса, на первом этапе которого проводится разделение компонентов в замкнутом контуре при циркуляции через колонку фазы “х”, а на втором этапе контур размыкается, и через колонку в обратном направлении движется фаза “у” (рис. 8). Установлено, что при разделении сложных смесей компоненты с низкими коэффициентами распределения могут быть разделены на первом этапе, а компоненты с высокими коэффициентами распределения – на втором. Для условий импульсного ввода пробы получены следующие теоретические зависимости для моделирования процесса разделения:
(11)
$\begin{gathered} {{X}_{n}}(t) = \frac{{aN\sqrt {{{N}_{{ec}}}} }}{{\sqrt {2\pi } }} \times \\ \times \,\,\sum\limits_{i = 1}^n {\frac{{\exp \left[ { - \frac{{{{N}_{{ec}}}{{{[Ni + abN(i - 1) - aNt]}}^{2}}}}{{2{{N}_{{ec}}}Ni + 2(i - 1){{{\left( {Nab} \right)}}^{2}}}}} \right]}}{{\sqrt {{{N}_{{ec}}}Ni + (i - 1){{{\left( {Nab} \right)}}^{2}}} }}} , \\ \end{gathered} $(12)
$\begin{gathered} Y(t) = {{K}_{D}}{{e}^{{ - {{K}_{D}}aNt}}}\sum\limits_{i = 1}^{50} {\frac{{{{{({{K}_{D}}aNt)}}^{{k - 1}}}}}{{(k - 1)!}}} {{X}_{n}}(k) + \\ + \,\,{{K}_{D}}\sum\limits_{k = 51}^N {\frac{{{{{({{K}_{D}}aNt)}}^{{k - 1}}}}}{{(k - 1)}}} \frac{{{{e}^{{k - 1 - {{K}_{D}}aNt}}}}}{{\sqrt {2\pi (k - 1)} }}{{X}_{n}}(k), \\ \end{gathered} $(13)
$\begin{gathered} {{X}_{n}}(k) = \frac{{aN\sqrt {{{N}_{{ec}}}} }}{{\sqrt {2\pi } }} \times \\ \times \,\,\sum\limits_{i = 1}^n {\frac{{\exp \left[ { - \frac{{{{N}_{{ec}}}{{{[N(i - 1)(1 + ab) + k - aN{{t}_{x}}]}}^{2}}}}{{2{{N}_{{ec}}}[N(i - 1) + k] + 2(i - 1){{{\left( {Nab} \right)}}^{2}}}}} \right]}}{{\sqrt {{{N}_{{ec}}}[N(i - 1) + k] + (i - 1){{{\left( {Nab} \right)}}^{2}}} }}} . \\ \end{gathered} $
Уравнение (11) описывает выходной профиль концентрации компонента с коэффициентом распределения KD на первом этапе, а уравнения (12), (13) – на втором этапе процесса разделения. Член Xn(k) в формуле (12) описывает (уравнение (13)) распределение компонента KD в колонке после первого этапа процесса при продолжительности циркуляции x-фазы tx (tx = τxFx/Vc). Отсчет времени на втором этапе начинается с t = 0. Как и ранее, уравнения (11)–(13) описывают нормированные хроматограммы: $X = {x \mathord{\left/ {\vphantom {x {\bar {x}}}} \right. \kern-0em} {\bar {x}}}$ – t = τFx/Vc; $Y = {y \mathord{\left/ {\vphantom {y {\bar {x}}}} \right. \kern-0em} {\bar {x}}}$ – t = τFy/Vc.
Несмотря на кажущуюся сложность уравнения, (11)–(13) можно легко использовать с любой стандартной компьютерной программой для моделирования двухэтапного процесса разделения. В электронной версии статьи [78] приведены примеры моделирования различных вариантов рассматриваемого метода разделения в программе Mathcad.
Элюентно-экструзивная хроматография (elution–extrusion counter-current chromatography). Данный метод сочетает классическое элюирование подвижной фазы с последующим вытеснением содержания колонки потоком неподвижной фазы [79, 80]. Когда смесь содержит компоненты с большими величинами коэффициентов распределения (KD), такой метод позволяет существенно сократить расход растворителей (реагентов) и время разделения. Компоненты смеси с малыми величинами KD разделяются при элюировании подвижной фазы, а компоненты с более высоким величинами KD разделяются внутри колонки и выталкиваются из нее потоком неподвижной фазы (во время экструзии). В работе [79] проведен приближенный анализ процесса элюентно-экструзивной хроматографии. Более точный анализ и математическое описание процесса на основе модели равновесных ступеней представлены в работе [80]; получены уравнения хроматограмм компонентов, выходящих из колонки с подвижной фазой в течение периода элюирования, и хроматограмм компонентов, выталкиваемых из колонки неподвижной фазой в течение периода экструзии.
Непрерывные стационарные методы разделения. Существует несколько способов увеличения производительности установок ЖХСНФ: 1) повышение концентрации компонентов в пробе; 2) выбор растворителей с высокой емкостью [81]; 3) увеличение масштабов колонки [82, 83]; 4) оптимизация геометрии колонки [84]; 5) использование многократного ввода пробы [83, 85]. Возможности первого способа ограничены растворимостью компонентов пробы. Наиболее перспективным представляется последний способ при реализации его в стационарном режиме. Особенность стационарных методов заключается в полунепрерывной загрузке пробы в колонку: хроматографическая установка имеет два резервуара с подвижной фазой – один с “чистой” подвижной фазой, а второй с раствором пробы в этой фазе; насос периодически переключается с одного резервуара на другой. Раствор компонентов разделяемой смеси периодически в течение определенного промежутка времени в чередующейся последовательности с потоком “чистой” подвижной фазы подается в колонку. Оба потока подаются с одинаковой объемной скоростью в колонку. Разделенные фракции компонентов периодически отбираются на выходе из колонки.
При использовании стационарных методов необходимо определить продолжительности периодов загрузки и интервалы, через которые раствор компонентов должен вводиться в колонку. Для моделирования таких процессов достаточно иметь теоретическое описание выходных концентрационных профилей после двух последовательных загрузок.
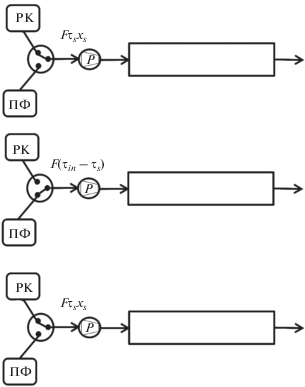
Схема стационарного процесса обычной хроматографии показана на рис. 9. Для компонента j с коэффициентом распределения KDj профили концентраций после двух последовательных загрузок раствора компонентов можно рассчитать по следующим уравнениям [86]:
(14)
${{X}_{{j1}}}(t) = \frac{1}{{\sqrt {\frac{{2\pi }}{{{{N}_{j}}a_{j}^{2}}} + \frac{{\pi t_{s}^{2}}}{6}} }}\exp \left[ { - \frac{{{{{\left( {\frac{1}{{{{a}_{j}}}} + \frac{{{{t}_{s}}}}{2} - t} \right)}}^{2}}}}{{\frac{2}{{{{N}_{j}}a_{j}^{2}}} + \frac{{t_{s}^{2}}}{6}}}} \right],$(15)
${{X}_{{j2}}}(t) = \frac{1}{{\sqrt {\frac{{2\pi }}{{{{N}_{j}}a_{j}^{2}}} + \frac{{\pi t_{s}^{2}}}{6}} }}\exp \left[ { - \frac{{{{{\left( {\frac{1}{{{{a}_{j}}}} + \frac{{{{t}_{s}}}}{2} + {{t}_{{in}}} - t} \right)}}^{2}}}}{{\frac{2}{{{{N}_{j}}a_{j}^{2}}} + \frac{{t_{s}^{2}}}{6}}}} \right],$Рис. 9.
Схема стационарного процесса обычной хроматографии: РК – раствор компонентов смеси; ПФ – подвижная фаза.
Уравнения (14) и (15) описывают профили концентраций, соответствующие первой и второй загрузкам. Уравнение (17) описывает результирующий профиль концентрации после двух загрузок раствора компонентов.
В методе стационарной рециркуляционной хроматографии, как и в случае обычной, раствор компонентов периодически с заданным интервалом τin (tin) непрерывно загружается в колонку в течение постоянного времени τs (ts). Первая загрузка раствора компонентов начинается при t = 0. Во время загрузки раствора в колонку циркуляционный контур открыт. После завершения первой загрузки раствора контур закрывается, и смесь циркулирует в системе до тех пор, пока не будет достигнута желаемая степень разделения компонентов. После этого контур открывается, чистая подвижная фаза закачивается в колонку, и начинается элюирование разделенных фракций компонентов. При τ = τin (t = tin) вторая порция раствора компонентов в течение времени τs непрерывно подается в колонку. После окончания второй загрузки раствора компонентов свежая подвижная фаза закачивается в колонку до тех пор, пока не будет завершено элюирование компонентов первой загрузки. После этого контур закрывается, и смесь циркулирует в системе до тех пор, пока не будет достигнута требуемая степень разделения компонентов второй загрузки. Далее указанные выше операции повторяются. Во время всех операций не прекращается движение потока подвижной фазы в колонке, так как переключения регулирующих клапанов на входе и выходе из колонки и насоса подвижной фазы от резервуара раствора компонентов к резервуару чистой подвижной фазы и наоборот происходят одновременно. В работе [86] приведены аналитические зависимости для моделирования стационарных процессов рециркуляционной хроматографии. Для удобства пользователей в электронной версии [86] в программе Mathcad представлены примеры моделирования различных вариантов рассматриваемого метода разделения.
В стационарных процессах противоточной хроматографии первый полупериод каждого цикла состоит из двух стадий: стадии загрузки раствора компонентов в первой фазе и стадии подачи “чистой” первой фазы. Режимными параметрами, определяющими эффективность процесса (степень разделения компонентов и производительность), являются продолжительность периодов подачи (движения) потоков фаз и раствора компонентов. Продолжительность периодов движения потоков фаз и подачи раствора компонентов постоянны во всех циклах. При работе в этом режиме после определенного числа циклов наступает квазистационарный режим, когда концентрационные профили в колонке и выходные концентрации в потоках фаз изменяются во времени, но остаются постоянными (повторяются) в каждом цикле процесса. Такой режим обеспечивает высокую производительность и чистоту получаемых фракций компонентов. Теория стационарных процессов противоточной хроматографии изложена в работах [87–89]; в электронной версии [89] размещен калькулятор для численных исследований стационарных противоточных процессов хроматографического разделения.
Выделение из растворов концентрированных фракций целевых компонентов. В аналитике нередко возникают проблемы, связанные с определением компонентов, содержащихся в очень малых количествах в сложных смесях. Например, при анализе лекарственных препаратов необходимо определять побочные продукты вплоть до чрезвычайно низкого уровня их содержания в лекарственном веществе. В таких случаях актуальной становится задача выделения этих компонентов в концентрированном виде. Применение техники многократных загрузок пробы, используемой в изложенных выше методах стационарной хроматографии, может помочь решить эту проблему. При этом, в отличие от стационарных методов, новые порции раствора смеси подаются в колонку через различные промежутки времени. Такой подход применим ко всем рассмотренным выше методам хроматографии. Теория нестационарных процессов противоточной жидкостно-жидкостной хроматографии с многократной загрузкой раствора смеси компонентов в колонку развита в работах [87, 89]. В электронной версии работы [89] размещен калькулятор для моделирования и анализа различных вариантов таких противоточных процессов.
В работах [90, 91] проведен анализ нестационарной рециркуляционной хроматографии с многократным импульсным вводом пробы в колонку; получены аналитические зависимости для моделирования различных вариантов и определения оптимальных условий процесса разделения; приведены примеры моделирования. На рис. 10 приведены схема и результаты моделирования процесса, когда из смеси раствора компонентов KD1 = 0.2, KD2 = 0.6 и KD3 = 1 целевой компонент KD2 выделяется в концентрированном виде в колонне с N = 500 с использованием трех вводов пробы. Побочные продукты удаляются из системы после каждого цикла. Как видно, после второго цикла (второй ввод пробы) может быть выделена концентрированная фракция компонента KD2; после третьего цикла целевой компонент концентрируется еще больше (высота пиков резко увеличивается, а их ширина практически не изменяется), но чистота его немного уменьшается. Моделирование процесса позволяет определить количество и время вводов пробы для достижения необходимой чистоты и концентрации целевого продукта.
Рис. 10.
Выделение концентрированной фракции промежуточно элюирующего компонента (KD2) из смеси компонентов KD1 = 0.2, KD2 = 0.6 и KD3 = 1 методом нестационарной рециркуляционной хроматографии [91]. A – ввод пробы; B – вывод компонентов. Параметры процесса: N = 500, b = 0.1, Nec = 300, Sf = 0.5.
Выбор метода разделения. Рассмотренные выше методы разделения смесей веществ основываются на нестационарных процессах жидкостной экстракции, поэтому их можно также назвать экстракционно-хроматографическими методами разделения. Выбор метода зависит от конкретной задачи и имеющегося хроматографического оборудования. Если хроматограф не обладает достаточной эффективностью для разделения заданной смеси веществ, то для повышения качества разделения можно использовать наиболее простой и легко реализуемый метод рециркуляционной хроматографии. Этот метод не требует существенной реконструкции экспериментальной установки. В случае разделения смесей, содержащих компоненты с высокими коэффициентами распределения, для сокращения продолжительности эксперимента и расхода растворителей рекомендуется элюентно-экструзивная хроматография, также не требующая реконструкции экспериментальной установки. Если эти простые методы не позволяют решить имеющуюся задачу разделения, следует использовать более сложные в техническом отношении противоточные и комбинированные методы. Эти методы можно рекомендовать для разделения сложных смесей, содержащих компоненты с близкими и сильно различающимися коэффициентами распределения. Когда требуется высокая производительность, например при разделении смесей для препаративных и технических целей, наиболее подходящими являются методы стационарной хроматографии. Задачи выделения концентрированных фракций отдельных компонентов из смесей веществ можно решать методами нестационарной противоточной и рециркуляционной хроматографии с многократным вводом пробы.
Аппаратурное оформление. За рубежом практическое применение нашли два вида аппаратов для осуществления процессов жидкостно-жидкостной хроматографии: гидростатические – последовательно соединенные камеры для контакта фаз, расположенные на поверхности цилиндра или пакета дисков и размещенные в обычной центрифуге, и гидродинамические – хроматографическая колонка в виде змеевика, намотанного в один или несколько слоев на барабан (или барабаны) планетарной центрифуги. Достоинством второй группы аппаратов является отсутствие подвижных сальников и более высокая эффективность межфазного массообмена, обусловленная интенсивным перемешиванием жидкостей в переменном (пульсирующем) поле центробежных сил. В обоих случаях неподвижная жидкая фаза удерживается в колонке в свободном состоянии с помощью центробежных сил. Недостатком центробежных хроматографических аппаратов является сложность конструкции и обусловленная этим высокая стоимость, а также проблемы, возникающие при увеличении масштабов процессов разделения.
В ИОНХ РАН разрабатываются высокопроизводительные хроматографические аппараты без использования центрифуг [92–97]. Предложено два варианта аппаратурного оформления: 1) в каскаде многоступенчатых колонн [92, 93, 95]: подвижной фазой служит дисперсная фаза, а сплошная фаза является неподвижной фазой; 2) в каскаде центробежных смесительно-отстойных экстракторов [96, 97]: подвижной и неподвижной могут быть как дисперсная, так и сплошная фазы.
В первом случае неподвижная фаза удерживается в колоннах с помощью сил вязкости и поверхностного натяжения. Хроматографическое устройство состоит из ряда последовательно соединенных вертикальных колонн, разделенных на ячейки горизонтальными перфорированными перегородками [92, 95]. После заполнения колонн неподвижной фазой через устройство в пульсационно-циклическом режиме прокачивается подвижная фаза. С помощью дозатора устанавливаются параметры прокачки, обеспечивающие циклическую подачу жидкости в форме отдельных импульсов. При истечении из отверстий горизонтальных перегородок происходит диспергирование подвижной фазы в объеме сплошной неподвижной фазы и пульсационное перемешивание фаз в ячейках колонн. В периоды отсутствия подачи подвижной фазы происходит гравитационное разделение фаз в ячейках колонн, и таким образом достигается многократное перемешивание и разделение фаз по высоте колонн. Достоинствами этого хроматографического устройства являются простота и отсутствие серьезных препятствий для увеличения масштабов. Работа многоколоночного хроматографического устройства иллюстрируется на видео в Supplementary data электронной версии статьи [88].
Удерживание неподвижной фазы в каскаде смесительно-отстойных центробежных экстракторов достигается путем ее рециркуляции между камерами смешения и разделения в каждой ступени каскада [96, 97]. В отличие от противоточной экстракции, ступени каскада соединены только по потоку водной (подвижной) фазы. В камере смешения центробежного экстрактора благодаря работе мешалки обеспечивается интенсивный контакт фаз, а в камере разделения, представляющей собой центробежный сепаратор, достигается быстрое разделение фаз. Поэтому в каждой ступени каскада достигается эффективность разделения, соответствующая одной теоретической тарелке.
* * *
Возможность усовершенствования хроматографических установок для процессов жидкостно-жидкостной хроматографии позволяет реализовывать различные способы разделения смесей веществ. Технически относительно простая модернизация может значительно, в некоторых случаях многократно, повысить эффективность процессов разделения и производительность установок жидкостно-жидкостной хроматографии. Повышение эффективности разделения достигается за счет виртуального удлинения колонки в методах рециркуляционной и противоточной хроматографии. Повышение производительности обеспечивается заменой импульсного ввода пробы на ее продолжительную загрузку в колонку. Дальнейшее повышение производительности может быть достигнуто методами стационарной хроматографии. Многократная загрузка пробы в нестационарном режиме позволяет выделить концентрированные фракции целевых компонентов из сложных смесей. Применение принципов жидкостно-жидкостной хроматографии может существенно упростить экстракционные технологии разделения в гидрометаллургии. Для этого потребуется не связанная с большими техническими проблемами модернизация экстракционного оборудования, в частности, каскада смесительно-отстойных экстракторов или многоступенчатых пульсационных колонн.
Список литературы
Марютина Т.А., Спиваков Б.Я., Шпигун Л.К., Павленко И.В., Золотов Ю.А. Концентрирование и разделение орто- и пирофосфат-ионов методом жидкостной хроматографии со свободной неподвижной фазой // Журн. аналит. химии. 1990. Т.45. № 4. С. 665.
Чмутова М.К., Марютина Т.А., Спиваков Б.Я., Мясоедов Б.Ф. Разделение америция (III) и европия (III) в системах с нейтральными бидентатными фосфороорганическими экстрагентами методом жидкостной хроматографии со свободной неподвижной фазой // Радиохимия. 1992. № 6. С. 56.
Федотов П.С., Марютина Т.А., Пичугин А.А., Спиваков Б.Я. Влияние кинетических свойств экстракционных систем на разделение некоторых элементов методом жидкостной хроматографии со свободной неподвижной фазой // Журн. неорг. химии. 1993. Т. 38. № 11. С. 1878.
Марютина Т.А., Федотов П.С., Спиваков Б.Я. Использование метода жидкостной хроматографии со свободной неподвижной фазой для концентрирования и разделения неорганических веществ. Двухфазные жидкостные системы // Журн. аналит. химии. 1997. Т. 52. № 12. С. 1263. (Maryutina T.A., Fedotov P.S., Spivakov B.Ya. Application of countercurrent chromatography to preconcentration and separation of inorganic substances: Two-phase liquid systems // J. Analyt. Chem. 1997. V. 52. P. 1138.)
Федотов П.С., Марютина Т.А., Гребнева О.Н., Кузьмин Н.М., Спиваков Б.Я. Использование метода жидкостной хроматографии со свободной неподвижной фазой для концентрирования и разделения неорганических веществ. Групповое выделение Zr, Hf, Nb, Ta для их последующего атомно-эмиссионного (с индуктивно связанной плазмой) определения // Журн. аналит. химии. 1997. Т. 52. № 11. С. 1141. .(Fedotov P.S., Maryutina T.A., Grebneva, O.N., Kuz’min, N.M., Spivakov B.Ya. Use of countercurrent patition chromatography for the preconcentration and separation of inorganic compounds: Group extraction of Zr, Hf, Nb, and Ta for their subsequent determination by inductively coupled plasma atomic emission spectrometry // J. Analyt. Chem. 1997. V. 52. P. 1034.)
Спиваков Б.Я., Марютина Т.А., Федотов П.С., Игнатова С.Н., Катасонова О.Н., Дамен И., Венрих Р. Разделение веществ во вращающихся спиральных колонках: от микроэлементов до микрочастиц // Журн. аналит. химии. 2002. Т. 57. № 10. С. 1096. (Spivakov B.Ya. Maryutina T.A., Fedotov P.S., Ignato-va S.N., Katasonova O.N., Dahmen J., Wennrich R. Separation of substances in rotating coiled columns:From trace elements to microparticles // J. Analyt. Chem. 2002. V. 57. P. 928.)
Ito Y., Bowman R.L. Countercurrent chromatography with the flow-throug coil planet centrifuge // J. Chromatogr. Sci. 1973. V. 11. P. 284.
Zolotov Yu.A., Spivakov B.Ya., Maryutina T.A., Bashlov V.L., Pavlenko I.V. Partition countercurrent chromatography in inorganic analysis // Fresenius J. Anal. Chem. 1989. V. 335. № 8. P. 938.
Conway W.D. Countercurrent chromatography: apparatus, theory and applications. N.Y.: VCH Publishers Inc., 1990. P. 475.
Maryutina T.A., Spivakov B.Ya., Tschopel P. Application of countercurrent chromatography to purification of chemical reagents // Fresenius J. Anal. Chem. 1996. V. 356. P. 430
Countercurrent Chromatography, Chromatographic Science Series. V. 82 / Eds. Menet J.M., Thiebaut D., N.Y.: Marcel Dekker, Inc., 1999.
Ignatova S.N., Maryutina T.A., Spivakov B.Ya., Karandashev V.K. Group separation of trace rare earth elements by countercurrent chromatography for their determination in high-purity calcium chloride // Fresenius J. Anal. Chem. 2001. V. 370. P. 1109.
Fedotov P.S. Untraditional applications of countercurrent chromatography // J. Liq. Chromatogr. Relat.Technol. 2002. V. 25. № 13-15. P. 2065.
Ito Y. Golden rules and pitfalls in selecting optimum conditions for high-speed counter-current chromatography // J. Chromatogr. A. 2005. V. 1065. P. 145.
Friesen J.B., Pauli G.F., Reciprocal symmetry plots as a representation of countercurrent chromatograms // Anal. Chem. 2007. V. 79. P. 2320.
Conway W.D. Counter-current chromatography: Simple process and confusing terminology // J. Chromatogr. A. 2011. V. 1218. P. 6015.
Hopmann E., Minceva M. Separation of a binary mixture by sequential centrifugal partition chromatography // J. Chromatogr. A. 2012. V. 1229. P. 140.
Hopmann E., Goll J., Minceva M. Sequential centrifugal partition chromatography: A new continuous chromatographic technology // Chem. Eng. Technol. 2012. V. 35. P. 72.
Ignatova S., Sutherland I. The 8th International Conference on Counter-current Chromatography held at Brunel University, London, UK, July 23–25, 2014 // J. Chromatogr. A. 2015. V. 1425. P. 1.
Friesen J.B., McAlpine J.B., Chen S.-N., Pauli G.F., The 9th International Countercurrent Chromatography Conference held at Dominican University, Chicago, USA, August 1–3, 2016 // J. Chromatogr. A. 2017. V. 1520. P. 1.
Bezold F., Minceva M. A water-free solvent system containing an L-menthol-based deep eutectic solvent for centrifugal partition chromatography applications // J. Chromatogr. A. 2019. V. 1587. P. 166.
Костанян А.Е. Общие закономерности процессов жидкостной хроматографии и противоточной экстракции // Теорет. основы хим. технол. 2006. Т. 40. № 6. С. 1. (Kostanyan A.E. General regularities of liquid chromatography and countercurrent extraction // Theor. Found. Chem. Eng. 2006. V. 40. P. 587.)
Kostanian A.E. Modelling counter-current chromatography: A chemical engineering perspective // J. Chromatogr. A. 2002. V. 973. P. 39.
Kostanian A.E., Berthod A., Ignatova S.N., Maryutina T.A., Spivakov B.Ya., Sutherland I.A. Countercurrent chromatographic separation: a hydrodynamic approach developed for extraction columns // J. Chromatogr. A. 2004. V. 1040. P. 63.
Kostanyan A.E. Controlled-cycle counter-current chromatography // J. Chromatogr. A. 2008. V. 1211. P. 55.
Костанян А.Е., Вошкин А.А. Анализ циклического процесса жидкостной хроматографии // Теорет. основы хим. технол. 2011. Т. 45. № 1. С. 68. (Kostanyan A.E., Voshkin A.A. Analysis of cyclical liquid chromatography // Theor. Found. Chem. Eng. 2011. V. 45. P. 68.)
Belter P.A., Speaker S.M. Controlled-cycle operations applied to extraction processes. // Ind. Eng. Chem. Process Design and Development. 1967. V. 6. P. 36.
Gerster J.A., Scull H.M. Performance of tray columns operated in cyclic mode // A.I.Ch.E. J. 1970. V. 16. P. 108.
Конобеев Б.И., Арутюнян Г.Р., Назаров П.С. Исследование циклической ректификации в тарельчатой колонне. // Теорет. основы хим. технол. 1977. Т. 11. № 4. С. 491.
Конобеев Б.И., Арутюнян Г.Р., Малюсов В.А. Описание циклической ректификации в насадочных колоннах с помощью математических моделей // Теорет. основы хим. технол. 1979. Т. 13. № 4. С. 499.
Колтунова Л.Н. Влияние циклической подачи фаз на эффективность массообменных аппаратов // Теорет. основы хим. технол. 1980. Т. 14. № 5. С. 643.
Kostanyan A.E., Belova V.V., Kholkin A.I. Modelling CCC and dual CCC using longitudinal mixing cell and eluting countercurrent distribution models // J. Chromatogr. A. 2007. V. 1151. № 1–2. P. 142.
Kostanyan A.E., Voshkin A.A. Analysis of new CCC operating modes // J. Chromatogr. A. 2007. V. 1151. № 1–2. P. 126
Альдерс А. Жидкостная экстракция. М.: Иностранная литература, 1962. С. 207.
Craig L.C. Identification of small amounts of organic compounds by distribution studies. II. Separation by counter-current distribution // J. Biol. Chem. 1944. V. 155 P. 519.
Berthod A., Billardello B., Geoffroy S. Polyphenols in countercurrent chromatography, an example of large scale separation // Analysis. 1999. V. 27. P. 750.
Marchal L., Intes O., Foucault A., Legrand J., Nuzillard J.-M., Renault J.-H. Rational improvement of centrifugal partition chromatographic settings for the production of 5-n-alkylresorcinols from wheat bran lipid extract I. Flooding conditions – optimising the injection step // J. Chromatogr. A. 2003. V. 1005. P. 51
Zhao C.-X., He C.-H. Sample capacity in preparative high-speed counter-current chromatography // J. Chromatogr. A. 2007. V. 1146. P. 186.
Peng A., Hewitson P. Ye H., Zu L., Garrard I., Sutherland I., Chen L., Ignatova S. Sample injection strategy to increase throughput in counter-current chromatography: Case study of Honokiol purification // J. Chromatogr. A. 2016. V. 1476. P. 19.
Kostanyan A.E. On influence of sample loading conditions on peak shape and separation efficiency in preparative isocratic counter-current chromatography // J. Chromatogr. A. 2012. V. 1254. P. 71.
Kostanyan A.E. Modeling of preparative closed-loop recycling liquid-liquid chromatography with specified duration of sample loading // J. Chromatogr. A. 2016. V. 1471. P. 94.
Porter R.S., Johnson J.F. Circular gas chromatography // Nature. 1959. V. 183. P. 391.
Seidel-Morgenstern A., Guiochon G. Theoretical study of recycling in preparative chromatography // AIChE J. 1993. V. 39. P. 809.
Dingenen J. Preparative chromatographic resolution of racemates on chiral stationary phases on laboratory and production scales by closed-loop recycling chromatography // J. Chromatogr. A. 1994. V. 666. P. 627.
Chartor F., Bailly M., Guiochon G. Recycling in preparative liquid chromatography // J. Chromatogr. A. 1994. V. 687. P. 13.
Heuer C., Seidel-Morgenstern A., Hugo P. Experimental investigation and modeling of closed-loop recycling in preparative chromatography // Chem. Eng. Sci. 1995. V. 50. P. 1115.
Du Q.Z., Ke C.Q., Ito Y. Recycling high-speed countercurrent chromatography for separation of taxol and cephalomannine // J. Liq. Chromatogr. Relat. Technol. 1998. V. 21. P. 157.
Oka H., Iwaya M., Harada K., Suzuki M., Ito Y. Recycling foam countercurrent chromatography // Anal. Chem. 2000. V. 72. P. 1490.
Han Q.B., Song J.Z., Qiao C.F., Wong L., Xu H.X. Preparative separation of gambogic acid and its C-2 epimer using recycling high-speed counter-current chromatography // J. Chromatogr. A. 2006. V. 1127. P. 298.
Ye H., Ignatova S., Luo H., Li Y., Peng A., Chen L., Sutherland I. Preparative separation of a terpenoid and alkaloids from Tripterygium wilfordii Hook. f. using high-performance counter-current chromatography. Comparison of various elution and operating strategies // J. Chromatogr. A. 2008. V. 1213. P. 145.
Xie J., Deng J., Tan F., Su J. Separation and purification of echinacoside from Penstemon barbatus (Can.) Roth by recycling high-speed counter-current chromatography // J. Chromatogr. B. 2010. V. 878. P. 2665.
Tong S., Guan Y.-X, Yan J., Zheng B., Zhao L. Enantiomeric separation of (R,S)-naproxen by recycling high speed counter-current chromatography with hydroxypropyl-β-cyclodextrin as chiral selector // J. Chromatogr. A. 2011. V. 1218. P. 5434.
Yang J., Ye H., Lai H., Li S., He S., Zhong S., Chen L., Peng A. Separation of anthraquinone compounds from the seed of Cassia obtusifolia L. using recycling counter-current chromatography // J. Sep. Sci. 2012. V. 35. P. 256.
Meng J., Yang Z., Liang J., Zhou H., Wu S., Multi-channel recycling counter-current chromatography for natural product isolation: tanshinones as examples // J. Chromatogr. A. 2014 V. 1327. P. 27.
Friesen J.B., McAlpine J.B., Chen S.-N., Pauli G.F. Countercurrent separation of natural products: An update // J. Nat. Prod. 2015. V. 78. P. 1765.
Chen Y., Yan X., Lu F., Jiang X., Friesen J.B., Pauli G.F., Chen S.-N., Li D.-P. Preparation of flavone di-C-glycoside isomers from Jian-Gu injection (Premna fulva Craib.) using recycling counter-current chromatography // J. Chromatogr. A. 2019. V. 1599. P. 180.
Kostanyan A.E., Modeling of closed-loop recycling liquid-liquid chromatography: Analytical solutions and model analysis // J. Chromatogr. A. 2015. V. 1406. P. 156.
Kostanyan A.E. Simple equations to simulate closed-loop recycling liquid–liquid chromatography: Ideal and non-ideal recycling models // J. Chromatogr. A. V. 2015. V. 1423. P. 71.
Kostanyan A.E., Erastov A. Theoretical study of closed-loop recycling liquid-liquid chromatography and experimental verification of the theory // J. Chromatogr. A. 2016. V. 1462 P. 55.
Lee Y.W. Dual counter-current chromatography – Its applications in natural products research // J. Chromatogr. A. 1991. V. 538. P. 37.
Delannay E., Toribio A., Boudesocque L., Nuzillard J.-M., Zeches-Hanrot M., Dardennes E., Dour G. Le, Sapi J., Renault J.-H. Multiple dual-mode centrifugal partition chromatography, a semi-continuous development mode for routine laboratory-scale purifications // J. Chromatogr. A. 2006. V. 1127. P. 45.
Hewitsona P, Ignatova S, Ye H, Chen L, Sutherland I. Intermittent counter-current extraction as an alternative approach to purification of Chinese herbal medicine // J. Chromatogr. A. 2009. V. 1216. P. 4187.
Rubioa N., Ignatova S., Minguillóna C., Sutherland I. Multiple dual-mode countercurrent chromatography applied to chiral separations using a (S)-naproxen derivative as chiral selector // J. Chromatogr. A. 2009. V. 1216. P. 8505.
Yang Y., Aisa H.A., Ito Y. Mathematical model of computer-programmed intermittent dual countercurrent chromatography applied to hydrostatic and hydrodynamic equilibrium systems // J. Chromatogr. A. 2009. V. 1216. P. 6310.
Aihua P., Haoyu Y., Jie S., Shichao H., Shijie Z., Shucai L., Lijuan C. Separation of honokiol and magnolol by intermittent counter-current extraction // J. Chromatogr. A. 2010. V. 1217. P. 5935.
Ignatova S., Hewitson P., Mathews B., Sutherland I. Evaluation of dual flow counter-current chromatography and intermittent counter-current extraction // J. Chromatogr. A. 2011. V. 1218. P. 6102.
Hewitson P., Ignatova S., Sutherland I. Intermittent counter-current extraction – Effect of the key operating parameters on selectivity and throughput // J. Chromatogr. A, 2011. V. 1218. P. 6053.
Mekaoui N., Berthod A. Using the liquid nature of the stationary phase. VI. Theoretical study of multi-dual mode countercurrent chromatography // J. Chromatogr. A. 2011. V. 1218. P. 6061.
Goll J., Frey A., Minceva M. Study of the separation limits of continuous solid support free liquid–liquid chromatography: Separation of capsaicin and dihydrocapsaicin by centrifugal partition chromatography // J. Chromatogr. A. 2013. V. 1284. P. 59.
Huang X.-Y., Ignatova S., Hewitson P., Di D.-L. An overview of recent progress in elution mode of counter current chromatography // Trends Anal. Chem. 2016. V. 77. P. 214.
Goll J., Morley R., Minceva M. Trapping multiple dual mode centrifugal partition chromatography for the separation of intermediately-eluting components: Operating parameter selection // J. Chromatogr. A. 2017. V. 1469. P. 68.
Morley R., Minceva M. Operating mode selection for the separation of intermediately-eluting components with countercurrent and centrifugal partition chromatography // J. Chromatogr. A. 2019. V. 1594. P. 140.
Hewitson P., Sutherland I., Kostanyan A.E., Voshkin A.A., Ignatova S. Intermittent counter-current extraction – Equilibrium cell model, scaling and an improved bobbin design // J. Chromatogr. A. 2013. V. 1303. P. 18.
Kostanyan A.E., Ignatova S., Sutherland I., Hewitson P., Zakhodjaeva Y.A., Erastov A.A. Steady-state and non-steady state operation of counter-current chromatography devices // J. Chromatogr. A. 2013. V. 1314. P. 94.
Костанян А.Е., Холькин А.И., Ерастов А.А., Белова В.В. Противоточно-циклический способ многоступенчатого экстракционного разделения смеси компонентов. Патент № 2014144416 РФ. 2015.
Kostanyan A.E., Erastov A.A., Shishilov O.N. Multiple dual mode counter-current chromatography with variable duration of alternating phase elution steps // J. Chromatogr. A. 2014. V. 1347. P. 87.
Kostanyan A.E., Belova V.V. Closed-loop recycling dual-mode counter-current chromatography. A theoretical study // J. Chromatogr. A. 2019. V. 1588. P. 174.
Kostanyan A.E., Galieva Z.N. Modeling of closed-loop recycling dual-mode counter-current chromatography based on non-ideal recycling model // J. Chromatogr. A. 2019. V. 1603. P. 240.
Berthod A., Friesen J.B., Inui T., Pauli G.F. Elution–extrusion countercurrent chromatography: Theory and concepts in metabolic analysis // Anal. Chem. 2007. V. 79. P. 3371.
Kostanyan A.E. Modelling of elution–extrusion counter-current chromatography using perfect replacement approach // J. Chromatogr. A. 2011. V. 1218. P. 6412.
Ma T., Dong H., Lu H., Zhao H., Guo L., Wang X. Preparative separation of caffeoylquinic acid isomers from Lonicerae japonicae Flos by pH-zone-refining counter-current chromatography and a strategy for selection of solvent systems with high sample loading capacities // J. Chromatogr. A. 2018. V. 1578. P. 61.
Costa F.D.N., Vieira M.N., Garrard I., Hewitson P., Jerz G., Leitão G.G., Ignatova S. Schinus terebinthifolius countercurrent chromatography (Part II): Intra-apparatus scale-up and inter-apparatus method transfer // J. Chromatogr. A. 2016. V. 1466. P. 76.
Müller M., Wasmer K., Vetter W. Multiple injection mode with or without repeated sample injections: Strategies to enhance productivity in countercurrent chromatography // J. Chromatogr. A. 2018. V. 1556. P. 88.
Peng A., Hewitson P., Sutherland I., Chen L., Ignatova S. How changes in column geometry and packing ratio can increase sample load and throughput by a factor of fifty in counter-current chromatography // J. Chromatogr. A. 2018. V. 1580. P. 120.
Delannay E., Toribio A., Boudesocque L., Nuzillard J.M., Zeches-Hanrot M., Dardennes E., Le Dour G., Sapi J., Renault J.-H. Multiple dual-mode centrifugal partition chromatography, a semi-continuous development mode for routine laboratory-scale purifications // J. Chromatogr. A. 2006. V. 1127. P. 45.
Kostanyan A., Martynova M. Modeling of two semi-continuous methods in liquid-liquid chromatography: Comparing conventional and closed-loop recycling modes // J. Chromatogr. A. 2020. V. 1614. Article 460 735.
Kostanyan A.E. Multiple dual mode counter-current chromatography with periodic sample injection: Steady-state and non-steady-state operation // J. Chromatogr. A. 2014. V. 1373. P. 81.
Kostanyan A.E., Erastov A.A. Steady state preparative multiple dual mode counter-current chromatography: Productivity and selectivity. Theory and experimental verification // J. Chromatogr. A . 2015. V. 1406. P. 118.
Kostanyan A.E., Shishilov O.N. An easy-to-use calculating machine to simulate steady state and non-steady-state preparative separations by multiple dual mode counter-current chromatography with semi-continuous loading of feed mixtures // J. Chromatogr. A. 2018. V. 1552 P. 92.
Kostanyan A.E. Theoretical study of separation and concentration of solutes by closed-loop recycling liquid-liquid chromatography with multiple sample injection // J. Chromatogr. A. 2017. V. 1506. P. 82.
Kostanyan A., Martynova M., Erastov A., Belova V. Simultaneous concentration and separation of target compounds from multicomponent mixtures by closed-loop recycling countercurrent chromatography // J. Chromatogr. A. 2018. V. 1560. P. 26.
Костанян А.Е., Вошкин А.А., Холькин А.И., Белова В.В. Пульсационно-циклический способ экстракционного разделения смеси компонентов и устройство для его осуществления. // Патент № 2403949 РФ, 2010. (PCT appl. Pub No WO/2011/043687. 14.04.2011).
Kostanyan A.E., Voshkin A.A., Kodin N.V. Pulsed cyclic device for liquid countercurrent chromatography // Theor. Found. Chem. Eng. 2011. V. 45. № 5. P. 779.
Kostanyan A.E., Voshkin A.A. Support-free pulsed liquid-liquid chromatography // J. Chromatogr. A. 2009. V. 1216. P. 7761.
Kostanyan A.E., Voshkin A.A., Kodin N.V. Controlled-cycle pulsed liquid–liquid chromatography. A modified version of Craig’s counter-current distribution // J. Chromatogr. A. 2011. V. 1218. P. 6135.
Костанян А.Е. Экстракционно-хроматографическое разделение редкоземельных металлов в каскаде центробежных экстракторов // Журн. неорг. химии. 2017. Т. 62. № 12. С. 1666. (Kostanyan A.E. Extraction chromatographic separation of rare-earth metals in a cascade of centrifugal extractors // J. Inorg. Chem. 2017. V. 63. P. 287.)
Kostanyan A.E., Erastov A.A. Industrial countercurrent chromatography separations based on a cascade of centrifugal mixer-settler extractors // J. Chromatogr. A. 2018. V. 1572. P. 212.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии