

Журнал аналитической химии, 2020, T. 75, № 5, стр. 413-421
Процедура осаждения железа, хрома, молибдена, вольфрама при определении мышьяка и сурьмы в легированной стали
А. В. Майорова a, b, *, А. А. Белозерова a, b, Т. Г. Окунева a, К. Ю. Шуняев a, b
a Институт металлургии Уральского отделения Российской академии наук
620016 Екатеринбург, ул. Амундсена, 101, Россия
b Уральский федеральный университет имени первого Президента России Б.Н. Ельцина
620016 Екатеринбург, ул. Мира, 19, Россия
* E-mail: imeturoran@mail.ru
Поступила в редакцию 18.02.2019
После доработки 22.05.2019
Принята к публикации 06.12.2019
Аннотация
Исследованы возможность разделения микропримесей As и Sb и макрокомпонентов Fe, Cr, Mo, W осаждением последних, а также закономерности соосаждения As и Sb. Ингибирование соосаждения As и Sb при осаждении Fe, Cr, W, Mo возможно с помощью введения определенного количества фтороводородной кислоты. Это приводит к уменьшению удельной поверхности и пористости осадков и ингибированию процесса соосаждения. Разработан способ одновременного определения As и Sb в легированной стали методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой с предварительным отделением основных компонентов Fe, Cr, Mo, W.
Присутствие небольших количеств As и Sb в легированных сталях приводит к охрупчиванию материала [1]. Содержание этих примесей в сталях строго регламентировано действующими государственными стандартами (например, [2, 3]). В ГОСТах [4, 5] для определения As и Sb в данных объектах рекомендуются спектрофотометрия, потенциометрия, электротермическая атомно-абсорбционная спектрометрия. Соответствующие методики анализа продолжительны и трудоемки, требуют предварительного отделения As и Sb от основных компонентов материала (Fe, Cr, Ni, Mo, W и т.д.) и не позволяют проводить их одновременное определение. Целесообразно разработать простую в исполнении методику одновременного определения As и Sb с использованием современного аналитического оборудования.
Метод атомно-эмиссионной спектрометрии с индуктивно связанной плазмой (АЭС-ИСП) характеризуется экспрессностью измерений, широким линейным диапазоном определяемых концентраций и возможностью одновременного многоэлементного анализа. Эти достоинства обусловили внедрение метода АЭС-ИСП в практику работы многих лабораторий [6]. Однако разработка АЭС-ИСП-методики определения микропримесей As и Sb – достаточно сложная задача, так как данные элементы имеют высокие значения потенциалов возбуждения спектральных линий [7, 8]. Сведения о методиках прямого АЭС-ИСП-определения As и Sb в сложных металлургических объектах (стали, сплавы) отсутствуют из-за спектральных помех от присутствующих в составе материала макрокомпонентов.
В лабораторной практике при АЭС-ИСП-определении As и Sb используют процедуры отделения от матрицы, например, выделяют аналиты генерацией их гидридов [9–11], отделяют мышьяк экстракцией от железосодержащей основы [12]. Указанные процедуры имеют недостатки: экстракционное отделение от железосодержащей матрицы [12] трудоемко, его эффективность зависит от концентрации азотной кислоты, которую необходимо строго регулировать; для получения легколетучих гидридов AsH3, SbH3 требуется наличие гидридного генератора, также необходимо ингибировать интерференции от переходных металлов [13–15]. В работе [15] показано, что присутствие Cr, Fe, Mo, W, Ni и Si негативно влияет на образование гидридов аналитов. Для ингибирования интерференций применяют различные способы. Например, в анализируемый раствор вводят винную, этилендиаминтетрауксусную кислоты, L-цистеин либо используют дополнительные процедуры отделения матрицы от аналитов [13, 14].
Ранее установлено [16, 17], что присутствие в анализируемом растворе макроколичеств W, Cr и Fe негативно влияет на точность АЭС-ИСП-определения As и Sb. Нами обнаружено, что подобное влияние оказывает присутствие молибдена в растворе. В работах [18, 19] описана возможность полного осаждения макроколичеств Fe, Cr, W, и Mo с помощью фторида натрия или ацетата свинца, однако поведение As и Sb в этих условиях не изучено.
Таким образом, разработка простого, экспрессного и экономичного способа предварительного отделения As и Sb от основных компонентов стали (Fe, Cr, Mo, W) остается актуальной задачей. Ее решение позволит одновременно определять методом АЭС-ИСП малые количества указанных аналитов с высокой точностью без использования гидридного генератора.
Цель настоящей работы – разработать процедуру осаждения и отделения Fe, Cr, Mo, W от As и Sb, позволяющую одновременно определять аналиты методом АЭС-ИСП.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление растворов. В термостойкие фторопластовые стаканы добавляли одновременно количество раствора ионов Cr(III), Fe(III), W(VI), Mo(VI), As(III) и Sb(III), соответствующее следующему составу стали, мас. %: Cr 15, W 15, Mo 15, Fe 55, As 0.002 и Sb 0.002, а для построения изотерм адсорбции содержание аналитов варьировали от 0.001 до 0.1 мас. %. Добавляли 20 мл смеси смеси конц. HCl и HNO3 (3 : 1, по объему) и определенный объем конц. HF (37.5 мас. %, от 0 до 40 мл). Нагревали на электроплите до начала кипения раствора (75°C), затем для отделения матрицы использовали две процедуры: 1) добавляли порциями при постоянном перемешивании 15 г NaF для создания мольного избытка осадителя NaF/Fe(III), Cr(III) > 32; 2) приливали порциями при постоянном перемешивании 75 мл CH3COOH (95 мас. %), 30 мл раствора Pb(CH3COO)2 с концентрацией 50.0 г/л для создания мольного избытка осадителя Pb(II)/W(VI), Mo(VI) > 2.5. По индикаторной бумаге (рН 4.0–5.6) доводили pH до значения 4.3, приливая порциями при постоянном перемешивании 2 М раствор NaOH. Образовавшийся осадок выдерживали в течение 15 мин при температуре кипения раствора, отфильтровывали через фильтр “белая лента” и промывали дистиллированной водой, уксусной кислотой. Полученные таким образом осадки отбрасывали или сохраняли для дальнейших исследований. Фильтрат переносили в мерную колбу из полипропилена, разбавляли дистиллированной водой до метки и перемешивали. При необходимости использовали процедуру разбавления. Полученные растворы анализировали на содержание As, Sb, Fe, Cr, Mo и W методом АЭС-ИСП.
Одновременно с пробами готовили холостой раствор, содержащий все компоненты, кроме ионов Mo(VI), W(VI), Cr(III), Fe(III), As(III) и Sb(III), с аналогичными концентрациями, используемыми при приготовлении анализируемых растворов.
Условия измерения атомной эмиссии W, Cr, Fe, As и Sb. Использовали АЭС-ИСП-спектрометр Optima 2100 DV (Perkin Elmer) с кварцевой горелкой. Операционные параметры спектрометра: высокочастотная мощность – 1500 Вт, пробоподающий поток аргона – 0.75 л/мин, вспомогательный поток аргона – 0.2 л/мин, плазмообразующий поток аргона – 15.0 л/мин, способ наблюдения плазмы – радиальный, высота наблюдения – 15 мм, скорость подачи раствора – 0.9 мл/мин, время распыления образца – 40 с, число измерений прибором одной пробы – 2. Применяли распылительную систему, устойчивую к агрессивному воздействию фтороводородной кислоты. Аналитические спектральные линии: As I 189.042, Sb I 206.836, Fe I 302.107, Сr II 206.158, W II 207.912 и Mo II 202.031 нм.
Градуировка спектрометра. Растворы для градуировки спектрометра готовили разбавлением государственных стандартных образцов состава растворов ионов W, Cr, Fe, As и Sb. При этом в них добавляли аликвоты холостого раствора так, чтобы концентрации кислот, ацетата свинца и ионов натрия, которые содержатся в холостом растворе, соответствовали их концентрациям в анализируемых растворах. Концентрации определяемых элементов в растворах для градуировки представлены в табл. 1.
Таблица 1.
Концентрации (мг/л) ионов As, Sb (микрокомпоненты), Cr, Fe, W и Mo (макрокомпоненты) в градуировочных растворах
| № раствора для градуировки спектрометра | As | Sb | Mo | Fe | Cr | W |
|---|---|---|---|---|---|---|
| 1 | 1.00 | 0.50 | – | – | – | – |
| 2 | 0.50 | 0.10 | – | – | – | – |
| 3 | 0.10 | 1.00 | – | – | – | – |
| 4 | – | – | 10.0 | 100.0 | 50.0 | 50.0 |
| 5 | – | – | 50.0 | 10.0 | 100.0 | 10.0 |
| 6 | – | – | 100.0 | 50.0 | 10.0 | 100.0 |
Определение параметров поверхности и пористости. Значения удельной поверхности и пористости осадков определяли методом низкотемпературной адсорбции–десорбции азота на приборе Nova1200e (Quantachrome, США). Порошки осадков предварительно прокаливали на воздухе при 500°C в течение 5 ч для удаления остаточной влаги.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Исследовали осаждение макрокомпонентов W, Mo, Cr и Fe и их отделение от As и Sb. На рис. 1 представлены спектры содержащих As, Sb, W, Mo, Cr и Fe растворов, полученных с использованием двух процедур отделения матрицы и без отделения.
Рис. 1.
Аналитические спектральные линии As 189.042 (а), Sb 206.836 (б), W II 207.912 (в), Mo II 202.031 (г), Fe I 302.107 (д), Сr II 206.158 нм (е) для градуировочного раствора, содержащего As, Sb – 1.0, Fe, Mo, W, Сr – 100 мг/л (1); исходного раствора, имитирующего состав стандартного образца состава легированной стали (2); исходного раствора с последующим осаждением мешающих компонентов реагентом NaF (3) и реагентом Pb(CH3COO)2 и NaОН в присутствии CH3COOH (4).
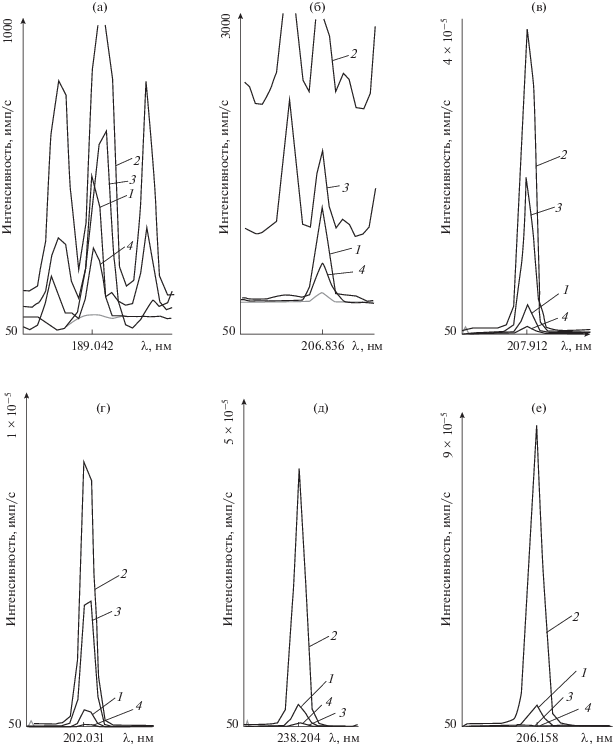
Установлено, что применение процедуры осаждения матрицы с помощью NaF приводит к успешному отделению макроколичеств Cr и Fe. По результатам рентгенофазового анализа осаждается кристаллический осадок состава Na3FeF6–Na3CrF6 [20], см. уравнения (1), (2).. Однако в фильтрате в большом количестве остаются W и Mo, а из-за спектральных помех от линий данных элементов точное определение As и Sb невозможно (рис. 1а, 1б). Использование процедуры отделения макроколичеств Fe, Cr, W и Mo с помощью Pb(CH3COO)2 и NaОН приводит к полному их осаждению в виде осадка Na3FeF6–Na3CrF6–PbMoO4–PbWO4, см. уравнения (3)–(6) [20, 21].
(1)
${\text{F}}{{{\text{e}}}^{{{\text{3}} + }}} + {\text{6NaF}} = {\text{N}}{{{\text{a}}}_{{\text{3}}}}{\text{Fe}}{{{\text{F}}}_{{\text{6}}}}{\kern 1pt} \downarrow + {\text{ 3N}}{{{\text{a}}}^{ + }},$(2)
${\text{C}}{{{\text{r}}}^{{{\text{3}} + }}} + {\text{6NaF}} = {\text{N}}{{{\text{a}}}_{{\text{3}}}}{\text{Cr}}{{{\text{F}}}_{{\text{6}}}}{\kern 1pt} \downarrow + {\text{ 3N}}{{{\text{a}}}^{ + }},$(3)
${\text{P}}{{{\text{b}}}^{{{\text{2}} + }}} + {\text{WO}}_{{\text{4}}}^{{{\text{2}} - }} = {\text{PbW}}{{{\text{O}}}_{{\text{4}}}}{\kern 1pt} \downarrow ,$(4)
${\text{P}}{{{\text{b}}}^{{{\text{2}} + }}} + {\text{MoO}}_{{\text{4}}}^{{{\text{2}} - }} = {\text{PbMo}}{{{\text{O}}}_{{\text{4}}}} \downarrow ,$(5)
${\text{3N}}{{{\text{a}}}^{ + }} + {\text{F}}{{{\text{e}}}^{{{\text{3}} + }}} + {\text{6}}{{{\text{F}}}^{--}} = {\text{N}}{{{\text{a}}}_{{\text{3}}}}{\text{Fe}}{{{\text{F}}}_{{\text{6}}}}{\kern 1pt} \downarrow ,~$(6)
${\text{3N}}{{{\text{a}}}^{ + }} + {\text{C}}{{{\text{r}}}^{{{\text{3}} + }}} + {\text{6}}{{{\text{F}}}^{--}} = {\text{N}}{{{\text{a}}}_{{\text{3}}}}{\text{Cr}}{{{\text{F}}}_{{\text{6}}}}{\kern 1pt} \downarrow {\text{.}}$Установлено, однако, что при отделении макрокомпонентов этими способами наблюдаются потери аналитов из-за их соосаждения. Таким образом, при условии ингибирования процесса соосаждения As и Sb на осадках Na3FeF6–Na3CrF6 и Na3FeF6–Na3CrF6–PbMoO4–PbWO4 можно рекомендовать процедуры осаждения макрокомпонентов легированных сталей Fe и Cr реагентом NaF и Fe, Cr, W и Mo реагентом Pb(CH3COO)2, NaОН (см. марки стали [2]).
Применение различных моделей изотерм адсорбции для описания процесса соосаждения мышьяка и сурьмы. Для интерпретации полученных экспериментальных данных по соосаждению As и Sb использовали модели равновесных изотерм адсорбции: Ленгмюра, Фрейндлиха, Дубинина–Радушкевича, Брунауэра–Эмметта–Теллера (БЭТ). Подробное описание используемых моделей и алгоритма построения изотерм сорбции даны в работах [17, 22–25]. Рассчитанные нами значения адсорбционных параметров приведены в табл. 2.
Таблица 2.
Рассчитанные значения адсорбционных параметров моделей Ленгмюра, Фрейндлиха, Дубинина–Радушкевича и Брунауэра–Эмметта–Теллера
| Модель | Параметр модели | Процедуры осаждения с реагентами | |||
|---|---|---|---|---|---|
| NaF | Pb(CH3COO)2, NaOH | ||||
| Sb | As | Sb | As | ||
| Ленгмюра [22] | KL, л/ммоль | 0.193 | 2.584 | 0.254 | 1.654 |
| am, ммоль/г | 13.7 | 4.0 | 6.9 | 7.0 | |
| R2 | 0.9811 | 0.9324 | 0.7651 | 0.7478 | |
| Фрейндлиха [23] | KF, (ммоль/г)·(л/ммоль)1/n | 0.0417 | 0.526 | 0.110 | 2.44 |
| 1/n | 0.7 | 0.8 | 0.9 | 1.7 | |
| R2 | 0.9953 | 0.9349 | 0.9835 | 0.9651 | |
| Дубинина–Радушкевича [24] | k, моль2/кДж2 | 0.005 | 0.005 | 0.007 | 0.007 |
| Е, кДж/моль | 9.7 | 10.0 | 8.7 | 8.5 | |
| R2 | 0.9972 | 0.9574 | 0.9829 | 0.9890 | |
| БЭТ [25] | KБЭТ, г/ммоль | 1346 | 3986 | 830 | 291 |
| am, ммоль/г | 0.014 | 0.064 | 0.042 | 0.75 | |
| R2 | 0.9812 | 0.9325 | 0.7444 | 0.7944 | |
Видно, что для описания процесса соосаждения As и Sb на осадках макрокомпонентов для обеих процедур осаждения лучше всего подходит модель Дубинина–Радушкевича и Фрейндлиха (наибольшие значения R2). В случае модели Ленгмюра для обоих аналитов R2 имеет наименьшее значение. Согласно теории [22–25] на поверхности осадков находится много активных центров с разной энергией, что служит доказательством ее гетерогенности. Таким образом, соосаждение мышьяка и сурьмы можно рассматривать как процесс заполнения объема микропор осадка, содержащего макроколичества Fe, Cr либо Fe, Cr, W и Mo.
Модель Дубинина–Радушкевича позволяет рассчитать среднюю свободную энергию адсорбции (7) [26]:
По значению величины E в уравнении изотермы Дубинина–Радушкевича можно судить о природе сил взаимодействия между As и Sb с активными центрами поверхности осадка, выяснить, имеют они физический или химический характер. Значения Е для As и Sb при использовании процедуры осаждения с NaF составляет 10.0 и 9.7 соответственно, а для процедуры с Pb(CH3COO)2, NaОН – 8.5 и 8.7, т.е. для обоих аналитов находятся в диапазоне 8–16 кДж/моль. В этом случае согласно теории [27] закрепление мышьяка и сурьмы в микропорах осадка происходит в результате химической ионообменной реакции.
Влияние фтороводородной кислоты на соосаждение мышьяка и сурьмы при отделении матричных элементов. Поскольку соосаждение As и Sb на осадках макрокомпонентов, полученных при осаждении с реагентами NaF или Pb(CH3COO)2, NaОН, обусловлено хемосорбцией по ионообменному механизму, процедуры разбавления, перемешивания, увеличения температуры осаждения не приведут к ингибированию процесса. При хемосорбционном механизме необходимо предотвратить эффект перенасыщения раствора и ингибировать процесс образования большого количества зародышей кристаллов во время формирования осадка, тем самым укрупняя его и уменьшая количество пор [28]. Для уменьшения относительного перенасыщения необходимо увеличить растворимость образующегося осадка и снизить концентрации осаждаемых ионов (Fe, Cr, W, Mo) в растворе. Очень эффективным способом понижения концентрации осаждаемого иона является связывание его в комплексное соединение средней прочности. В нашем случае целесообразно применить в качестве комплексообразующего агента фтороводородную кислоту, поскольку ее используют при кислотном разложении вольфрамсодержащих материалов и для предотвращения осаждения вольфрамовой кислоты.
Исследовано влияние фтороводородной кислоты на процесс соосаждения As и Sb при отделении Fe, Cr, W и Mo. В табл. 3 показаны изменения значений параметров удельной поверхности и пористости осадков, полученных в присутствии и в отсутствие фтороводородной кислоты при осаждении макрокомпонентов Fe, Cr, W и Mo.
Таблица 3.
Результаты определения параметров поверхности и пористости осадков макрокомпонентов, полученные методом низкотемпературной адсорбции–десорбции азота
| Параметр поверхности | Осаждение с реагентами | |||
|---|---|---|---|---|
| NaF + V(HF), мл | Pb(CH3COO)2, NaOH + V(HF), мл | |||
| 0 | 15 | 5 | 15 | |
| Удельная поверхность по методу БЭТ, м2/г | 1.9 | 1.4 | 3.8 | 1.7 |
| Удельная поверхность мезопор (t-метод), м2/г | 1.9 | 1.3 | 3.8 | 1.7 |
| Удельная поверхность микропор (t-метод), м2/г | 0.06 | 0.15 | 0 | 0 |
| Суммарный объем пор (в диапазоне от 0 до 154 нм), см3/г | 0.005 | 0.002 | 0.020 | 0.005 |
| Средний диаметр пор, нм | 10.4 | 6.4 | 21.2 | 11.3 |
| Диаметр доминирующего количества пор, нм | 3.1 | 3.9 | 4.0 | 3.1 |
| Структура пор (на основании анализа петли гистерезиса) | Открытая | Открытая | Открытая | Открытая |
Для всех образцов характерен относительно низкий уровень удельной поверхности и пористости. Значения удельной поверхности и пористости осадков, содержащих Fe, Cr, W и Mo, больше, чем содержащих Fe и Cr, следовательно, степень соосаждения аналитов на таких осадках выше. Значения удельной поверхности осадков, полученных в условиях эксперимента при использовании HF, в 1.4 раза (NaF) и в 2.2 раза (Pb(CH3COO)2 и NaОН) меньше, чем без ее использования (или минимального количества). При осаждении с NaF и Pb(CH3COO)2, NaОН в присутствии 15 мл HF (37.5 мас. %) средний диаметр пор осадков снижается в 1.6 и 1.9 раз, а суммарный объем пор – в 2.2 и 4 раза соответственно. Также установлено, что осадки макрокомпонентов обладают открытой пористостью. Таким образом, доказано, что при осаждении макрокомпонентов с помощью NaF или Pb(CH3COO)2, NaОН в присутствии HF получаются осадки с меньшими удельной поверхностью и пористостью, чем без ее использования (или при минимальном количестве).
Результаты АЭС-ИСП-анализа фильтратов, полученных после процедуры отделения макрокомпонентов, представлены на рис. 2а, 2б. Видно, что увеличение в условиях эксперимента объема фтороводородной кислоты до 15 мл для обоих способов осаждения макрокомпонентов позволяет существенно ингибировать потерю аналитов из анализируемого раствора. Относительное содержание As и Sb в растворе более 95 (NaF) и 90% (Pb(CH3COO)2, NaОН) соответственно. Последующее увеличение в условиях эксперимента объема HF с 15 до 20 мл приводит к частичному растворению осадка. При использовании реагента NaF в анализируемый раствор переходит хром, а при использовании Pb(CH3COO)2, NaОН переходят вольфрам и хром. Присутствие в фильтрате W и Cr с концентрацией более 100 и 1 мг/мл соответственно приводит к спектральным помехам и снижает точность АЭС-ИСП-определения As и Sb, а именно относительное содержание аналитов становится больше 100% (см. рис. 2, кривые для As и Sb при объеме HF 20–40 мл).
Рис. 2.
Зависимости относительного содержания (%) элементов в жидкой фазе (относительно начального) от объема HF (37.5 мас. %) после отделения осадка реагентами NaF (а), Pb(CH3COO)2 и NaОН (б).
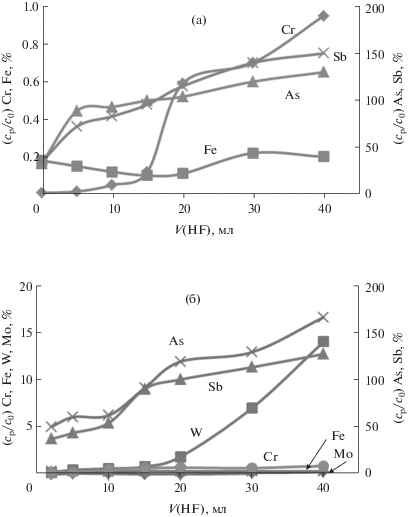
Таким образом, при отделении макроколичеств Fe, Cr, W, Mo для нивелирования процесса соосаждения мышьяка и сурьмы в максимальной степени необходимо введение 15 мл HF (37.5 мас. %).
Одновременное АЭС-ИСП-определение мышьяка и сурьмы в легированной стали. Для экспериментальной проверки эффективности применения найденных условий ингибирования процесса соосаждения As и Sb при отделении от Fe, Cr, W, Mo использовали государственные стандартные образцы состава легированной стали.
Пробоподготовка стандартных образцов к АЭС-ИСП-определению мышьяка и сурьмы включала две стадии: 1) растворение в присутствии 15 мл HF; 2) осаждение и отделение мешающих элементов реагентами NaF или Pb(CH3COO)2,NaОН (как описано в “Экспериментальной части”). Результаты одновременного АЭС-ИСП-определения мышьяка и сурьмы в стандартных образцах приведены в табл. 4. Погрешность анализа рассчитывали как ${{{{t}_{{\left( {0.{\text{95}};\,{\text{4}}} \right)}}}s} \mathord{\left/ {\vphantom {{{{t}_{{\left( {0.{\text{95}};\,{\text{4}}} \right)}}}s} {\sqrt n }}} \right. \kern-0em} {\sqrt n }},$ где t – коэффициент Стьюдента (для двухстороннего распределения), равный 2.78 при количестве измерений n = 5 и доверительной вероятности P = 0.95, s – стандартное отклонение. Из данных табл. 4 видно, что аттестованные и найденные значения массовой доли As и Sb практически во всех случаях совпадают в пределах случайной погрешности. Разница между аттестованными и найденными значениями содержания As и Sb не превышает нормативов, приведенных в ГОСТах [4, 5]. Полученные результаты доказывают эффективность предложенной методики определения As и Sb при отделении от макроколичеств Fe, Cr или Fe, Cr, W, Mo.
Таблица 4.
Результаты определения содержания мышьяка и сурьмы в стандартных образцах методом атомно-эмиссионной пектрометрии с индуктивно связанной плазмой
| Стандартный образец |
As | Sb | ||
|---|---|---|---|---|
| аттестовано | найдено | аттестовано | найдено | |
| Осаждение Fe, Cr реагентом NaF | ||||
| ГСО 9289-2009 (УГ87) |
0.116 ± 0.002 | 0.117 ± 0.004 | 0.0012 ± 0.0002 | 0.0013 ± 0.0004 |
| ГСО 9289-2009 (УГ 88) |
0.0007 ± 0.0001 | 0.0007 ± 0.0001 | 0.0003 ± 0.0001 | 0.0003 ± 0.0002 |
| ГСО 9289-2009 (УГ 89) |
0.0043 ± 0.0003 | 0.0044 ± 0.0004 | 0.0011 ± 0.0001 | 0.0011 ± 0.0002 |
| ГСО 9289-2009 (УГ 90) |
0.0044 ± 0.0003 | 0.0043 ± 0.0005 | 0.0011 ± 0.0001 | 0.0011 ± 0.0004 |
| ГСО 9289-2009 (УГ 91) |
0.0004 ± 0.0001 | 0.0004 ± 0.0002 | 0.00009 ± 0.00002 | н/н* |
| ГСО 9289-2009 (УГ 92) |
0.0027 ± 0.0003 | 0.0029 ± 0.0006 | 0.0005 ± 0.0001 | 0.0006 ± 0.0002 |
| Осаждение Fe, Cr, W, Mo реагентом Pb(CH3COO)2, NaОН | ||||
| ГСО 1134-93П (С42Г) |
~0.005** | 0.005 ± 0.001 | н/а*** | н/н |
| ГСО 1416-89П (С43Г) |
~0.004** | 0.003 ± 0.001 | н/а | н/н |
| ГСО 1052-91П (С28Д) |
~0.002** | 0.0020 ± 0.0002 | 0.0004 ± 0.0001 | 0.0005±0.0001 |
| ГСО 1495-89П (С27Г) |
~0.006** | 0.0070 ± 0.0005 | н/а | н/н |
Благодарим к. х. н. М.А. Машковцева за определение параметров поверхности и пористости образцов и ценные замечания, сделанные при обсуждении работы. Работа выполнена в рамках Государственного задания ИМЕТ УрО РАН с использованием оборудования центра коллективного пользования “Урал-М”, при финансовой поддержке постановления № 211 Правительства Российской Федерации, контракт № 02.A03.21.0006.
Список литературы
Гудремон Э.А. Специальные стали. Т. 2. М.: Металлургия, 1966. 540 с.
ГОСТ 5632-2014. Легированные нержавеющие стали и сплавы коррозионно-стойкие, жаростойкие и жаропрочные. Марки. М.: Стандартинформ, 2015. 49 с.
ГОСТ 19281-89. Прокат из стали повышенной прочности. Общие технические условия. М.: Стандартинформ, 2009. 14 с.
ГОСТ 12358-2002. Стали легированные и высоколегированные. Методы определения мышьяка. М.: Стандартинформ, 2008. 8 с.
ГОСТ 12362-79. Стали легированные и высоколегированные. Методы определения микропримесей сурьмы, свинца, олова, цинка и кадмия. М.: Издательство стандартов, 1981. 41 с.
Пупышев А.А., Данилова Д.А. Использование атомно-эмиссионной спектрометрии с индуктивно-связанной плазмой для анализа материалов и продуктов черной металлургии // Аналитика и контроль. 2007. Т. 11. № 2–3. С. 131.
Sansonetti J.E., Martin W.C. Handbook of basic atomic spectroscopic data // J. Phys. Chem. Ref. Data. 2005. V. 34. P. 1559.
Schierle C., Thorne A.P. Inductively coupled plasma Fourier transform spectrometry: A study of element spectra and a table of inductively coupled plasma lines // Spectrochim. Acta B. 1995. V. 50. P. 27.
Ozaki E.A., Oliveira E. Simultaneous determination of arsenic, bismuth and antimony in steels and nickel alloys by inductively coupled plasma atomic emission spectrometry with hydride generation // J. Anal. At. Spectrom. 1993. V. 8. P. 367.
Imakita T., Inui M., Hamada K., Taniguchi M., Nakahara T. Determination of trace amounts of arsenic, bismuth and antimony in iron and steel by continuous hydride generation and ICP-AES or ICP-MS // Tetsu-to-Hagane. 1999. V. 85. № 10. P. 724.
Matsumoto A., Takeuchi H., Nakahara T. Simultaneous determination of arsenic and antimony in steels and pure coppers by high power nitrogen microwave induced plasma atomic emission spectrometry coupled with hydride generation technique // Tetsu-to-Hagane. 2002. V. 88. № 5. P. 270.
Xu Yu., Zhou J., Wang G., Zhou J., Tao G. Determination of trace amounts of lead, arsenic, nickel and cobalt in high-purity iron oxide pigment by inductively coupled plasma atomic emission spectrometry after iron matrix removal with extractant-contained resin // Anal. Chim. Acta. 2007. V. 584. P. 204.
Kumar A.R., Riyazuddin P. Chemical interferences in hydride-generation atomic spectrometry // Trends Anal. Chem. 2010. V. 29. P. 166.
Chanvaivit S., Brindle I.D. Matrix independent determination of hydride-forming elements in steels by hydride generation-inductively coupled plasma atomic emission spectrometry // J. Anal. At. Spectrom. 2000. V. 15. P. 1015.
Kujirai O., Kohri M., Yamada K., Okochi H. Simultaneous determination of arsenic, bismuth, antimony, selenium and tellurium in molybdenum by continuous hydride generation and inductively coupled plasma-atomic emission spectrometry // Anal. Sci. 1990. V. 6. № 3. P. 379.
Белозерова А.А., Майорова А.В., Печищева Н.В., Боярникова Н.Г., Шуняев К.Ю. Методика определения мышьяка, сурьмы и висмута в материалах с высоким содержанием вольфрама и меди методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой // Заводск. лаборатория. Диагностика материалов. 2016. Т. 82. № 6. С. 10.
Майорова А.В., Мельчаков С.Ю., Окунева Т.Г., Воронцова К.А., Машковцев М.А. Изучение процесса соосаждения мышьяка и сурьмы при отделении макроколичеств железа, хрома в виде Na3FeF6, Na3CrF6 // Аналитика и контроль. 2017. Т. 21. № 3. С. 281.
Анализ минерального сырья / Под ред. Книпович Ю.Н., Морачевского Ю.В. Л.: ГХИ, 1959. 1055 с.
Тананаев И.В., Дейчман Э.Н. Новый метод отделения малых количеств Zn, Ni, Co, Mn, Cd, Cu и Ti от больших количеств железа // Заводск. лаборатория. 1946. № 12. С. 30.
Maiorova A.V., Pechishcheva N.V., Shunyaev K.Yu. Separation of iron and chromium in the form of insoluble fluorides for ICP-AES determination of trace arsenic in nickel heat-resistant alloys / 15th Israeli-Russian Bi-national Workshop “The Optimization of Composition, Structure and Properties of Metals, Oxides, Composites, Nano-and Amorphous Materials”. Екатеринбург, 26–30 сентября 2016 г. P. 92.
Майорова А.В., Белозерова А.А., Мельчаков С.Ю., Машковцев М.А., Суворкина А.С., Шуняев К.Ю. Определение содержания мышьяка и сурьмы в ферровольфраме методом атомно-эмиссионной спектрометрии с индуктивно связанной плазмой // Журн. аналит. химии. 2019. Т. 74. № 7. Приложение. С. S24.
Langmuir I. The constitution and fundamental properties of solids and liquids // J. Am. Chem. Soc. 1916. V. 38. P. 2221.
Freundlich H.M.F. Over the adsorption in solution // J. Phys. Chem. 1906. V. 57. P. 385.
Дубинин М.М., Радушкевич Л.В. К вопросу об уравнении характеристической кривой для активных углей // Доклады Академии наук СССР. 1947. № 4. С. 331.
Brunauer S., Emmett P.H., Teller E. Adsorption of gases in multimolecular layers // J. Am. Chem. Soc. 1938. V. 60. № 2. P. 309.
Hobson J.P. Physical adsorption isotherms extending from ultrahigh vacuum to vapor pressure // J. Phys. Chem. 1969. V. 75. № 8. P. 2720.
Helfferich F.G. Ion Exchange. New York, USA: Dover publications Inc., 1962. 624 p.
Алексеев В.Н. Количественный анализ. М.: Химия, 1972. 504 с.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии