

Журнал аналитической химии, 2020, T. 75, № 6, стр. 483-495
Пробоподготовка биологических тканей и клеток для их анализа методом времяпролетной вторично-ионной масс-спектрометрии
А. А. Гулин a, b, *, В. А. Надточенко a, b, В. Н. Погорелова c, М. Я. Мельников b, А. Г. Погорелов c
a Федеральный исследовательский центр химической физики им. Н.Н. Семенова Российской академии наук
(ФИЦ ХФ РАН)
119991 Москва, ул. Косыгина, 4, Россия
b Московский государственный университет имени М.В. Ломоносова, химический факультет
19991 Москва, Ленинские горы, 1, стр. 3, Россия
c Институт теоретической и экспериментальной биофизики Российской академии наук
142290 Московская обл., Пущино, ул. Институтская, 3, Россия
* E-mail: aleksandr.gulin@phystech.edu
Поступила в редакцию 26.03.2019
После доработки 04.07.2019
Принята к публикации 25.12.2019
Аннотация
Обзор посвящен применениям времяпролетной вторично-ионной масс-спектрометрии (ВИМС) для анализа биологических тканей и клеток. Использование кластерных первичных ионов существенно повысило чувствительность метода, особенно в диапазоне молекулярных масс выше нескольких сотен Да, что сделало доступным исследование липидов, аминокислот, нуклеиновых кислот, метаболитов и лекарственных препаратов. Как и в других методах, основанных на облучении поверхности образца потоком частиц, измерения в ВИМС проводят в условиях сверхвысокого вакуума, что делает пробоподготовку биологических препаратов критически важным этапом работы. Разработано несколько методик подготовки изолированных клеток, клеточных культур и тканей для такого анализа. Большинство из них основаны на классических методиках, используемых в методах гистологии и цитологии, включая оптическую, электронную и зондовую микроскопию, а также электронно-зондовый микроанализ. Однако ряд аналитических особенностей ВИМС делает необходимым адаптацию этих методик. Тем не менее, универсального решения не существует, и основной задачей данного обзора была систематизация и сравнение разработанных методик пробоподготовки для ВИМС. Кроме того, рассматривается влияние инструментальных факторов на результаты анализа биологических препаратов методом ВИМС.
Времяпролетная вторично-ионная масс-спектрометрия – один из чувствительных методов изучения состава объектов и его пространственного распределения в них путем анализа вторичных ионов, полученных при бомбардировке поверхности пучком первичных ионов. Одним из главных достоинств метода является отсутствие необходимости проведения дополнительных манипуляций с образцом: использования различных меток и красителей, нанесения матрицы, напыления тонкого слоя металла и др. Это обеспечило широкое применение метода для биофизических и прикладных медицинских исследований. Методом ВИМС исследуют срезы тканей животных и человека [1–4], прокариотические [5, 6] и эукариотические [7–9] клетки, семена и ткани растений [10–13] и т.д.
Механизм генерации вторичных ионов обусловлен ионным распылением и сильно зависит как от состояния объекта исследования (пробоподготовки), так и от параметров ионизации (типа первичных ионов, их энергии, угла наклона пучка относительно поверхности и т.д.). ВИМС позволяет рутинно получать масс-спектры, карты распределения ионов (режим имиджинга и профили распределения веществ по глубине образца (рис. 1) [14, 15]. Минимальный размер зонда первичных ионов (фокуса) в ВИМС варьируется в зависимости от вида первичных ионов и составляет от 50 нм до нескольких микрон. Оценки пространственного разрешения для биологических образцов, основанные на анализе ионных изображений, показывают, что часто лимитирующим фактором является низкий уровень интенсивности вторичных ионов. Однако при высоком уровне сигнала оцениваемое пространственное разрешение соответствует размеру зонда первичных ионов и приблизительно составляет 100–200 нм [16, 17].

Отметим, что другие методы масс-спектрометрии (матрично-активированная лазерная десорбция/ионизация, десорбционная электрораспылительная ионизация, лазерная абляция с индуктивно связанной плазмой) также позволяют проводить химическое картирование состава, но они существенно ограничены в пространственном разрешении (десятки микрон и более) [18–20]. Учитывая, что характерный размер дифференцированной клетки составляет ~10 мкм и лишь для незначительного числа видов клеток превышает 50 мкм, ВИМС является единственным методом масс-спектрального картирования на субклеточном уровне [9, 21, 22].
Наряду с латеральным распределением ионов, ВИМС позволяет проводить исследование в сагиттальной плоскости при профилировании по глубине. Суть метода заключается в последовательном удалении поверхностных слоев образца (испарении) ионным пучком. Для данной процедуры используют как основной источник первичных ионов, так и дополнительный ионный источник. Послойное картирование лежит в основе трехмерной реконструкции клеток или тканей [23–26]. Морфологическое разрешение по глубине зависит от многих факторов: энергии первичных ионов, угла, под которым производится бомбардировка, плотности образца и т.д., и при оптимальных условиях может достигать ~1 нм [27, 28]. Примером реализации столь высокого разрешения может служить работа, в которой удалось получить профиль оксидной пленки толщиной 1–4 нм на поверхности нержавеющей стали [29]. Применительно к биологическим образцам такое разрешение означает потенциальную возможность детального картирования различий в распределении липидов во внутреннем и внешнем слоях клеточных мембран при профилировании по глубине.
Поскольку метод основан на бомбардировке исследуемого образца заряженными частицами с последующим анализом вторичных ионов во времяпролетном масс-анализаторе, для функционирования масс-спектрометра требуются условия сверхвысокого вакуума (от 10–9 мбар и менее). Следовательно, накладываются дополнительные ограничения на состояние биологических объектов, которые должны быть либо в дегидратированном, либо замороженном состоянии. В связи с этим критически важным этапом эксперимента становится пробоподготовка препарата. В отличие от методов микроскопии, для анализа ВИМС требуется сохранение не только морфологии, но и химического состава образца (и его распределения), поэтому традиционные подходы гистологии и цитологии, как правило, неприменимы. Таким образом, для реализации аналитических возможностей ВИМС применительно к изучению химического состава биологических клеток необходимо сочетание пробоподготовки и оптимальных условий возбуждения вторичных ионов.
ВЛИЯНИЕ ПАРАМЕТРОВ ПЕРВИЧНЫХ ИОНОВ
Традиционно метод ВИМС применяли для элементного анализа вещества, особенно полупроводников. Внедрение источников, генерирующих кластерные ионы, в начале XXI века позволило на порядок повысить выход органических ионов на один первичный ион [30–32], что привело к активному исследованию биологических препаратов. В табл. 1 представлена информация о типах используемых первичных ионов [15, 33, 34].
Таблица 1.
Первичные ионы, используемые в масс-спектрометрии вторичных ионов
| Источник ионов | Примеры ионов | Энергия, кэВ | Минимальный размер фокуса |
|---|---|---|---|
| Электронный удар | ${\text{Ar}}_{n}^{ + },{\text{O}}_{2}^{ + },{\text{SF}}_{5}^{ + },{\text{C}}_{{60}}^{{q + }}$ | 1−40 | 0.2−5 мкм |
| Ионизация полем | Ga+, In+, ${\text{Au}}_{n}^{ + },{\text{Bi}}_{n}^{{q + }}$ | 1−60 | 10 нм |
| Ионизация на поверхности | Cs+ | 1−16 | 50 нм |
| Радиочастотная ионизация | Ar+, O−, ${\text{O}}_{2}^{ + }$ | 1−17 | 200 нм |
На практике для исследований биологических клеток и тканей, как правило, используют полиатомные ионы: ${\text{Bi}}_{n}^{{q + }}$ (n = 1, 3, 5, 7; q = 1, 2), ${\text{C}}_{{60}}^{{q + }}$ (q = = 1–3), ${\text{Ar}}_{n}^{ + }$ (n = 1000–4000), обладающие меньшей энергией на один атом. В случае использования фуллерена и кластеров аргона уместно говорить не о первичных ионах, а о заряженных наночастицах. Расчеты методом молекулярной динамики показывают, что кластерные ионы передают бо́льшую часть энергии верхним (находящимся ближе к поверхности) слоям образца, что уменьшает глубину проникновения иона [35, 36]. Эксперименты по выявлению глубины проникновения первичных ионов для органических пленок подтвердили выявленную закономерность [37].
Для оценки возможностей источника первичных ионов в ВИМС принято использовать два параметра: выход вторичных ионов и эффективность ионизации [38, 39]. Выход вторичных ионов определяют как число детектированных вторичных ионов, деленное на число всех первичных ионов, бомбардировавших образец за время эксперимента. Эффективность ионизации определяют как выход вторичных ионов на единицу площади, повреждаемой одним первичным ионом. При исследовании срезов мозга крыс показано, что при ускоряющем напряжении 25 кВ для холестерина эффективность ионизации ионами ${\text{Bi}}_{3}^{ + }$ отличается в ~6 раз по сравнению с ${\text{Bi}}_{1}^{ + }$ [30]. Лучшую эффективность показали ионы ${\text{Bi}}_{5}^{{2 + }}$ (энергия 50 кэВ, поскольку кластер двухзарядный), но их доля в потоке ${\text{Bi}}_{n}^{{q + }}$ невелика, что ограничивает использование данного вида первичных ионов [38]. Таким образом, из ряда жидкометаллических источников ионы ${\text{Bi}}_{3}^{ + }$ и ${\text{Au}}_{3}^{ + }$ наиболее востребованы для исследования биологических образцов. Кластерные ионы на основе фуллерена, получаемые методом электронного удара, обладают лучшими параметрами выхода вторичных ионов и эффективности ионизации по сравнению с жидкометаллическими ионными источниками за счет меньшей энергии на атом [38, 40].
Для режима молекулярного картирования лимитирующим фактором является пространственное разрешение, которое зависит от размера зонда (фокуса) первичных ионов. Следует отметить, что данные о минимальном размере фокуса (табл. 1) приведены для источника, работающего в режиме непрерывного тока. Поскольку большинство серийных масс-спектрометров оснащено времяпролетным масс-анализатором, что предполагает импульсный режим работы, доступные для рутинного анализа значения размера фокуса могут отличаться на порядок. В настоящее время для источников на основе висмута пространственное разрешение достигает ~100 нм [17, 41], для источников на основе фуллерена ~1 мкм [42] и ~5 мкм для аргоновых кластеров [43]. С учетом размера клеток на данный момент жидкометаллические ионные источники остаются единственным вариантом для анализа на клеточном и субклеточном уровнях.
Тип первичных ионов влияет не только на общий выход ионов, но и на соотношение пиков фрагментарных ионов. В работе [44] сравнили масс-спектры пяти белков для восьми различных первичных ионов. Для выявления корреляций и различий использовали метод главных компонент. Большинство типов первичных ионов показало существенно различающиеся соотношения фрагментарных ионов (аминокислот) для одного белка. Показано, что спектры одного белка при бомбардировке разными ионами могут отличаться сильнее, чем спектры разных белков при бомбардировке одним типом ионов. По этой причине образцы целесообразно сравнивать между собой только при одном и том же источнике ионов.
Сравнительное моделирование ионного распыления кристалла Ag (1, 1, 1), проведенное для первичных ионов Ga+, ${\text{Au}}_{3}^{ + },$ ${\text{C}}_{{60}}^{ + },$ ${\text{Ar}}_{{872}}^{ + },$ показало, что чем больше масса первичного иона, тем больше ширина и меньше глубина кратера, образованного на поверхности образца под действием пучка первичных ионов [45]. Размер кластера ${\text{Ar}}_{{872}}^{ + }$ настолько велик, что он блокирует эмиссию значительной части вторичных ионов из центральной зоны. Вероятность ионизации для кластеров аргона ниже, чем для C60, что снижает их эффективность в качестве первичных ионов для анализа. Однако эффективность распыления ${\text{Ar}}_{n}^{ + }$ высока при его взаимодействии с образцом, поэтому источник такого типа применяют в процессе послойного травления органических материалов, биологических клеток и тканей. Травление с использованием многоатомных кластеров аргона приводит к сглаживанию рельефа поверхности. Показано, что после травления шероховатость поверхности слоя лейцина уменьшилась со 180 до 130 нм [46].
Для многослойной структуры из чередующихся слоев аминокислот фенилаланина и тирозина, полученной путем термического испарения в условиях высокого вакуума, использование ${\text{Ar}}_{{1700}}^{ + }$ обеспечивает лучшее разрешение по глубине по сравнению с ${\text{C}}_{{60}}^{ + }$ и тем более с Cs+ [47, 48]. В цитируемых работах не наблюдался эффект накопления сигнала углерода, регистрируемого для остальных первичных ионов. Эффект проявляется в постепенном исчезновении сигнала сложных органических молекул и накоплении сигнала углерода, а также неспецифических фрагментарных ионов (CH2, CH3, C2H5 и т.д.). Это означает, что практически весь поврежденный материал удаляется сразу же и не оседает на поверхность.
Отметим, что на данный момент не существует универсальных первичных ионов, которые бы одинаково хорошо подходили как для спектроскопических исследований, так и для химического картирования биологических препаратов. Перспективными считаются ионы на основе кластеров из нескольких тысяч молекул воды [49] и жидкометаллические ионные источники на основе многоатомных (сотни и тысячи атомов) кластеров золота с энергиями до 1 МэВ [50]. Интересным решением может оказаться использование газов смешанного состава для бомбардировки. Например, показано увеличение выхода некоторых вторичных ионов при добавлении метана к аргоновой смеси [51].
ВЛИЯНИЕ ФАКТОРА ПРОБОПОДГОТОВКИ
Поскольку анализ ВИМС проводят в условиях сверхвысокого вакуума, биологические образцы, содержащие воду, не могут быть исследованы данным методом непосредственно. Наиболее распространенными типами биологических объектов, исследуемых с помощью ВИМС, являются клеточные линии, выращенные на подложке, и срезы мягких тканей лабораторных животных. В целом методы пробоподготовки, используемые для клеток и тканей, основаны на одних и тех же принципах, но есть и некоторые различия.
Анализ культур клеток на подложке. Рис. 2 иллюстрирует различные варианты пробоподготовки, использовавшиеся применительно к образцам культуры клеток.
Рис. 2.
Схема возможных путей пробоподготовки, применяемых для анализа культуры клеток методом ВИМС: слева – протоколы, основанные на химической фиксации клеток, справа – протоколы, основанные на криофиксации образца; пунктирной линей выделены необязательные этапы.
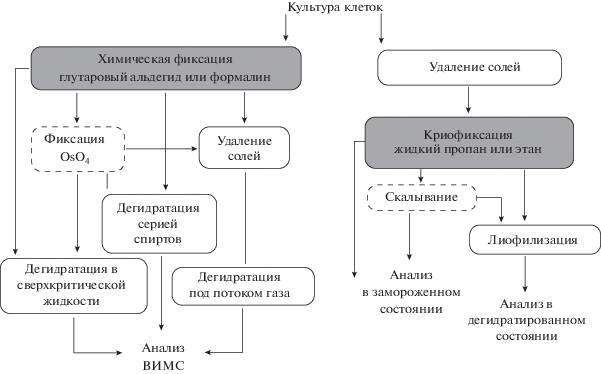
Фиксация. Ключевой момент здесь – способ фиксации клетки на начальном этапе. Наиболее распространен метод химической фиксации в глутаровом альдегиде или параформальдегиде, а также метод криофиксации в хладоагенте (рис. 2). Как правило, клетки фиксируются одним из этих двух способов, но возможно и их сочетание. Химическую фиксацию проводят путем замены на 10–30 мин инкубационной среды буферным раствором, содержащим 4% формалина или 2.5% глутарового альдегида. В результате образуется связь между аминными группами мембранных белков либо через метиленовые мостики (для формалина) [52], либо через иминную связь (глутаровый альдегид) [53]. При использовании глутарового альдегида могут образовываться небольшие (от 10 нм до нескольких мкм) поры в плазматической мембране клетки [54].
Криофиксацию осуществляют путем помещения подложки с клетками в жидкий пропан или этан, охлаждаемые до температуры жидкого азота (–196°С). Выбор хладоагента обусловлен высокой теплоемкостью вещества и относительно низкой температурой затвердевания, что обеспечивает высокую скорость замерзания внутриклеточной воды с образованием аморфного льда [55]. Важным фактором является и объем замораживаемого образца, поскольку чем он меньше, тем быстрее происходит полная заморозка препарата. По этой причине при криофиксации клеток необходимо минимизировать слой жидкости, покрывающей поверхность. В отличие от химической фиксации, криофиксация обеспечивает сохранность распределения легко диффундирующей компоненты цитоплазмы, соответствующей прижизненному состоянию клетки [54, 56, 57] и препятствует проявлению осмотических эффектов [58].
Удаление солей. ВИМС чувствителен к загрязнению анализируемой поверхности, поскольку вторичные ионы генерируются в поверхностном слое толщиной ~10 нм и менее. Клеточные культуры инкубируют в средах, содержащих электролиты и органические молекулы, поэтому обычная дегидратация оставляет на поверхности препарата слой осевших солей. Такой слой существенно затрудняет анализ, поскольку в присутствии солей сигнал биологических молекул разбивается на несколько каналов (например, [M + H], [M + Na], [M + K]), что затрудняет количественную интерпретацию спектров. Однако главная сложность заключается в том, что соли существенно уменьшают выход органических ионов, как это показано для срезов мозга крыс, липидных [59] и полипептидных пленок [60]. Сигнал вторичных ионов удавалось “восстановить” после промывки образца. Для промывки обычно используют либо изотонические растворы, либо дистиллированную воду. Наиболее подходящими изотоническими растворами являются ацетат аммония (150 мМ) [61] и формиат аммония (10 мМ, pH 7.4) [55, 60, 62]. Показано, что различные способы отмывки (NaCl, фосфатный буферный раствор, ацетат аммония) не приводят к трансмембранному перераспределению липидного состава [17].
Анализ в замороженном состоянии. Важным этапом пробоподготовки является перенос образца из криоагента в вакуумную камеру, где проводят замораживания–скалывания и/или анализ методом ВИМС. При манипуляциях с замороженным препаратом при комнатной температуре на его поверхности образуется конденсат, являющийся источником загрязнений. По этой причине образец переносят в парах жидкого азота в предварительно охлажденную до температуры жидкого азота камеру [23, 63]. Температура образца во время анализа обычно поддерживается на уровне (–130)–(–160)°C, однако широко применяют экспериментальную схему с предварительным нагревом образца до –80°C на 30 мин со скоростью 5 град/мин, предложенную в работах [64, 65] и использующуюся другими авторами [66, 67]. Достоинством предложенного протокола является удаление избытка конденсированной воды на поверхности образца, происходящее при –80°C. При этом не наблюдается изменение морфологии образца и рекристаллизации воды в верхних слоях. В результате последующего понижения температуры до –100°С возможна вторичная конденсация небольшого количества воды, что увеличивает выход положительно заряженных молекулярных ионов приблизительно в два раза [64]. Описанный эффект хорошо согласуется с модельными экспериментами, в которых лед использовали в качестве подложки для аминокислот и аденина [68], а также при нанесении льда на биоматериал [69]. Считается, что механизм усиления сигнала связан с образованием дополнительных протонов при разрушении молекул воды под действием первичных ионов. Таким образом, лед можно считать очень удачной естественной “заливочной средой”.
Интересно, что в работе [63] также были обнаружены пики этана или пропана (в зависимости от среды, в которой проводили криофиксацию) на поверхности клеток сразу после начала анализа, которые через некоторое время пропадали. Вероятно, это связано с сублимацией этана и пропана в вакууме.
Дегидратация. Альтернативой анализу в замороженном состоянии является дегидратация образца. Существует по крайней мере четыре метода дегидратации, применяющихся для исследований методом ВИМС (табл. 2).
Таблица 2.
Методы дегидратации, применявшиеся для исследований методом ВИМС
| Название | Условия | Использование растворителей |
|---|---|---|
| В потоке газа | Атмосфера, поток инертного газа | – |
| Лиофилизация | Вакуум | – |
| Серия спиртов | Атмосфера | + |
| В критической точке | Состояние фазового перехода “жидкость–газ” | + |
При дегидратации в сверхкритической жидкости и дегидратации через серию спиртов с повышающейся концентрацией используют растворители – ацетон, этанол, ксилол. Показано, что использование таких растворителей приводит к значительному снижению сигнала липидов, поэтому их применение нежелательно [54, 67]. Предложено использовать обдув аргоном с объемной скоростью 12–15 см3/с [61]. Вместо аргона можно использовать любой другой химически чистый газ (например, азот). Предварительно следует отмыть инкубационный раствор; необходимость этого обсуждалась выше. Дегидратацию в потоке газа, как правило, применяют в комбинации с химической фиксацией при атмосферном давлении и комнатной температуре, тогда как лиофилизацию проводят в вакууме после криофиксации. Суть процесса заключается в сублимации воды из твердого состояния (лед) в пар, минуя жидкую фазу. В процессе лиофилизации важно не допустить повышения температуры образца выше –80°C, что приведет к рекристаллизации льда и бо́льшему повреждению внутриклеточных структур.
Следует отметить, что дегидратация приводит к некоторому изменению толщины и формы клетки [70], поскольку удаляется основной компонент клетки – вода. Данный фактор несколько ограничивает применение метода для исследований органелл, содержащих большое количество воды, например цитоплазмы. Для подобных исследований предпочтительнее использовать замороженные образцы или методы, позволяющие прижизненный анализ.
Замораживание–скалывание. После замораживания–скалывания образца анализ поверхности методом ВИМС проводят в замороженном состоянии или в лиофилизированной форме, однако в обоих случаях удаляют внешний слой плазматической мембраны клетки. Для этого клетки выращивают на кремниевой подложке, пока они не заполнят порядка 70% поверхности. Затем на подложку наносят полистирольные шарики, соразмерные клеткам, и клетки накрывают второй кремниевой пластиной. Полученный “сэндвич” замораживают в жидком пропане или этане, после чего разъединяют. В результате разделения часть клеток остается на одной подложке, часть на другой. Замороженные клетки разламываются достаточно произвольным образом и их части остаются на обеих подложках. Липидный бислой плазматической мембраны клетки, как правило, разламывается на две комплементарные поверхности [17, 71]. Таким образом, внутренняя часть бислоя остается вместе с клеткой, а наружная – на противоположной пластине. Скалывание можно проводить в вакууме или в среде криоагента, а также при помощи специальных устройств в камере предподготовки образца масс-спектрометра [72].
Сравнение методов и новые подходы. Одной из первых публикаций, посвященных сравнению различных методов пробоподготовки для ВИМС, была работа [54] по сравнению фибробластов, приготовленных при помощи криофиксации с последующей лиофилизацией и фиксацией глутаровым альдегидом с дегидратацией через серию спиртовых растворов с повышающейся концентрацией. Оба метода фиксации не привели к изменению морфологии клетки, однако при криофиксации распределение легко диффундирующих ионов (Na+, K+) оказалось ближе к прижизненному состоянию. Кроме того, сигнал фосфатидилхолина (одного из главных липидных компонентов плазматической мембраны клетки) уменьшился в ~70 раз после дегидратации через серию спиртов.
Для фибробластов сравнили три протокола: химической фиксации с дегидратацией в потоке воздуха, криофиксации с последующей лиофилизацией, анализа в замороженном состоянии [66]. Сигнал органических ионов в лиофилизированных клетках в среднем был в 2.6 раза меньше, чем в фиксированных формалином. Авторы предположили, что в процессе отмывки клеток от солей и формальдегида из клеток вымываются хлорид-ионы, которые “подавляют” выход положительных вторичных ионов. Тем не менее, значимых отличий в масс-спектрах не обнаружено, т.е. вымывания внутриклеточных органических ионов не происходило. Напротив, при сравнении лиофилизированных и замороженных клеток наблюдаются значительные отличия, поскольку в замороженном состоянии регистрировали больше пиков, большинство из которых относится к кластерам воды и внутриклеточных солей. Для ионов с низкой молекулярной массой различия в интенсивности сигнала незначительны, в то время как для положительно заряженных молекулярных ионов с высокой массой выход ионов выше в замороженном состоянии, чем в дегидратированном. По-видимому, это объясняется тем, что, во-первых, при криогенных температурах снижается уровень повреждений, наносимых ионной бомбардировкой, а во-вторых, лед является источником протонов для образования протонированной молекулы [M + H]+. При этом обнаружено существенное (в разы) падение интенсивности отрицательных ионов по сравнению с лиофилизированными клетками. По сравнению с лиофилизированным препаратом у замороженного образца обнаружено существенное падение интенсивности отрицательных ионов (в разы), что соответствует выводам теоретических исследований, согласно которым лед уменьшает выход отрицательно заряженных вторичных ионов [73, 74].
В работе [67] проанализированы масс-спектры мезенхимальных стволовых клеток для большого числа различных методов пробоподготовки при помощи метода главных компонент. Авторы использовали три возможных варианта химической фиксации: 4%-ным формалином, 2.5%-ным глутаровым альдегидом и смесью данных альдегидов с концентрациями 2 и 1% соответственно. Выходы вторичных ионов липидов оказались близкими, но наблюдались и некоторые отклонения для разных ионов. Показано, что использование растворителей при дегидратации значительно снижает выход липидных ионов. Вторичная фиксация липидов тетраоксидом осмия не увеличивает выход вторичных ионов липидов при использовании растворителей. Кроме того, обработка OsO4 приводит к появлению пиков с m/z > 300, которые интерферируют с пиками органических компонентов клетки. Авторы пришли к заключению о нецелесообразности использования оксида осмия. Выход ионов липидов при анализе в замороженном состоянии оказался значительно ниже, чем для лиофилизированных и химически фиксированных образцов, высушенных в потоке газа, что несколько противоречит выводам работы [66].
Предложен модифицированный протокол, использующий дегидратацию через серию спиртов [70]. Кроме того, оценивали изменение профиля клеток меланомы при помощи атомно-силовой микроскопии в зависимости от стадии протокола. Форма клетки оставалась неизменной, тогда как высота клетки изменялась. Высота клетки увеличилась на 30–40% после фиксации и промывки деионизированной водой. При фиксации формальдегидом наблюдали и некоторые изменения морфологии клетки, в частности, исчезали видимые актиновые филаменты. После дегидратации через серию спиртов высота клетки уменьшилась до прижизненного значения. Последующее испарение растворителя вызвало сжатие клетки в несколько раз, в результате через плазматическую мембрану стали отчетливо видны контуры ядра.
Предложен и охарактеризован метод получения ионных изображений срезов одиночных клеток на примере GV-ооцитов мыши [16, 75, 76]. Ключевой особенностью данного метода является возможность параллельно исследовать один и тот же образец методами ВИМС, оптической, электронной и зондовой микроскопии, а также электронно-зондового микроанализа. Схематично последовательность пробоподготовки представлена на рис. 3. В основу метода положена криофиксация в жидком пропане с последующей низкотемпературной дегидратацией в вакууме. Высушенные клетки заключали в заливочную среду Epon 812. После ее полимеризации при помощи ультрамикротома получали серию последовательных срезов ооцита толщиной 2 мкм, что позволяет отбирать срез, плоскость которого проходит через нужную структуру клетки. Следовательно, нет необходимости проводить процедуру ионного травления для доступа к внутриклеточным органеллам. Поверхность среза можно считать достаточно плоской, шероховатость поверхности варьируется в зависимости от зоны внутри клетки, но не превышает 100 нм [16, 75]. Как показывают исследования артефактов, возникающих вследствие рельефа поверхности, перепадами высот менее 1 мкм можно пренебречь [77, 78], в отличие от случая, когда клетки высушиваются на поверхности подложки. К негативным моментам метода стоит отнести присутствие ионов заливочной среды в масс-спектрах, что затрудняет их интерпретацию.

Анализ срезов тканей. Ключевым фактором пробоподготовки биологических тканей является размер образца, что вынуждает использовать микротомию или криомикротомию замороженного сегмента исследуемого образца [79]. Остановимся более подробно на некоторых деталях данного протокола.
Микротомия и заливка. Микротомия сегмента органа или органа необходима по ряду причин. Во-первых, ВИМС наиболее эффективно работает в случае плоской поверхности образцов, что особенно критично для проведения химического картирования [77, 78]. Во-вторых, в отличие от клетки, образцы органов и тканей существенно больше по размеру, поэтому ионное травление образца до нужной глубины займет много времени. В-третьих, тонкие срезы обеспечивают существенно лучший сток статического заряда, накапливающегося в результате бомбардировки первичными ионами.
Для получения среза наиболее распространена криотомия образца ткани в криостате при низкой температуре. Криосрезы обычно получают при ‒20°C. Толщина криосреза варьирует от 5 до 50 мкм. При кажущейся простоте метода затруднение вызывает механическое закрепление замороженной ткани на держателе криомикротома. Для этого на поверхность держателя наносят каплю специального состава, в которую погружают образец ткани. Затем весь комплекс замораживают в криоагенте. Наиболее распространенными являются следующие заливочные среды: OCT [80], карбоксиметилцеллюлоза [81], желатин [82, 83] или лед [84]. Карбоксиметилцеллюлозу чаще применяют для более объемных образцов, а желатин, напротив, для мелких [85]. К сожалению, при криотомии перечисленные вещества плавятся на кромке лезвия ножа, что приводит к их размазыванию по поверхности криосреза [80, 86].
Дегидратация. Как и заливка, дегидратация является необязательным этапом. Образец можно анализировать и в замороженном виде без удаления воды. Более того, лед является очень удачной “естественной заливочной средой”, поскольку молекулы воды служат источником протонов, способных повысить выход протонированных молекул [87]. Однако сложность манипуляций с замороженными недегидратированными препаратами и криосрезами ограничивает их применение. Наибольшую опасность представляет риск конденсации влаги из воздуха на поверхности образца. В связи с этим распространение получили лиофилизированные криосрезы. Лиофилизацию криосреза проводят в вакуумной камере в течение 1–3 ч. Важно обеспечить при этом максимально медленное изменение температуры образца. В результате низкотемпературной дегидратации толщина среза может уменьшиться по-разному в зависимости от типа ткани, в том числе и значительно (в 10 раз) [88, 89]. Как и для клеточных культур, другими вариантами дегидратации могут быть использование серии спиртов с повышающейся концентрацией и обдув газом при комнатной температуре.
Нанесение криосреза на подложку. Одним из самых распространенных методов закрепления замороженного среза на охлажденную до той же температуры подложку является плавление зоны криосреза вследствие локального разогрева, вызванного теплом приложенного пальца к обратной стороне подложки. Лед плавится, а затем замерзает заново, что способствует адгезии среза к поверхности. Другой подход основан на использовании клейкой ленты, на которую помещают образец [90]. Даже такой на первый взгляд незначительный элемент протокола приводит к значительным изменениям биохимического состава препарата. При исследовании лиофилизированных срезов мозга крыс при помощи травления ионами ${\text{Ar}}_{{1500}}^{ + }$ показано, что сигнал холестерина уменьшается с увеличением глубины после локального разогрева криосреза [4]. При использовании клейкой ленты для механической фиксации среза сигнал остается примерно на постоянном уровне. Таким образом, локальный разогрев приводит к перераспределению холестерина в поверхностные слои ткани. Интересно, что некоторые другие липиды, в частности, фосфатидилхолин, сохраняют распределение в обоих случаях. Перераспределение холестерина в поверхностные слои может приводить к изменению интенсивности других вторичных ионов липидов. Например, в модельных экспериментах отмечено его влияние на ионы фосфатидилхолина, сфингомиелина, церамида, триацилглицеридов и витамина Е [89, 91].
В сравнительном исследовании срезов мозга Drosophila [92] использовали три типа образцов: два аналогичные работе со срезами мозга крысы и препарат в замороженном виде, приклеенный на клейкую ленту. Установлено различное распределение триацилглицеридов, диацилглицеридов и жирных кислот в латеральной плоскости при разных методах нанесения срезов. Кроме того, после локального разогрева, вызванного приложением пальца к обратной стороне подложки, размер мозга на срезе уменьшился на 30% по сравнению с креплением криосреза на клейкую ленту. По мнению авторов, из-за локального разогрева влага могла накопиться на поверхности ткани, а при последующей повторной заморозке целостность клеточных мембран была нарушена и влага с поверхности проникла внутрь поврежденных клеток. По этой причине сигнал триацилглицеридов и других молекул обнаружен в тех тканях, в которых его не было изначально. Вместе с тем распределение положительных ионов в лиофилизированном и замороженных срезах, приклеенных на клейкую ленту, аналогично. Данный факт свидетельствует об отсутствии влияния лиофилизации на локализацию липидов.
Таким образом, параметры ионизации и пробоподготовка являются двумя важнейшими факторами, определяющими применимость ВИМС для анализа клеток и тканей. К сожалению, универсальных решений, одинаково хорошо подходящих для всех типов образцов и решаемых задач, не существует. Что касается выбора источника ионизации для серийных аппаратов, то, вероятно, лучшим выбором для спектроскопических исследований будет ${\text{C}}_{{60}}^{ + },$ для химического картирования –${\text{Bi}}_{3}^{ + },$ для ионного травления –${\text{Ar}}_{n}^{ + }.$ Неоднозначная ситуация наблюдается и для пробоподготовки образцов. Препараты клеток и тканей сохраняют параметры, близкие к прижизненному состоянию, после криофиксации. Однако в этом случае манипуляции с замороженным объектом и его анализ при низких температурах требуют определенной квалификации и весьма трудоемки. Альтернативой представляется лиофилизациия криофиксированного материала, после чего можно работать с препаратом при комнатной температуре. Сравнение лиофилизированных образцов культур клеток и тканей с замороженными показывает, что химический состав и его распределение в процессе низкотемпературной дегидратации сохраняются. Другим альтернативным вариантом подготовки клеточных культур является химическая фиксация формалином или глутаровым альдегидом с последующей дегидратацией в потоке газа. Следует иметь в виду, что, скорее всего, распределение и внутриклеточная концентрация легко диффундирующих катионов и анионов (Na+, K+, Cl–) при этом не сохранятся. С другой стороны, вымывание внутриклеточных солей приведет к усилению сигналов липидно-белковых компонентов клетки. Напротив, использование для дегидратации растворителей, в частности, спиртов, нежелательно ввиду негативного влияния на интенсивность сигналов вторичных ионов липидов. Применение оксида осмия(VIII) также не показало практической целесообразности. Таким образом при выборе методических приемов и параметров при анализе ВИМС следует исходить из приоритетов конкретного исследования, его цели и свойств анализируемого вещества.
Работа выполнена при поддержке гранта РФФИ № 18-33-00940, а также гранта РНФ № 17‑76-20014 (раздел “Анализ срезов тканей”) в рамках государственного задания “Химико–физические механизмы взаимодействия интенсивного лазерного излучения с биологическими системами” (АААА-А19-119012990175-9).
Список литературы
Tian H., Sparvero L.J., Amoscato A.A., Bloom A., Bayir H., Kagan V.E., Winograd N. Gas cluster ion beam time-of-flight secondary ion mass spectrometry high-resolution imaging of cardiolipin speciation in the brain: Identification of molecular losses after traumatic injury // Anal. Chem. 2017. V. 89. № 8. P. 4611.
Veith L., Vennemann A., Breitenstein D., Engelhard C., Wiemann M., Hagenhoff B. Detection of SiO2 nanoparticles in lung tissue by ToF-SIMS imaging and fluorescence microscopy // Analyst. 2017. V. 142. № 14. P. 2631.
Pour M.D., Jennische E., Lange S., Ewing A.G., Malmberg P. Food-induced changes of lipids in rat neuronal tissue visualized by ToF-SIMS imaging // Sci. Rep. 2016. V. 6. Article 32797.
Shon H.K., Kim S.H., Yoon S., Shin C.Y., Lee T.G. Molecular depth profiling on rat brain tissue sections prepared using different sampling methods // Biointerphases. 2018. V. 13. № 3. Article 03B411.
Wehrli P.M., Angerer T.B., Farewell A., Fletcher J.S., Gottfries J. Investigating the role of the stringent response in lipid modifications during the stationary phase in e. Coli by direct analysis with time-of-flight-secondary ion mass spectrometry // Anal. Chem. 2016. V. 88. № 17. P. 8680.
Гулин А.А., Кокшарова О.А., Попова А.А., Хмель И.А., Астафьев А.А., Шахов А.М., Надточенко В.А. Визуализация серебра в клетках цианобактерий Anabaena sp. PCC 7120 методами времяпролетной масс-спектроскопии вторичных ионов и двухфотонной люминесцентной микроскопии // Российские нанотехнологии. 2016. Т. 11. № 5–6. С. 72.
Angerer T.B., Magnusson Y., Landberg G., Fletcher J.S. Lipid heterogeneity resulting from fatty acid processing in the human breast cancer microenvironment identified by GCIB-ToF-SIMS imaging // Anal. Chem. 2016. V. 88. № 23. P. 11946.
Newman C.F., Hayelund R., Passarelli M.K., Marshall P.S., Francis I., West A., Alexander M.R., Gilmore I.S., Dollery C.T. Intracellular drug uptake a comparison of single cell measurements using tof-sims imaging and quantification from cell populations with LC/MS/MS // Anal. Chem. 2017. V. 89. № 22. P. 11944.
Sheng L.F., Cai L.S., Wang J., Li Z.P., Mo Y.X., Zhang S.C., Xu J.J., Zhang X.R., Chen H.Y. Simultaneous imaging of newly synthesized proteins and lipids in single cell by TOF-SIMS // Int. J. Mass Spectrom. 2017. V. 421. P. 238.
Converso V., Fearn S., Ware E., McPhail D.S., Flemming A.J., Bundy J.G. Analysis and imaging of biocidal agrochemicals using ToF-SIMS // Sci. Rep. 2017. V. 7. Article 10728.
Kyriacou B., Moore K.L., Paterson D., de Jonge M.D., Howard D.L., Stangoulis J., Tester M., Lombi E., Johnson A.A.T. Localization of iron in rice grain using synchrotron X-ray fluorescence microscopy and high resolution secondary ion mass spectrometry // J. Cereal Sci. 2014. V. 59. № 2. P. 173.
Saito K., Watanabe Y., Shirakawa M., Matsushita Y., Imai T., Koike T., Sano Y., Funada R., Fukazawa K., Fukushima K. Direct mapping of morphological distribution of syringyl and guaiacyl lignin in the xylem of maple by time-of-flight secondary ion mass spectrometry // Plant J. 2012. V. 69. № 3. P. 542.
Moore K.L., Chen Y., van de Meene A.M.L., Hughes L., Liu W.J., Geraki T., Mosselmans F., McGrath S.P., Grovenor C., Zhao F.J. Combined NanoSIMS and synchrotron X-ray fluorescence reveal distinct cellular and subcellular distribution patterns of trace elements in rice tissues // New Phytol. 2014. V. 201. № 1. P. 104.
Vickerman J.C. Molecular Surface Mass Spectrometry by SIMS. Singapore: John Wiley & Sons, 2009. P. 113.
Breitenstein D., Hagenhoff B., Schnieders A. Biomolecular Analysis by Time-of-Flight Secondary Ion Mass Spectrometry (ToF-SIMS). Cham, Switzerland: Springer, 2014. P. 35.
Gulin A., Nadtochenko V., Astafiev A., Pogorelova V., Rtimi S., Pogorelov A. Correlating microscopy techniques and ToF-SIMS analysis of fully grown mammalian oocytes // Analyst. 2016. V. 141. № 13. P. 4121.
Draude F., Korsgen M., Pelster A., Schwerdtle T., Muthing J., Arlinghaus H.F. Characterization of freeze-fractured epithelial plasma membranes on nanometer scale with ToF-SIMS // Anal. Bioanal. Chem. 2015. V. 407. № 8. P. 2203.
Touboul D., Laprevote O., Brunelle A. Micrometric molecular histology of lipids by mass spectrometry imaging // Curr. Opin. Chem. Biol. 2011. V. 15. № 5. P. 725.
Touboul D., Brunelle A. What more can TOF-SIMS bring than other MS imaging methods? // Bioanalysis. 2016. V. 8. № 5. P. 367.
Vickerman J.C. Molecular imaging and depth profiling by mass spectrometry-SIMS, MALDI or DESI? // Analyst. 2011. V. 136. № 11. P. 2199.
Chini C.E., Fisher G.L., Johnson B., Tamkun M.M., Kraft M.L. Observation of endoplasmic reticulum tubules via TOF-SIMS tandem mass spectrometry imaging of transfected cells // Biointerphases. 2018. V. 13. № 3. Article 03B409.
Henss A., Otto S.K., Schaepe K., Pauksch L., Lips K.S., Rohnke M. High resolution imaging and 3D analysis of Ag nanoparticles in cells with ToF-SIMS and delayed extraction // Biointerphases. 2018. V. 13. № 3. Article 03B410.
Robinson M.A., Graham D.J., Castner D.G. ToF-SIMS depth profiling of cells: z-correction, 3d imaging, and sputter rate of individual NIH/3T3 fibroblasts // Anal. Chem. 2012. V. 84. № 11. P. 4880.
Vanbellingen Q.P., Castellanos A., Rodriguez-Silva M., Paudel I., Chambers J.W., Fernandez-Lima F.A. Analysis of chemotherapeutic drug delivery at the single cell level using 3D-MSI-TOF-SIMS // J. Am. Soc. Mass Spectrom. 2016. V. 27. № 12. P. 2033.
Fornai L., Angelini A., Klinkert I., Giskes F., Kiss A., Eijkel G., Hove E.A.A.-v., Klerk L.A., Fedrigo M., Pieraccini G., Moneti G., Valente M., Thiene G., Heeren R.M.A. Three-dimensional molecular reconstruction of rat heart with mass spectrometry imaging // Anal. Bioanal. Chem. 2012. V. 404. № 10. P. 2927.
Masaki N., Ishizaki I., Hayasaka T., Fisher G.L., Sanada N., Yokota H., Setou M. Three-dimensional image of cleavage bodies in nuclei is configured using gas cluster ion beam with time-of-flight secondary ion mass spectrometry // Sci. Rep. 2015. V. 5. Article 1000.
Dowsett M.G. Depth profiling using ultra-low-energy secondary ion mass spectrometry // Appl. Surf. Sci. 2003. V. 203–204. P. 5.
Liu R., Ng C.M., Wee A.T.S. Surface roughening effect in sub-keV SIMS depth profiling // Appl. Surf. Sci. 2003. V. 203–204. P. 256.
Rees E.E., McPhail D.S., Ryan M.P., Kelly J., Dowsett M.G. Low energy SIMS characterisation of ultra thin oxides on ferrous alloys // Appl. Surf. Sci. 2003. V. 203–204. P. 660.
Touboul D., Kollmer F., Niehuis E., Brunelle A., Laprevote O. Improvement of biological time-of-flight-secondary ion mass spectrometry imaging with a bismuth cluster ion source // J. Am. Soc. Mass Spectrom. 2005. V. 16. № 10. P. 1608.
Kersting R., Hagenhoff B., Kollmer F., Mollers R., Niehuis E. Influence of primary ion bombardment conditions on the emission of molecular secondary ions // Appl. Surf. Sci. 2004. V. 231–232. P. 261.
Гулин А.А., Павлюков М.С., Гусев С.А., Малахова Ю.Н., Бузин А.И., Чвалун С.Н., Алдаров К.Г., Клинов Д.В., Гуларян С.К., Надточенко В.А. Возможности применения метода TOF-SIMS для оценки липидного состава мембранных структур клетки // Биологические мембраны. 2017. Т. 34. № 3. С. 215.
Van der Heide P. Secondary ion mass spectrometry: an introduction to principles and practices. Canada: John Wiley & Sons, 2014. 365 p.
van Hove E.R.A., Smith D.F., Heeren R.M.A. A concise review of mass spectrometry imaging // J. Chromatogr. A. 2010. V. 1217. № 25. P. 3946.
Postawa Z., Czerwinski B., Szewczyk M., Smiley E.J., Winograd N., Garrison B.J. Enhancement of sputtering yields due to c60 versus ga bombardment of ag{111} as explored by molecular dynamics simulations // Anal. Chem. 2003. V. 75. № 17. P. 4402.
Paruch R., Rzeznik L., Russo M.F., Garrison B.J., Postawa Z. Molecular dynamics study of the effect of surface topography on sputtering induced by 20 kev au3 and c60 clusters // J. Phys. Chem. C. 2010. V. 114. № 12. P. 5532.
Muramoto S., Brison J., Castner D.G. Exploring the surface sensitivity of tof-secondary ion mass spectrometry by measuring the implantation and sampling depths of bi-n and c-60 ions in organic films // Anal. Chem. 2012. V. 84. № 1. P. 365.
Kollmer F. Cluster primary ion bombardment of organic materials // Appl. Surf. Sci. 2004. V. 231–232. P. 153.
Brunelle A., Touboul D., Laprevote O. Biological tissue imaging with time-of-flight secondary ion mass spectrometry and cluster ion sources // J. Mass Spectrom. 2005. V. 40. № 8. P. 985.
Mahoney C.M. Cluster Secondary Ion Mass Spectrometry: Principles and Applications. Hoboken, New Jersey: John Wiley & Sons, 2013. 350 p.
Kubicek M., Holzlechner G., Opitz A.K., Larisegger S., Hutter H., Fleig J. A novel ToF-SIMS operation mode for sub 100nm lateral resolution: Application and performance // Appl. Surf. Sci. 2014. V. 289. P. 407.
Fletcher J.S., Lockyer N.P., Vaidyanathan S., Vickerman J.C. TOF-SIMS 3D biomolecular imaging of Xenopus laevis oocytes using buckminsterfullerene C60 primary ions // Anal. Chem. 2007. V. 79. № 6. P. 2199.
Angerer T.B., Blenkinsopp P., Fletcher J.S. High energy gas cluster ions for organic and biological analysis by time-of-flight secondary ion mass spectrometry // Int. J. Mass Spectrom. 2015. V. 377. P. 591.
Muramoto S., Graham D.J., Wagner M.S., Lee T.G., Moon D.W., Castner D.G. ToF-SIMS analysis of adsorbed proteins: principal component analysis of the primary ion species effect on the protein fragmentation patterns // J. Phys. Chem. C. 2011. V. 115. № 49. P. 24 247.
Garrison B.J., Postawa Z. Molecular Dynamics Simulations, the Theoretical Partner to Dynamic Cluster SIMS Experiments. Chichester, UK: IM Publications, 2013. P 732.
Ichiki K., Ninomiya S., Nakata Y., Honda Y., Seki T., Aoki T., Matsuo J. High sputtering yields of organic compounds by large gas cluster ions // Appl. Surf. Sci. 2008. V. 255. № 4. P. 1148.
Wehbe N., Mouhib T., Delcorte A., Bertrand P., Moellers R., Niehuis E., Houssiau L. Comparison of fullerene and large argon clusters for the molecular depth profiling of amino acid multilayers // Anal. Bioanal. Chem. 2014. V. 406. № 1. P. 201.
Wehbe N., Tabarrant T., Brison J., Mouhib T., Delcorte A., Bertrand P., Moellers R., Niehuis E., Houssiau L. TOF-SIMS depth profiling of multilayer amino-acid films using large Argon cluster Ar-n(+), C-60(+) and Cs+ sputtering ions: A comparative study // Surf. Interface Anal. 2013. V. 45. № 1. P. 178.
Rabbani S.S.N., Razo I.B., Kohn T., Lockyer N.P., Vickerman J.C. Enhancing ion yields in time-of-flight-secondary ion mass spectrometry: a comparative study of argon and water cluster primary beams // Anal. Chem. 2015. V. 87. № 4. P. 2367.
Eller M.J., Vinjamuri A., Tomlin B.E., Schweikert E.A. Molecular colocalization using massive gold cluster secondary ion mass spectrometry // Anal. Chem. 2018. V. 90. № 21. P. 12692.
Wucher A., Tian H., Winograd N. A mixed cluster ion beam to enhance the ionization efficiency in molecular secondary ion mass spectrometry // Rapid Commun. Mass Spectrom. 2014. V. 28. № 4. P. 396.
Puchtler H., Meloan S.N. On the chemistry of formaldehyde fixation and its effects on immunohistochemical reactions // Histochemistry. 1985. V. 82. № 3. P. 201.
Walt D.R., Agayn V.I. The chemistry of enzyme and protein immobilization with glutaraldehyde // Trends Anal. Chem. 1994. V. 13. № 10. P. 425.
Malm J., Giannaras D., Riehle M.O., Gadegaard N., Sjovall P. Fixation and drying protocols for the preparation of cell samples for time-of-flight secondary ion mass spectrometry analysis // Anal. Chem. 2009. V. 81. № 17. P. 7197.
Passarelli M.K., Winograd N. Lipid imaging with time-of-flight secondary ion mass spectrometry (ToF-SIMS) // Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2011. V. 1811. № 11. P. 976.
Zierold K. Cryofixation methods for ion localization in cells by electron-probe microanalysis – A review // J. Microsc. (Oxford, U. K.). 1991. V. 161. P. 357.
Pogorelov A.G., Katkov I.I., Pogorelova V.N. Influence of exposure to vitrification solutions on 2-cell mouse embryos: I. Intracellular potassium and sodium content // Cryoletters. 2007. V. 28. № 6. P. 403.
Studer D., Hennecke H., Muller M. High-pressure freezing of soybean nodules leads to an improved preservation of ultrastructure // Planta. 1992. V. 188. № 2. P. 155.
Piwowar A.M., Lockyer N.P., Vickerman J.C. Salt effects on ion formation in desorption mass spectrometry: an investigation into the role of alkali chlorides on peak suppression in time-of-flight-secondary ion mass spectrometry // Anal. Chem. 2009. V. 81. № 3. P. 1040.
Jones E.A., Lockyer N.P., Vickerman J.C. Depth profiling brain tissue sections with a 40 keV C-60(+) primary ion beam // Anal. Chem. 2008. V. 80. № 6. P. 2125.
Berman E.S.F., Fortson S.L., Checchi K.D., Wu L., Felton J.S., Wu K.J.J., Kulp K.S. Preparation of single cells for imaging/profiling mass spectrometry // J. Am. Soc. Mass Spectrom. 2008. V. 19. № 8. P. 1230.
Nygren H., Eriksson C., Malmberg P., Sahlin H., Carlsson L., Lausmaa J., Sjovall P. A cell preparation method allowing subcellular localization of cholesterol and phosphocholine with imaging TOF-SIMS // Colloids Surf., B. 2003. V. 30. № 1–2. P. 87.
Piwowar A.M., Keskin S., Delgado M.O., Shen K., Hue J.J., Lanekoff I., Ewing A.G., Winograd N. C60-ToF SIMS imaging of frozen hydrated HeLa cells // Surf. Interface Anal. 2013. V. 45. № 1. P. 302.
Piehowski P.D., Kurczy M.E., Willingham D., Parry S., Heien M.L., Winograd N., Ewing A.G. Freeze-etching and vapor matrix deposition for ToF-SIMS imaging of single cells // Langmuir. 2008. V. 24. № 15. P. 7906.
Kurczy M.E., Piehowski P.D., Parry S.A., Jiang M., Chen G., Ewing A.G., Winograd N. Which is more important in bioimaging SIMS experiments-The sample preparation or the nature of the projectile? // Appl. Surf. Sci. 2008. V. 255. № 4. P. 1298.
Robinson M.A., Castner D.G. Characterization of sample preparation methods of NIH/3T3 fibroblasts for ToF-SIMS analysis // Biointerphases. 2013. V. 8. Article 15.
Schaepe K., Kokesch-Himmelreich J., Rohnke M., Wagner A.S., Schaaf T., Wenisch S., Janek J. Assessment of different sample preparation routes for mass spectrometric monitoring and imaging of lipids in bone cells via ToF-SIMS // Biointerphases. 2015. V. 10. № 1. Article 019016.
Conlan X.A., Lockyer N.P., Vickerman J.C. Is proton cationization promoted by polyatomic primary ion bombardment during time-of-flight secondary ion mass spectrometry analysis of frozen aqueous solutions? // Rapid Commun. Mass Spectrom. 2006. V. 20. № 8. P. 1327.
Aoki N., Komatsu M., Kyogaku M., Hashimoto H. Enhancing peptide molecular imaging by depositing ice in time-of-flight secondary ion mass spectrometry // Int. J. Mass Spectrom. 2013. V. 351. P. 1.
Bobrowska J., Pabijan J., Wiltowska-Zuber J., Jany B.R., Krok F., Awsiuk K., Rysz J., Budkowski A., Lekka M. Protocol of single cells preparation for time of flight secondary ion mass spectrometry // Anal. Biochem. 2016. V. 511. P. 52.
Draude F., Galla S., Pelster A., Tentschert J., Jungnickel H., Haase A., Mantion A., Thunemann A.F., Taubert A., Luch A., Arlinghaus H.F. ToF-SIMS and Laser-SNMS analysis of macrophages after exposure to silver nanoparticles // Surf. Interface Anal. 2013. V. 45. № 1. P. 286.
Lanekoff I., Kurczy M.E., Adams K.L., Malm J., Karlsson R., Sjovall P., Ewing A.G. An in situ fracture device to image lipids in single cells using ToF-SIMS // Surf. Interface Anal. 2011. V. 43. № 1–2. P. 257.
Wojciechowski I.A., Kutliev U., Sun S.X., Szakal C., Winograd N., Garrison B.J. Emission of ionic water clusters from water ice films bombarded by energetic projectiles // Appl. Surf. Sci. 2004. V. 231. P. 72.
Wojciechowski I.A., Sun S.X., Szakal C., Winograd N., Garrison B.J. Ion emission from water ice due to energetic particle bombardment // J. Phys. Chem. A. 2004. V. 108. № 15. P. 2993.
Astafiev A.A., Gulin A.A., Osychenko A.A., Solodina A.E., Syrchina M.S., Titov A.A., Shakhov A.M., Pogorelov A.G., Pogorelova V.N., Panait A.I., Nadtochenko V.A. Structural features of the nucleolus in the mouse germinal vesicle oocyte revealed by AFM, SEM, and ToF-SIMS // Russ. Nanotechnol. 2017. V. 12. № 7–8. P. 444.
Погорелов А.Г., Гулин А.А., Погорелова В.Н., Панаит А.И., Погорелова М.А., Надточенко В.А. Особенности применения метода TOF-SIMS для анализа биоорганических образцов // Биофизика. 2018. Т. 63. № 2. С. 303.
Rangarajan S., Tyler B.J. Topography in secondary ion mass spectroscopy images // J. Vac. Sci. Technol., A. 2006. V. 24. № 5. P. 1730.
Lee J.L.S., Gilmore I.S., Seah M.P., Fletcher I.W. Topography and field effects in secondary ion mass spectrometry - part i: conducting samples // J. Am. Soc. Mass Spectrom. 2011. V. 22. № 10. P. 1718.
Yoon S., Lee T.G. Biological tissue sample preparation for time-of-flight secondary ion mass spectrometry (ToF-SIMS) imaging // Nano Converg. 2018. V. 5. Article 24.
Zaima N., Hayasaka T., Goto-Inoue N., Setou M. Matrix-assisted laser desorption/ionization imaging mass spectrometry // Int. J. Mol. Sci. 2010. V. 11. № 12. P. 5041.
Hansen H.T., Janfelt C. Aspects of quantitation in mass spectrometry imaging investigated on cryo-sections of spiked tissue homogenates // Anal. Chem. 2016. V. 88. № 23. P. 11513.
Chen R.B., Hui L.M., Sturm R.M., Li L.J. Three dimensional mapping of neuropeptides and lipids in crustacean brain by mass spectral imaging // J. Am. Soc. Mass Spectrom. 2009. V. 20. № 6. P. 1068.
Gemperline E., Jayaraman D., Maeda J., Ane J.M., Li L.J. Multifaceted investigation of metabolites during nitrogen fixation in medicago via high resolution MA-LDI-MS imaging and ESI-MS // J. Am. Soc. Mass Spectrom. 2015. V. 26. № 1. P. 149.
Khatib-Shahidi S., Andersson M., Herman J.L., Gillespie T.A., Caprioli R.M. Direct molecular analysis of whole-body animal tissue sections by imaging MALDI mass spectrometry // Anal. Chem. 2006. V. 78. № 18. P. 6448.
Goodwin R.J.A. Sample preparation for mass spectrometry imaging: Small mistakes can lead to big consequences // J. Proteomics. 2012. V. 75. № 16. P. 4893.
Schwartz S.A., Reyzer M.L., Caprioli R.M. Direct tissue analysis using matrix-assisted laser desorption/ionization mass spectrometry: practical aspects of sample preparation // J. Mass Spectrom. 2003. V. 38. № 7. P. 699.
Jones E.A., Lockyer N.P., Kordys J., Vickerman J.C. Suppression and enhancement of secondary ion formation due to the chemical environment in static-secondary ion mass spectrometry // J. Am. Soc. Mass Spectrom. 2007. V. 18. № 8. P. 1559.
Anthony A., Colurso G.J., Bocan T.M.A., Doebler J.A. Interferometric analysis of intrasection and intersection thickness variability associated with cryostat microtomy // Histochem. J. 1984. V. 16. № 1. P. 61.
Bich C., Havelund R., Moellers R., Touboul D., Kollmer F., Niehuis E., Gilmore I.S., Brunelle A. Argon cluster ion source evaluation on lipid standards and rat brain tissue samples // Anal. Chem. 2013. V. 85. № 16. P. 7745.
Kawamoto T. Use of a new adhesive film for the preparation of multi-purpose fresh-frozen sections from hard tissues, whole-animals, insects and plants // Arch. Histol. Cytol. 2003. V. 66. № 2. P. 123.
Kutuzov N., Gulin A., Lyaskovskiy V., Nadtochenko V., Maksimov G. ATP-mediated compositional change in peripheral myelin membranes: a comparative raman spectroscopy and time-of-flight secondary ion mass spectrometry study // PLoS ONE. 2015. V. 10. № 11. Article e0142084.
Le M.U.T., Son J.G., Shon H.K., Park J.H., Lee S.B., Lee T.G. Comparison between thaw-mounting and use of conductive tape for sample preparation in ToF-SIMS imaging of lipids in Drosophila microRNA-14 model // Biointerphases. 2018. V. 13. Article 03B414.
Дополнительные материалы отсутствуют.
Инструменты
Журнал аналитической химии